Abstract
Obstructive sleep apnea (OSA) is a highly prevalent disease characterized by recurrent closure of the upper airway during sleep. It has a complex pathophysiology involving four main phenotypes. An abnormal upper airway anatomy is the key factor that predisposes to sleep-related collapse of the pharynx, but it may not be sufficient for OSA development. Non-anatomical traits, including (1) a compromised neuromuscular response of the upper airway to obstruction, (2) an unstable respiratory control (high loop gain), and (3) a low arousal threshold, predict the development of OSA in association with anatomical abnormalities. Current therapies for OSA, such as continuous positive airway pressure (CPAP) and oral appliances, have poor adherence or variable efficacy among patients. The search for novel therapeutic approaches for OSA, including pharmacological agents, has been pursued over the past years. New insights into OSA pharmacotherapy have been provided by preclinical studies, which highlight the importance of appropriate use of animal models of OSA, their applicability, and limitations. In the present review, we discuss potential pharmacological targets for OSA discovered using animal models.
1. Introduction
Obstructive sleep apnea (OSA) is characterized by cyclical obstruction of the upper airway causing intermittent cessation or reduction of airflow during sleep []. Epidemiological studies have shown that moderate to severe OSA affects up to 33% to 50% of the adult population [,]. Continuous positive airway pressure (CPAP) is the first-line therapy for OSA []. However, OSA patients frequently do not tolerate CPAP therapy, showing a poor adherence to treatment []. Approximately 50% of OSA patients do not use CPAP during sleep or use it insufficiently (less than 4 h/night) [,]. Second-line treatment approaches, including oral appliances and surgery of the upper airway, can be alternatives for CPAP, but their efficacy is variable. Low compliance with CPAP and the lack of efficacious alternatives highlight the importance of pharmacotherapy development for OSA. Although a great effort towards a pharmacological approach has been made over the recent years, there is still no approved medication for OSA.
At least four major traits contribute to the development of OSA [,,,,]: (1) Impaired upper airway anatomy, (2) impaired upper airway neuromuscular response, (3) respiratory control instability (loop gain), and (4) low respiratory arousal threshold (Figure 1). Anatomic predisposition is necessary but not sufficient for OSA development [], since obstructive events do not occur during wakefulness. This highlights the role of non-anatomical traits, and the potential to target these traits with pharmacotherapy. Animal models have been useful for an understanding of OSA pathogenesis and identification of potential pharmacological targets. Investigators have focused either on the development of animal models of upper airway obstruction during sleep, or modeling one of the predisposing traits (upper airway neuromuscular responses, respiratory control, or sleep continuity—Figure 1). In this review, we will discuss current insights into potential pharmacotherapy of OSA based on animal models.
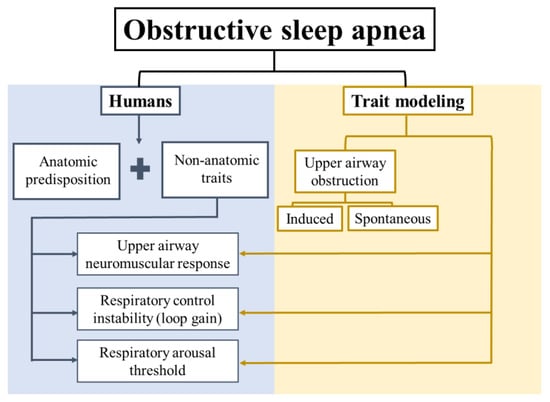
Figure 1.
Schematic representation of the anatomical and non-anatomical traits of obstructive sleep apnea (OSA) pathophysiology in humans, and a general classification of OSA traits modeling.
2. Upper Airway Anatomy or Collapsibility
2.1. Definition and Human Evidence
Anatomic predisposition is one of the key factors for the development of pharyngeal collapse and consequent OSA [,,,,]. Anatomic factors include craniofacial abnormalities and obesity. Several studies have shown that OSA patients have craniofacial morphology that predisposes to a narrow, crowded, and collapsible upper airway. Reduced pharyngeal airway space, inferiorly placed hyoid bone, and increased anterior facial height distinguished adults with OSA from controls []. Volumetric magnetic resonance imaging (MRI) showed that patients with OSA have enlarged lateral pharyngeal walls, tongue, and total soft tissues surrounding the upper airway compared to control patients []. Consequently, the average airway area and lateral and anterior–posterior dimensions were significantly smaller in OSA patients []. Shortened cranial base and altered size and position of the maxilla and mandible are also associated with narrowing of the pharyngeal area and a higher risk of OSA [,,].
Measurement of upper airway dimensions using imaging techniques is often performed during wakefulness and does not necessarily imply the functional impairment of the upper airway during sleep []. Rather, the luminal pressure at which the upper airway collapses (Pcrit) is the gold standard metric to assess of the propensity of the upper airway to close during sleep [,,,]. A negative Pcrit implies that the airway remains open, even when negative nasal pressure is applied; while a positive Pcrit implies that the airway opens only when positive airway pressure is applied. In patients with normal upper airway anatomy, Pcrit values below −5 cmH2O are necessary to protect from pharyngeal airway collapse and OSA [,]. Higher Pcrit values reflect a more collapsible upper airway and OSA patients with severe anatomical compromise generally show Pcrit values > 0–2 cmH2O [,]. Pcrit values differ depending on body position and sleep state, with increased collapsibility in the supine position and in rapid eye movement (REM) sleep [,]. Pcrit can be further divided into two more specific parameters. First, the passive Pcrit quantifies collapsibility when airway reflexes are not engaged. Passive Pcrit is solely predicated upon static upper airway anatomy. Second, the active Pcrit quantifies collapsibility when airway reflexes are engaged by flow limitation, leading to increased motor tone. The Johns Hopkins and Harvard laboratories developed a set of research techniques to measure passive and active Pcrit [].
Obesity contributes to pharyngeal collapse during sleep and is a well-known risk factor for OSA []. The adipose tissue deposition in structures surrounding the upper airway may reduce the pharyngeal airspace. Obese patients with OSA showed more fat deposition in the soft palate compared to weight-matched controls without OSA []. Patients with OSA have larger pharyngeal fat pad areas compared to control subjects [,,]. Increased tongue volumes and greater fat deposition at the base of the tongue can also be observed in obese patients with OSA compared to obese controls []. Central adiposity can also contribute to the anatomical predisposition of the upper airway to OSA. The abdominal accumulation of adipose tissue reduces lung volumes and promotes caudal traction of the pharynx, increasing the collapsibility of the upper airway [,,,]. Conversely, weight loss diminishes upper airway collapsibility. A body mass index (BMI) reduction by 17% decreased the Pcrit from 3.1 ± 4.2 to −2.4 ± 4.4 cmH2O in OSA patients [].
Although many OSA therapies attempt to reverse the dysfunction of upper airway, the anatomy of the upper airway is generally not affected by pharmacologic manipulation [], with the exception of weight loss [] as well as diuretics in heart failure patients, which can reduce rostro-caudal fluid shifts []. Nevertheless, animal models with different patterns of pharyngeal anatomic impairment have been used to test potential pharmacotherapies for OSA.
2.2. Animal Models Focused on Upper Airway Anatomic Predisposition to OSA
Spontaneous OSA has been identified in several animals []. The English bulldog has a narrow upper airway with a large soft palate and was one of the first animals exhibiting a breathing pattern similar to that of human OSA []. OSA in bulldogs was present predominantly in REM sleep. However, it was not associated with obesity. By contrast, Yucatan minipigs develop obesity-related OSA []. In addition, several animal models used mechanical upper airway obstruction [,,,,,]. An interesting feline model of OSA observed in the supine position with the neck flexed mirrors positional apnea in humans []. However, the utility of these models for pharmacotherapy is uncertain.
Obese rodents exhibit features of sleep apnea, including impaired upper airway anatomy or function, and changes in breathing patterns during sleep. Obese Zucker rats (body weight ~600 g) have reduced pharyngeal cross-sectional area during both expiration and inspiration [], and a greater amount of fat infiltration in the tongue muscle compared to non-obese Zucker rats []. New Zealand obese mice (NZO), which exhibit leptin resistance and metabolic syndrome, have increased visceral fat compared to lean mice. NZO mice have fat deposits in the pharyngeal soft tissues, tongue, soft palate, and upper airway walls []. Recently, Baum and colleagues [] showed that NZO mice had a larger number of spontaneous apneas and hypopneas than lean New Zealand black mice. However, the study did not distinguish between obstructive and central events since sleep studies were conducted without the assessment of respiratory effort.
Our group developed a system to measure Pcrit in anesthetized mice by inducing ramp decreases in nasal pressure while measuring expiratory airflow obstruction, obviating the need for monitoring tracheal pressure []. Obese mice showed a higher passive Pcrit than lean mice []. Our group also developed a plethysmography system initially described by Hernandez and colleagues [], which has the capacity of recording sleep and breathing in freely behaving mice. We have shown that both leptin-deficient ob/ob mice, diet-induced obesity mice (DIO), and NZO mice exhibit sleep disordered breathing, characterized by marked increases in inspiratory flow limitation (IFL) during REM sleep [,,]. ob/ob and DIO mice also exhibited hypoventilation, with a higher partial pressure of carbon dioxide (PaCO2) during wakefulness than lean mice [,]. Taken together with the imaging findings described above, we can conclude that obese rodents have compromised upper airway anatomy and increased collapsibility, leading to OSA. However, these rodent models may not be ideal models of OSA due to their mild phenotype (primarily flow-limited breathing and obstructive hypopneas, without complete obstructive apneas), which is predominantly expressed in REM sleep [,]. Obese rodent models have yet to be leveraged for the development of pharmacotherapy.
3. Upper Airway Neuromuscular Response
3.1. Definition and Human Evidence
OSA develops when there is inadequate neuromuscular response to pharyngeal obstruction during sleep. One of the major determinants of upper airway patency is tongue muscle tone. The tongue is composed of extrinsic and intrinsic muscle groups, both of which are innervated by the hypoglossal nerve []. Extrinsic muscles are largely responsible for the tongue movements, whereas the intrinsic muscles maintain its shape. The extrinsic tongue muscles fall in two main categories: Protrudors that move the tongue forward, including the genioglossus (GG), a major pharyngeal dilator; and retractors that move the tongue backwards towards the pharyngeal wall []. Hypoglossal motor neuron activity initiates and maintains contraction, while its absence causes relaxation, of nearly all the muscle fibers in the tongue. Pivotal work by Remmers and colleagues [] clarified the role of neuromuscular input to the GG in the pathophysiology of human OSA. The lack of GG motor input favored tongue prolapse and oropharynx occlusion while motor activation of the GG counteracted pharyngeal closure.
Lingual muscles work in synergy with the GG to maintain airway patency. In rodents, hypercapnic stimulation increased retractor (styloglossus and hypoglossus) muscle tone, suggesting that both muscle groups play a role in stabilizing tongue structures when ventilatory drive is high []. In humans, investigators documented different activation patterns between wakefulness and sleep []. At sleep onset, tongue protrudor and retractor activity decreases []. In response to airway obstruction, GG muscle activity increases above wakefulness level, and retractor activity decreases []. Studies in rats showed that tongue protrudor and retractor co-activation is superior to protrudor activation alone to maintain pharyngeal patency [,]. The role of GG tone on upper airway patency is also illustrated by hypoglossal nerve stimulation therapy, which uses an electrical impulse generator to recruit upper airway regional muscles and treat OSA [].
As in humans, the rodent tongue functions as a muscular hydrostat controlled by axonic projections of motor neurons arranged in a similar fashion in the hypoglossal nucleus. These similarities enable us to utilize rodent models to understand the role of neuromuscular responses in the pathophysiology of OSA, and to develop pharmacotherapies. Authors have focused on two main outcomes to characterize upper airway neuromuscular responses: (1) Electromyography (EMG) and (2) airflow.
3.2. Animal Models of Neuromuscular Response: EMG Outcomes
Several seminal studies have examined central control of upper airway tone in animals, with GG EMG as the primary outcome. The first evidence of hypoglossal motor neuron modulation was described in an anesthetized feline with pontine carbachol injections, a cholinergic receptor agonist, to mimic REM sleep atonia []. Reduction in hypoglossal motoneuronal activity was mediated by reduced serotonergic excitation due to the decreased activity of medullary raphe neurons []. Serotonin (5-hydroxytryptamine (5-HT)) exerted excitatory effects on hypoglossal motoneurons in anesthetized rats, whereas the withdrawal of serotonergic input caused the loss of neuromuscular input under anesthesia []. However, Sood and colleagues [] later demonstrated that inhibition of the serotonin axis failed to modulate genioglossus responses during natural non-rapid eye movement (NREM) and REM sleep.
Richard Horner’s laboratory examined the effects of noradrenergic and cholinergic drugs on GG activation during natural NREM and REM sleep. In rats, Chan and colleagues [] examined the effects of noradrenergic hypoglossal antagonism or stimulation mediated by terazosin and phenylephrine, respectively (Table 1). Perfusion of the α 1 receptor antagonist terazosin into the hypoglossal nucleus decreased GG activity while the α 1 receptor agonist phenylephrine increased GG activity during wakefulness and NREM sleep but not during REM sleep. Subsequently, Grace and colleagues [,] showed that GG muscle tone in REM sleep was maintained by muscarinic receptors, and muscarinic inhibition was functionally linked to inwardly rectifying potassium channel (GIRK) activation. The blockade of GIRK channels in the hypoglossal motor pool with barium augmented tonic and respiratory-related GG activity in rats, reversing the upper airway hypotonia during NREM and REM sleep [] (Table 1).

Table 1.
Summary of findings about potential pharmacotherapy for obstructive sleep apnea from studies using animal models.
Song and Poon [] provided another line of evidence that noradrenergic stimulation plays an important role in GG muscle control. The investigators used yohimbine, an antagonist of presynaptic α2-adrenerereceptor (i.e., a noradrenergic agonist). Yohimbine significantly increased GG muscle tone in anesthetized rats (Table 1). Taken together with the data from Horner’s lab in sleeping rats, the study provides credible evidence that noradrenergic drugs may be useful in OSA. These experiments laid the foundation for a recent human clinical trial utilizing a combination of the noradrenergic agonist, atomoxitine, and a muscarinic blocker, oxybutinine []. Nevertheless, both classes of drugs have significant systemic adverse effects on cardiovascular, gastrointestinal, and urogenital systems, limiting their clinical use.
In more recent experiments, activation of the hypoglossal motor pool was accomplished by introduction of bioengineered receptors, called designer receptors exclusively activated by designer drugs (DREADDs) []. DREADDs are G-protein-coupled human cholinergic receptors that have been genetically modified to be recognized only by specifically engineered ligands []. Fleury Curado and colleagues stereotactically injected adeno-associated viruses carrying DREADDS into the hypoglossal motor pool []. Eight weeks after virus administration, activation of these DREADDS induced significant pharynx dilation throughout the respiratory cycle (Table 1). These results re-emphasize the principle that selective modulation of the hypoglossal motor pool may maintain upper airway stability, but the use of gene therapy as treatment for OSA remains uncertain.
The reliance on EMG responses as the primary outcome may be problematic. First, GG EMG activation is not tantamount to airway patency; airflow measurements are required to determine whether GG recruitment could improve OSA. Second, other muscles innervated by the hypoglossal nerve (tongue retractors and intrinsic muscles) as well as other cranial nerves (glossopharyngeal and vagus) are likely to be involved and have not been sufficiently studied. Successful drug development for OSA will require attention to both of these limitations.
3.3. Animal Models of Neuromuscular Response: Airflow Outcomes
Translational models have been used to demonstrate the impact of anatomical or neuromuscular interventions on airflow. In rabbits, Lee and colleagues [,] paralyzed the GG muscle with botulinum toxin type A and evaluated polysomnography and upper airway dynamics using computed tomography (CT) scans during drug-induced sleep. Two weeks after injection, the number of apneas and hypopneas increased and the diameter of the upper airway at the level of the palate and tongue base was diminished. In anesthetized animals, electrical stimulation of the hypoglossal nerve increased airflow and upper airway patency [,,,,,,,], laying the foundation for the development of electrical-stimulating devices for OSA. In 1993, Schwartz and colleagues [] utilized an isolated feline upper airway model to modulate GG activity. Hypoglossal nerve stimulation reduced Pcrit and increased flow. These translational models have advanced our understanding of the pathogenesis of OSA but have yet to be leveraged for pharmacotherapy.
A major limitation of translational OSA research is the need for anesthesia, which is likely to affect upper airway muscle tone and breathing. The Hopkins group developed a system for continuously recording polysomnographic signals in unanesthetized, unrestrained mice []. They modified a whole-body plethysmography chamber with features, including an open-circuit design and an air bladder upon which the mouse rests, to enable extended monitoring and assessment of tidal volume, airflow, and respiratory effort. Polotsky and colleagues used this system to measure Pcrit and associated GG EMG activity, airflow, and effort in a variety of mouse models, including leptin-deficient ob/ob [,], DIO [], and NZO mice []. DIO and NZO mice exhibited IFL during REM sleep [,,] and higher passive Pcrit values than lean mice []. A more severe OSA phenotype was proposed by Fleury Curado and colleagues [] by silencing hypoglossal motoneurons in lean C57BL/6J mice using inhibitory DREADDS. The DREADDS were delivered bilaterally to the hypoglossal nucleus, and inhibitory activity was then induced by intraperitoneal injection of the DREADD ligand, clozapine-N-oxide (CNO). These mice developed flow-limited breaths during both REM and NREM sleep, which would correspond to snoring or obstructive hypoventilation in children [,,].
Another rodent model of OSA is the leptin-deficient ob/ob mouse. Leptin is secreted by adipose tissue into the circulation. It crosses the blood–brain barrier (BBB) and acts in hypothalamic centers to increase metabolic rate and suppress food intake []. ob/ob mice and leptin receptor-deficient db/db mice are hyperphagic, hypometabolic, and severely obese []. It was previously shown that ob/ob mice have hypercapnia and reduced CO2 sensitivity, which resolves with leptin replacement [,] (Table 1). More recently, leptin has been shown to have a role in controlling upper airway patency [,,]. ob/ob mice exhibit inspiratory flow limitation, especially during REM sleep. Subcutaneous leptin improved upper airway patency and increased ventilatory drive, with no significant change in passive Pcrit [,] (Table 1). To localize the site of leptin’s activity, Yao and colleagues [] injected leptin into different brain regions. They took advantage of the unidirectional rostral–caudal flow of cerebrospinal fluid by administering leptin in the lateral as opposed to the fourth ventricle. Only leptin injections in the lateral ventricle relieved upper airway obstruction while both routes of leptin administration stimulated ventilation (Table 1). This suggests that leptin acts in the dorsomedial hypothalamus to relieve upper airway obstruction, and the hindbrain (nucleus of the solitary tract) to upregulate ventilation.
In subsequent work, our group have shown that DIO mice develop sleep disordered breathing, despite high circulating leptin levels and leptin resistance []. Moreover, these findings were consistent with reports in obese patients [,]. Leptin resistance has multiple mechanisms, but limited permeability of the BBB plays a key role [,,,]. Berger and colleagues [] administered leptin intranasally in mice in an effort to bypass the BBB. Intranasal leptin increased ventilation during NREM and REM sleep and decreased the number of oxygen desaturation events in REM sleep (Table 1). By contrast, intraperitoneal leptin did not significantly improve breathing during sleep. Thus, central leptin signaling may be a future target for OSA drug development.
4. Respiratory Control Instability (Loop Gain)
4.1. Definition and Human Evidence
During wakefulness, breathing is regulated by both conscious respiratory drive from supra-pontine brain structures, and autonomic chemoreflex control []. During sleep, respiratory drive is mainly governed by chemoreflexes, with the fluctuations in CO2 levels representing the major stimulus to breathe [,]. The magnitude of the ventilatory response to CO2 during sleep can drive oscillations in breathing, leading to arousals during respiratory disturbances []. “Loop gain” (an engineering concept) describes the amplitude of ventilation in response to a disturbance, as might occur with obstruction of the upper airway []. There are two major components that regulate loop gain: (1) A controller gain and (2) plant gain []. Controller gain predominantly refers to chemoreflex sensitivity to arterial blood gases while the plant gain reflects the capacity of the respiratory system to alter gas exchange. A delay also exists between the detection of blood gas fluctuations by chemoreceptors (controller gain) and ventilatory responses to these fluctuations (plant gain), which can initiate and propagate ventilatory stability [].
Different techniques have been developed to assess loop gain in OSA patients. Wellman and colleagues [,] developed a non-invasive method to measure OSA traits, including the gain of the ventilatory control system, by intermittently dropping CPAP from optimum pressures, in which the upper airway is completely opened, to several subtherapeutic pressures for 3 to 5 min during sleep. At suboptimum CPAP pressures, there is compensatory ventilation, in which the respiratory drive increases but does not reach eupneic levels, because of the upper airway obstruction. In this method, loop gain is quantified as the compensatory ventilatory response (ΔVentilation) divided by the increase in ventilatory drive (ΔVentilatory drive) [,]. However, all of the currently available techniques only quantify the general loop gain of OSA patients and cannot provide a clear estimation of controller and plant gains.
In patients with anatomical predisposition to upper airway collapse, high loop gain causes instability, thereby increasing the severity of OSA [,]. A high loop gain was observed in 36% of patients with high anatomic predisposition to OSA defined by a Pcrit between −2 and +2 cmH2O []. High loop gain might lead to an over-exuberant respiratory response to obstruction, triggering large negative inspiratory pressure swings and collapse of the pharyngeal upper airway. In addition, hyperventilation may diminish the activity of hypoglossal motoneurons, leading to upper airway collapse [,,,,]. However, it is still unclear if (1) OSA patients have a higher loop gain compared to non-OSA individuals and (2) whether a high loop gain is inherent to OSA or is induced by OSA traits. As extensively reviewed by Deacon-Diaz and Malhotra [], the findings comparing loop gain between OSA and non-OSA patients are still contradictory. However, evidence from studies controlling for key confounding factors (i.e., obesity and CPAP status) have suggested a higher loop gain in apneic individuals [].
4.2. Animal Models of Ventilatory Instability in OSA
Mice do not inherently have OSA but may develop sleep disordered breathing when their ventilatory control system is perturbed. Intermittent hypoxia caused by apneic events augments the ventilatory response to hypoxia (HVR) [,,]. Exposure to intermittent hypoxia can induce neuroplasticity of motoneurons involved in respiratory control, including the phrenic and hypoglossal neurons []. The sustained increase in ventilatory neural activity induced by intermittent hypoxia that persists after hypoxic stimulation has been termed long-term facilitation (LTF). LTF reduces CO2 levels below eupnea, leading to an increase in the controller gain [,,,]. Prabhakar’s laboratory has examined LTF and its contribution the development of an OSA phenotype in rodents. Rats exposed to nine episodes of hypoxia (5% O2) for 10 days (8 h/day) had a 48% increase in HVR, analyzed by efferent phrenic nerve activity [].
The carotid body is a critical organ of oxygen sensing and altering its function can lead to disordered breathing patterns. Prabhakar’s laboratory found that LTF was related to increased HVR (i.e., chemosensitivity) in carotid bodies []. Transcriptional activator hypoxia-inducible factor 1 alpha (HIF-1α) has been proposed as a key molecule in the regulation of carotid body plasticity and HVR. In wild-type mice, preconditioning to hypoxia for 3 days increased the HVR []. However, Hif1α+/− mice showed a reduced HVR and the absence of carotid body activity in response to hypoxia, suggesting the role of HIF-1α in the ventilatory adaptation and carotid body plasticity to chronic hypoxia. Peng and colleagues [] investigated the effects of the gasotransmitters, carbon monoxide (CO) and hydrogen sulfide (H2S), on carotid body activity, HVR, and induction of sleep apneas. They showed that the deficiency of CO augmented apneas and hypopneas during NREM and REM sleep in mice. On the other hand, lack of H2S by genetic ablation or pharmacological blockade of the H2S-synthesizing enzyme normalized breathing and prevented central and obstructive apneas in mice and rats (Table 1). These findings demonstrate that altered ventilatory control can lead to sleep apnea, and suggest that one approach to treating OSA may be blocking H2S release.
Recently, our lab discovered that the carotid bodies respond to leptin, with downstream effects on minute ventilation and HVR []. We showed that the long functional isoform of leptin receptor (LepRb) is expressed in approximately 74% of glomus cells in carotid body and that leptin infusion increased carotid sinus nerve activity in vivo. In lean C57BL/6J mice, subcutaneous infusion of leptin increased minute ventilation and HVR, and these leptin effects were abolished after carotid body denervation. In obese LepRb-deficient db/db mice, transfection of LepRb in the carotid body also increased minute ventilation and HVR (Table 1). Similar findings of leptin-modulating HVR were also observed in obese Zucker rats [] (Table 1). Overall, our results indicate that leptin is a potent ventilatory stimulant, increasing HVR by acting on the LepRb expressed in carotid bodies. Our data suggest that the modulation of leptin signaling could be a target for pharmacotherapy. However, excessive carotid body activity by leptin administration may in theory destabilize respiratory control, causing apneas. Leptin also has several peripheral effects, such as increasing blood pressure [,], which may limit its use for OSA.
5. Respiratory Arousal Threshold
5.1. Definition and Human Evidence
Respiratory arousals contribute to the pathophysiology of OSA, destabilizing both the control of breathing and upper airway function [,]. Thus, preventing respiratory arousals (i.e., increasing the arousal threshold) might be a pharmacological target for OSA. Studies in humans using sedative-hypnotic drugs to achieve this goal have yielded inconsistent results [,,,,,,]. Eckert and colleagues [] showed that Eszopliclone was effective in reducing the apnea-hypopnea index in individuals with a low arousal threshold (stage 2 arousal threshold between 0 and −15 cmH2O) but not in those with a higher arousal threshold (stage 2 arousal threshold = −40 ± 6; overall range −25 to −63 cmH2O). An arousal threshold in recent studies was determined based on the respiratory effort inducing arousal []. Thus, pharmacological agents that increase the arousal threshold may be used in patients with a low arousal threshold.
5.2. Animal Models Focusing on Arousal Threshold
Stimuli that provoke arousal in OSA include CO2 elevation and airway occlusion. Studies in rodents, dogs, and piglets utilized progressive hypercapnia during sleep to quantify the arousal threshold as a function of CO2 levels. Park and colleagues demonstrated that sedative hypnotics increased the arousal threshold and increased GG muscle activity when administered systemically, but decreased GG activity when the same drugs were administered locally to the hypoglossal nucleus [,] (Table 1).
Saper and his group [,,] described the neural circuitry responsible for respiratory arousals in mammals. Interestingly, all the chemosensory and mechanical stimuli converge upon the external lateral parabrachial nucleus (PBel) in the pons. By utilizing selective optogenetic activation and inhibition they were able to modulate hypercapnia-induced arousals and identify sites in the forebrain that receive input from the PBel [] (Table 1). Thus, the PBel and its network are promising cellular targets for interventions to promote respiratory stability during sleep.
6. Conclusions and Future Perspectives
OSA is a complex disorder caused by factors, including altered upper airway anatomy, upper airway neuromuscular responses, respiratory control, and respiratory arousal threshold. Animal models have advanced our understanding of how these factors contribute to the pathogenesis of OSA. These models have revealed potentially druggable targets for OSA, including (1) the role of leptin in the control of breathing, (2) the role of noradrenergic agonists and antimuscarinic agents in GG muscle control, (3) the role of serotoninergic input to the hypoglossal nucleus during sleep, and (4) the role of sedative hypnotics. Modern techniques, such as DREADDs and optogenetics, are promising tools to investigate the pathophysiology of OSA and may yield novel targets for OSA pharmacotherapy.
Author Contributions
Conceptualization, L.J.K., C.F., T.F.C., J.C.J., and V.Y.P.; Writing—original draft preparation, L.J.K., C.F., T.F.C., J.C.J., and V.Y.P.; Writing—review and editing, L.J.K., C.F., T.F.C., J.C.J., and V.Y.P.
Funding
This research was funded by National Institutes of Health (NIH R01s HL133100, R01 HL128970, and R01 HL138932 to V.Y.P.; and R01 HL135483 to J.C.J.).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dempsey, J.A.; Veasey, S.C.; Morgan, B.J.; O’Donnell, C.P. Pathophysiology of Sleep Apnea. Physiol. Rev. 2010, 90, 47–112. [Google Scholar] [CrossRef] [PubMed]
- Tufik, S.; Santos-Silva, R.; Taddei, J.A.; Bittencourt, L.R.A. Obstructive Sleep Apnea Syndrome in the Sao Paulo Epidemiologic Sleep Study. Sleep Med. 2010, 11, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Heinzer, R.; Vat, S.; Marques-Vidal, P.; Marti-Soler, H.; Andries, D.; Tobback, N.; Mooser, V.; Preisig, M.; Malhotra, A.; Waeber, G.; et al. Prevalence of sleep-disordered breathing in the general population: The HypnoLaus study. Lancet Respir. Med. 2015, 3, 310–318. [Google Scholar] [CrossRef]
- Sullivan, C.E.; Issa, F.G.; Berthon-Jones, M.; Eves, L. Reversal of obstructive sleep apnoea by continuous positive airway pressure applied through the nares. Lancet 1981, 1, 862–865. [Google Scholar] [CrossRef]
- Weaver, T.E.; Grunstein, R.R. Adherence to continuous positive airway pressure therapy: The challenge to effective treatment. Proc. Am. Thorac. Soc. 2008, 5, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Kribbs, N.B.; Pack, A.I.; Kline, L.R.; Smith, P.L.; Schwartz, A.R.; Schubert, N.M.; Redline, S.; Henry, J.N.; Getsy, J.E.; Dinges, D.F. Objective measurement of patterns of nasal CPAP use by patients with obstructive sleep apnea. Am. Rev. Respir. Dis. 1993, 147, 887–895. [Google Scholar] [CrossRef]
- Owens, R.L.; Eckert, D.J.; Yeh, S.Y.; Malhotra, A. Upper airway function in the pathogenesis of obstructive sleep apnea: A review of the current literature. Curr. Opin. Pulm. Med. 2008, 14, 519–524. [Google Scholar] [CrossRef]
- Wellman, A.; Eckert, D.J.; Jordan, A.S.; Edwards, B.A.; Passaglia, C.L.; Jackson, A.C.; Gautam, S.; Owens, R.L.; Malhotra, A.; White, D.P. A method for measuring and modeling the physiological traits causing obstructive sleep apnea. J. Appl. Physiol. 2011, 110, 1627–1637. [Google Scholar] [CrossRef]
- Eckert, D.J.; White, D.P.; Jordan, A.S.; Malhotra, A.; Wellman, A. Defining phenotypic causes of obstructive sleep apnea. Identification of novel therapeutic targets. Am. J. Respir. Crit. Care Med. 2013, 188, 996–1004. [Google Scholar] [CrossRef]
- Wellman, A.; Edwards, B.A.; Sands, S.A.; Owens, R.L.; Nemati, S.; Butler, J.; Passaglia, C.L.; Jackson, A.C.; Malhotra, A.; White, D.P. A simplified method for determining phenotypic traits in patients with obstructive sleep apnea. J. Appl. Physiol. 2013, 114, 911–922. [Google Scholar] [CrossRef]
- Eckert, D.J. Phenotypic approaches to obstructive sleep apnoea–New pathways for targeted therapy. Sleep Med. Rev. 2018, 37, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, B.C.; Kharbanda, O.P.; Sardana, H.K.; Balachandran, R.; Sardana, V.; Kapoor, P.; Gupta, A.; Vasamsetti, S. Craniofacial and upper airway morphology in adult obstructive sleep apnea patients: A systematic review and meta-analysis of cephalometric studies. Sleep Med. Rev. 2017, 31, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Schwab, R.J.; Pasirstein, M.; Pierson, R.; Mackley, A.; Hachadoorian, R.; Arens, R.; Maislin, G.; Pack, A.I. Identification of upper airway anatomic risk factors for obstructive sleep apnea with volumetric magnetic resonance imaging. Am. J. Respir. Crit. Care Med. 2003, 168, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Lowe, A.A.; Santamaria, J.D.; Fleetham, J.A.; Price, C. Facial morphology and obstructive sleep apnea. Am. J. Orthod. Dentofac. Orthop. 1986, 90, 484–491. [Google Scholar] [CrossRef]
- Miles, P.G.; Vig, P.S.; Weyant, R.J.; Forrest, T.D.; Rockette, H.E. Craniofacial structure and obstructive sleep apnea syndrome–a qualitative analysis and meta-analysis of the literature. Am. J. Orthod. Dentofac. Orthop. 1996, 109, 163–172. [Google Scholar] [CrossRef]
- Schwartz, A.R.; O’Donnell, C.P.; Baron, J.; Schubert, N.; Alam, D.; Samadi, S.D.; Smith, P.L. The hypotonic upper airway in obstructive sleep apnea: Role of structures and neuromuscular activity. Am. J. Respir. Crit. Care Med. 1998, 157, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Carberry, J.C.; Jordan, A.S.; White, D.P.; Wellman, A.; Eckert, D.J. Upper Airway Collapsibility (Pcrit) and Pharyngeal Dilator Muscle Activity are Sleep Stage Dependent. Sleep 2016, 39, 511–521. [Google Scholar] [CrossRef]
- Gold, A.R.; Schwartz, A.R. The pharyngeal critical pressure. The whys and hows of using nasal continuous positive airway pressure diagnostically. Chest 1996, 110, 1077–1088. [Google Scholar] [CrossRef]
- Kirkness, J.P.; Schwartz, A.R.; Schneider, H.; Punjabi, N.M.; Maly, J.J.; Laffan, A.M.; McGinley, B.M.; Magnuson, T.; Schweitzer, M.; Smith, P.L.; et al. Contribution of male sex, age, and obesity to mechanical instability of the upper airway during sleep. J. Appl. Physiol. 2008, 104, 1618–1624. [Google Scholar] [CrossRef]
- Schwartz, A.R.; Patil, S.P.; Laffan, A.M.; Polotsky, V.; Schneider, H.; Smith, P.L. Obesity and obstructive sleep apnea: Pathogenic mechanisms and therapeutic approaches. Proc. Am. Thorac. Soc. 2008, 5, 185–192. [Google Scholar] [CrossRef]
- Joosten, S.A.; Edwards, B.A.; Wellman, A.; Turton, A.; Skuza, E.M.; Berger, P.J.; Hamilton, G.S. The Effect of Body Position on Physiological Factors that Contribute to Obstructive Sleep Apnea. Sleep 2015, 38, 1469–1478. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.P.; Schneider, H.; Marx, J.J.; Gladmon, E.; Schwartz, A.R.; Smith, P.L. Neuromechanical control of upper airway patency during sleep. J. Appl. Physiol. 2007, 102, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Horner, R.L.; Mohiaddin, R.H.; Lowell, D.G.; Shea, S.A.; Burman, E.D.; Longmore, D.B.; Guz, A. Sites and sizes of fat deposits around the pharynx in obese patients with obstructive sleep apnoea and weight matched controls. Eur. Respir. J. 1989, 2, 613–622. [Google Scholar] [PubMed]
- Arens, R.; Sin, S.; Nandalike, K.; Rieder, J.; Khan, U.I.; Freeman, K.; Wylie-Rosett, J.; Lipton, M.L.; Wootton, D.M.; McDonough, J.M.; et al. Upper airway structure and body fat composition in obese children with obstructive sleep apnea syndrome. Am. J. Respir. Crit. Care Med. 2011, 183, 782–787. [Google Scholar] [CrossRef]
- Pahkala, R.; Seppä, J.; Ikonen, A.; Smirnov, G.; Tuomilehto, H. The impact of pharyngeal fat tissue on the pathogenesis of obstructive sleep apnea. Sleep Breath. 2014, 18, 275–282. [Google Scholar] [CrossRef]
- Kim, A.M.; Keenan, B.T.; Jackson, N.; Chan, E.L.; Staley, B.; Poptani, H.; Torigian, D.A.; Pack, A.I.; Schwab, R.J. Tongue fat and its relationship to obstructive sleep apnea. Sleep 2014, 37, 1639–1648. [Google Scholar] [CrossRef]
- Van de Graaff, W.B. Thoracic influence on upper airway patency. J. Appl. Physiol. 1988, 65, 2124–2131. [Google Scholar] [CrossRef]
- Heinzer, R.C.; Stanchina, M.L.; Malhotra, A.; Jordan, A.S.; Patel, S.R.; Lo, Y.-L.; Wellman, A.; Schory, K.; Dover, L.; White, D.P. Effect of increased lung volume on sleep disordered breathing in patients with sleep apnoea. Thorax 2006, 61, 435–439. [Google Scholar] [CrossRef]
- Kairaitis, K.; Byth, K.; Parikh, R.; Stavrinou, R.; Wheatley, J.R.; Amis, T.C. Tracheal traction effects on upper airway patency in rabbits: The role of tissue pressure. Sleep 2007, 30, 179–186. [Google Scholar] [CrossRef]
- Schwartz, A.R.; Gold, A.R.; Schubert, N.; Stryzak, A.; Wise, R.A.; Permutt, S.; Smith, P.L. Effect of weight loss on upper airway collapsibility in obstructive sleep apnea. Am. Rev. Respir. Dis. 1991, 144, 494–498. [Google Scholar] [CrossRef]
- White, D.P. Pharmacologic Approaches to the Treatment of Obstructive Sleep Apnea. Sleep Med. Clin. 2016, 11, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, K.; Lee, R.W.W.; Phillips, C.L.; Dungan, G.; Yee, B.J.; Magnussen, J.S.; Grunstein, R.R.; Cistulli, P.A. Effect of weight loss on upper airway size and facial fat in men with obstructive sleep apnoea. Thorax 2011, 66, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Bucca, C.B.; Brussino, L.; Battisti, A.; Mutani, R.; Rolla, G.; Mangiardi, L.; Cicolin, A. Diuretics in obstructive sleep apnea with diastolic heart failure. Chest 2007, 132, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Chopra, S.; Polotsky, V.Y.; Jun, J.C. Sleep Apnea Research in Animals. Past, Present, and Future. Am. J. Respir. Cell Mol. Biol. 2016, 54, 299–305. [Google Scholar] [CrossRef]
- Hendricks, J.C.; Kline, L.R.; Kovalski, R.J.; O’Brien, J.A.; Morrison, A.R.; Pack, A.I. The English bulldog: A natural model of sleep-disordered breathing. J. Appl. Physiol. 1987, 63, 1344–1350. [Google Scholar] [CrossRef]
- Lonergan, R.P.; Ware, J.C.; Atkinson, R.L.; Winter, W.C.; Suratt, P.M. Sleep apnea in obese miniature pigs. J. Appl. Physiol. 1998, 84, 531–536. [Google Scholar] [CrossRef]
- Farré, R.; Nácher, M.; Serrano-Mollar, A.; Gáldiz, J.B.; Alvarez, F.J.; Navajas, D.; Montserrat, J.M. Rat model of chronic recurrent airway obstructions to study the sleep apnea syndrome. Sleep 2007, 30, 930–933. [Google Scholar] [CrossRef]
- Neuzeret, P.-C.; Gormand, F.; Reix, P.; Parrot, S.; Sastre, J.-P.; Buda, C.; Guidon, G.; Sakai, K.; Lin, J.-S. A new animal model of obstructive sleep apnea responding to continuous positive airway pressure. Sleep 2011, 34, 541–548. [Google Scholar] [CrossRef]
- Crossland, R.F.; Durgan, D.J.; Lloyd, E.E.; Phillips, S.C.; Reddy, A.K.; Marrelli, S.P.; Bryan, R.M. A new rodent model for obstructive sleep apnea: Effects on ATP-mediated dilations in cerebral arteries. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R334–R342. [Google Scholar] [CrossRef]
- Lee, M.-C.; Lee, C.H.; Hong, S.-L.; Kim, S.-W.; Lee, W.-H.; Lim, J.Y.; Joe, S.; Yoon, I.-Y.; Kim, J.-W. Establishment of a rabbit model of obstructive sleep apnea by paralyzing the genioglossus. JAMA Otolaryngol. Head Neck Surg. 2013, 139, 834–840. [Google Scholar] [CrossRef][Green Version]
- Lu, H.; Dong, F.; Liu, C.; Wang, J.; Liu, Y.; Xiao, W. An animal model of obstructive sleep apnoea-hypopnea syndrome corrected by mandibular advancement device. Eur. J. Orthod. 2015, 37, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-C.; Rhee, C.-S.; Joe, S.; Yoon, I.-Y.; Kim, J.-W. A Single Primary Site Obstruction May Lead to Sleep-Disordered Breathing in Multiple Sites: An Animal Model. Ann. Otol. Rhinol. Laryngol. 2016, 125, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Brennick, M.J.; Pickup, S.; Cater, J.R.; Kuna, S.T. Phasic respiratory pharyngeal mechanics by magnetic resonance imaging in lean and obese zucker rats. Am. J. Respir. Crit. Care Med. 2006, 173, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- Brennick, M.J.; Delikatny, J.; Pack, A.I.; Pickup, S.; Shinde, S.; Zhu, J.-X.; Roscoe, I.; Kim, D.Y.; Buxbaum, L.U.; Cater, J.R.; et al. Tongue fat infiltration in obese versus lean Zucker rats. Sleep 2014, 37, 1095–1102. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Brennick, M.J.; Pack, A.I.; Ko, K.; Kim, E.; Pickup, S.; Maislin, G.; Schwab, R.J. Altered upper airway and soft tissue structures in the New Zealand Obese mouse. Am. J. Respir. Crit. Care Med. 2009, 179, 158–169. [Google Scholar] [CrossRef]
- Baum, D.M.; Morales Rodriguez, B.; Attali, V.; Cauhapé, M.; Arnulf, I.; Cardot, P.; Bodineau, L.; Fiamma, M.-N. New Zealand Obese Mice as a Translational Model of Obesity-related Obstructive Sleep Apnea Syndrome. Am. J. Respir. Crit. Care Med. 2018, 198, 1336–1339. [Google Scholar] [CrossRef]
- Nishimura, Y.; Arias, R.S.; Pho, H.; Pham, L.V.; Curado, T.F.; Polotsky, V.Y.; Schwartz, A.R. A Novel Non-invasive Approach for Measuring Upper Airway Collapsibility in Mice. Front. Neurol. 2018, 9, 985. [Google Scholar] [CrossRef]
- Polotsky, M.; Elsayed-Ahmed, A.S.; Pichard, L.; Richardson, R.A.; Smith, P.L.; Schneider, H.; Kirkness, J.P.; Polotsky, V.; Schwartz, A.R. Effect of age and weight on upper airway function in a mouse model. J. Appl. Physiol. 2011, 111, 696–703. [Google Scholar] [CrossRef]
- Hernandez, A.B.; Kirkness, J.P.; Smith, P.L.; Schneider, H.; Polotsky, M.; Richardson, R.A.; Hernandez, W.C.; Schwartz, A.R. Novel whole body plethysmography system for the continuous characterization of sleep and breathing in a mouse. J. Appl. Physiol. 2012, 112, 671–680. [Google Scholar] [CrossRef]
- Pho, H.; Hernandez, A.B.; Arias, R.S.; Leitner, E.B.; Van Kooten, S.; Kirkness, J.P.; Schneider, H.; Smith, P.L.; Polotsky, V.Y.; Schwartz, A.R. The effect of leptin replacement on sleep-disordered breathing in the leptin-deficient ob/ob mouse. J. Appl. Physiol. 2016, 120, 78–86. [Google Scholar] [CrossRef]
- Fleury Curado, T.; Pho, H.; Berger, S.; Caballero-Eraso, C.; Shin, M.-K.; Sennes, L.U.; Pham, L.; Schwartz, A.R.; Polotsky, V.Y. Sleep-disordered breathing in C57BL/6J mice with diet-induced obesity. Sleep 2018, 41, zsy089. [Google Scholar] [CrossRef] [PubMed]
- O’donnell, C.P.; Schaub, C.D.; Haines, A.S.; Berkowitz, D.E.; Tankersley, C.G.; Schwartz, A.R.; Smith, P.L. Leptin prevents respiratory depression in obesity. Am. J. Respir. Crit. Care Med. 1999, 159, 1477–1484. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.M.; O’Donnell, C.P. Rodent models of sleep apnea. Respir. Physiol. Neurobiol. 2013, 188, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, F.N.; Meadows, P.; Jacobowitz, O.; Davidson, T.M. Tongue anatomy and physiology, the scientific basis for a novel targeted neurostimulation system designed for the treatment of obstructive sleep apnea. Neuromodulation 2013, 16, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Remmers, J.E.; deGroot, W.J.; Sauerland, E.K.; Anch, A.M. Pathogenesis of upper airway occlusion during sleep. J. Appl. Physiol. Respir. 1978, 44, 931–938. [Google Scholar] [CrossRef]
- Fuller, D.; Mateika, J.H.; Fregosi, R.F. Co-activation of tongue protrudor and retractor muscles during chemoreceptor stimulation in the rat. J. Physiol. (Lond.) 1998, 507, 265–276. [Google Scholar] [CrossRef]
- Dotan, Y.; Pillar, G.; Tov, N.; Oliven, R.; Steinfeld, U.; Gaitini, L.; Odeh, M.; Schwartz, A.R.; Oliven, A. Dissociation of electromyogram and mechanical response in sleep apnoea during propofol anaesthesia. Eur. Respir. J. 2013, 41, 74–84. [Google Scholar] [CrossRef]
- Fuller, D.D.; Williams, J.S.; Janssen, P.L.; Fregosi, R.F. Effect of co-activation of tongue protrudor and retractor muscles on tongue movements and pharyngeal airflow mechanics in the rat. J. Physiol. (Lond.) 1999, 51, 601–613. [Google Scholar] [CrossRef]
- Schwartz, A.R.; Bennett, M.L.; Smith, P.L.; De Backer, W.; Hedner, J.; Boudewyns, A.; Van de Heyning, P.; Ejnell, H.; Hochban, W.; Knaack, L.; et al. Therapeutic electrical stimulation of the hypoglossal nerve in obstructive sleep apnea. Arch. Otolaryngol. Head Neck Surg. 2001, 127, 1216–1223. [Google Scholar] [CrossRef]
- Kubin, L.; Davies, R.O.; Pack, A.I. Control of Upper Airway Motoneurons during REM Sleep. News Physiol. Sci. 1998, 13, 91–97. [Google Scholar] [CrossRef]
- Veasey, S.C. Serotonin agonists and antagonists in obstructive sleep apnea: Therapeutic potential. Am. J. Respir. Med. 2003, 2, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Sood, S.; Liu, X.; Liu, H.; Horner, R.L. Genioglossus muscle activity and serotonergic modulation of hypoglossal motor output in obese Zucker rats. J. Appl. Physiol. 2007, 102, 2240–2250. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.; Steenland, H.W.; Liu, H.; Horner, R.L. Endogenous excitatory drive modulating respiratory muscle activity across sleep-wake states. Am. J. Respir. Crit. Care Med. 2006, 174, 1264–1273. [Google Scholar] [CrossRef] [PubMed]
- Grace, K.P.; Hughes, S.W.; Horner, R.L. Identification of the mechanism mediating genioglossus muscle suppression in REM sleep. Am. J. Respir. Crit. Care Med. 2013, 187, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Grace, K.P.; Hughes, S.W.; Shahabi, S.; Horner, R.L. K+ channel modulation causes genioglossus inhibition in REM sleep and is a strategy for reactivation. Respir. Physiol. Neurobiol. 2013, 188, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Grace, K.P.; Hughes, S.W.; Horner, R.L. Identification of a Pharmacological Target for Genioglossus Reactivation throughout Sleep. Sleep 2014, 37, 41–50. [Google Scholar] [CrossRef]
- Song, G.; Poon, C.-S. α2-Adrenergic blockade rescues hypoglossal motor defense against obstructive sleep apnea. JCI Insight 2017, 2, e91456. [Google Scholar] [CrossRef]
- Fleury Curado, T.; Fishbein, K.; Pho, H.; Brennick, M.; Dergacheva, O.; Sennes, L.U.; Pham, L.V.; Ladenheim, E.E.; Spencer, R.; Mendelowitz, D.; et al. Chemogenetic stimulation of the hypoglossal neurons improves upper airway patency. Sci. Rep. 2017, 7, 44392. [Google Scholar] [CrossRef]
- Polotsky, M.; Elsayed-Ahmed, A.S.; Pichard, L.; Harris, C.C.; Smith, P.L.; Schneider, H.; Kirkness, J.P.; Polotsky, V.; Schwartz, A.R. Effects of leptin and obesity on the upper airway function. J. Appl. Physiol. 2012, 112, 1637–1643. [Google Scholar] [CrossRef]
- Yao, Q.; Pho, H.; Kirkness, J.; Ladenheim, E.E.; Bi, S.; Moran, T.H.; Fuller, D.D.; Schwartz, A.R.; Polotsky, V.Y. Localizing Effects of Leptin on Upper Airway and Respiratory Control during Sleep. Sleep 2016, 39, 1097–1106. [Google Scholar] [CrossRef]
- Berger, S.; Pho, H.; Fleury-Curado, T.; Bevans-Fonti, S.; Younas, H.; Shin, M.-K.; Jun, J.C.; Anokye-Danso, F.; Ahima, R.S.; Enquist, L.W.; et al. Intranasal Leptin Relieves Sleep-disordered Breathing in Mice with Diet-induced Obesity. Am. J. Respir. Crit. Care Med. 2019, 199, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.-J.; Zhang, X.; Gridina, A.; Chupikova, I.; McCormick, D.L.; Thomas, R.J.; Scammell, T.E.; Kim, G.; Vasavda, C.; Nanduri, J.; et al. Complementary roles of gasotransmitters CO and H2S in sleep apnea. Proc. Natl. Acad. Sci. USA 2017, 114, 1413–1418. [Google Scholar] [CrossRef] [PubMed]
- Caballero-Eraso, C.; Shin, M.-K.; Pho, H.; Kim, L.J.; Pichard, L.E.; Wu, Z.-J.; Gu, C.; Berger, S.; Pham, L.; Yeung, H.-Y.B.; et al. Leptin acts in the carotid bodies to increase minute ventilation during wakefulness and sleep and augment the hypoxic ventilatory response. J. Physiol. (Lond.) 2019, 597, 151–172. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Wang, H.; Feng, J.; Wei, Z.; Yu, H.; Zhang, X.; Zhang, Y.; Wang, S. Leptin Signaling in the Carotid Body Regulates a Hypoxic Ventilatory Response Through Altering TASK Channel Expression. Front. Physiol. 2018, 9, 249. [Google Scholar] [CrossRef]
- Park, E.; Younes, M.; Liu, H.; Liu, X.; Horner, R.L. Systemic vs. central administration of common hypnotics reveals opposing effects on genioglossus muscle activity in rats. Sleep 2008, 31, 355–365. [Google Scholar] [CrossRef]
- Kaur, S.; Wang, J.L.; Ferrari, L.; Thankachan, S.; Kroeger, D.; Venner, A.; Lazarus, M.; Wellman, A.; Arrigoni, E.; Fuller, P.M.; et al. A Genetically Defined Circuit for Arousal from Sleep during Hypercapnia. Neuron 2017, 96, 1153–1167.e5. [Google Scholar] [CrossRef]
- Taranto-Montemurro, L.; Messineo, L.; Sands, S.A.; Azarbarzin, A.; Marques, M.; Edwards, B.A.; Eckert, D.J.; White, D.P.; Wellman, A. The Combination of Atomoxetine and Oxybutynin Greatly Reduces Obstructive Sleep Apnea Severity. A Randomized, Placebo-controlled, Double-Blind Crossover Trial. Am. J. Respir. Crit. Care Med. 2019, 199, 1267–1276. [Google Scholar] [CrossRef]
- Horton, G.A.; Fraigne, J.J.; Torontali, Z.A.; Snow, M.B.; Lapierre, J.L.; Liu, H.; Montandon, G.; Peever, J.H.; Horner, R.L. Activation of the Hypoglossal to Tongue Musculature Motor Pathway by Remote Control. Sci. Rep. 2017, 7, 45860. [Google Scholar] [CrossRef]
- Roth, B.L. DREADDs for Neuroscientists. Neuron 2016, 89, 683–694. [Google Scholar] [CrossRef]
- Miki, H.; Hida, W.; Shindoh, C.; Kikuchi, Y.; Chonan, T.; Taguchi, O.; Inoue, H.; Takishima, T. Effects of electrical stimulation of the genioglossus on upper airway resistance in anesthetized dogs. Am. Rev. Respir. Dis. 1989, 140, 1279–1284. [Google Scholar] [CrossRef]
- Schwartz, A.R.; Thut, D.C.; Russ, B.; Seelagy, M.; Yuan, X.; Brower, R.G.; Permutt, S.; Wise, R.A.; Smith, P.L. Effect of electrical stimulation of the hypoglossal nerve on airflow mechanics in the isolated upper airway. Am. Rev. Respir. Dis. 1993, 147, 1144–1150. [Google Scholar] [CrossRef] [PubMed]
- Oliven, A.; Odeh, M.; Schnall, R.P. Improved upper airway patency elicited by electrical stimulation of the hypoglossus nerves. Respiration 1996, 63, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Eisele, D.W.; Schwartz, A.R.; Hari, A.; Thut, D.C.; Smith, P.L. The effects of selective nerve stimulation on upper airway airflow mechanics. Arch. Otolaryngol. Head Neck Surg. 1995, 121, 1361–1364. [Google Scholar] [CrossRef] [PubMed]
- Bishara, H.; Odeh, M.; Schnall, R.P.; Gavriely, N.; Oliven, A. Electrically-activated dilator muscles reduce pharyngeal resistance in anaesthetized dogs with upper airway obstruction. Eur. Respir. J. 1995, 8, 1537–1542. [Google Scholar] [PubMed]
- Goding, G.S.; Eisele, D.W.; Testerman, R.; Smith, P.L.; Roertgen, K.; Schwartz, A.R. Relief of upper airway obstruction with hypoglossal nerve stimulation in the canine. Laryngoscope 1998, 108, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Yoo, P.B.; Durand, D.M. Effects of selective hypoglossal nerve stimulation on canine upper airway mechanics. J. Appl. Physiol. 2005, 99, 937–943. [Google Scholar] [CrossRef] [PubMed]
- Fleury Curado, T.A.; Pho, H.; Dergacheva, O.; Berger, S.; Lee, R.; Freire, C.; Asherov, A.; Sennes, L.U.; Mendelowitz, D.; Schwartz, A.R.; et al. Silencing of Hypoglossal Motoneurons Leads to Sleep Disordered Breathing in Lean Mice. Front. Neurol. 2018, 9, 962. [Google Scholar] [CrossRef]
- Marcus, C.L.; McColley, S.A.; Carroll, J.L.; Loughlin, G.M.; Smith, P.L.; Schwartz, A.R. Upper airway collapsibility in children with obstructive sleep apnea syndrome. J. Appl. Physiol. 1994, 77, 918–924. [Google Scholar] [CrossRef]
- Marcus, C.L. Sleep-disordered breathing in children. Am. J. Respir. Crit. Care Med. 2001, 164, 16–30. [Google Scholar] [CrossRef]
- Morris, D.L.; Rui, L. Recent advances in understanding leptin signaling and leptin resistance. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1247–E1259. [Google Scholar] [CrossRef]
- Friedman, J.M.; Halaas, J.L. Leptin and the regulation of body weight in mammals. Nature 1998, 395, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Breslow, M.J.; Min-Lee, K.; Brown, D.R.; Chacko, V.P.; Palmer, D.; Berkowitz, D.E. Effect of leptin deficiency on metabolic rate in ob/ob mice. Am. J. Physiol. 1999, 276, E443–E449. [Google Scholar] [PubMed]
- Ip, M.S.; Lam, K.S.; Ho, C.; Tsang, K.W.; Lam, W. Serum leptin and vascular risk factors in obstructive sleep apnea. Chest 2000, 118, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Phipps, P.R.; Starritt, E.; Caterson, I.; Grunstein, R.R. Association of serum leptin with hypoventilation in human obesity. Thorax 2002, 57, 75–76. [Google Scholar] [CrossRef] [PubMed]
- Maffei, M.; Halaas, J.; Ravussin, E.; Pratley, R.E.; Lee, G.H.; Zhang, Y.; Fei, H.; Kim, S.; Lallone, R.; Ranganathan, S. Leptin levels in human and rodent: Measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat. Med. 1995, 1, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Considine, R.V.; Sinha, M.K.; Heiman, M.L.; Kriauciunas, A.; Stephens, T.W.; Nyce, M.R.; Ohannesian, J.P.; Marco, C.C.; McKee, L.J.; Bauer, T.L. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N. Engl. J. Med. 1996, 334, 292–295. [Google Scholar] [CrossRef]
- Scarpace, P.J.; Zhang, Y. Leptin resistance: A prediposing factor for diet-induced obesity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R493–R500. [Google Scholar] [CrossRef]
- Wauman, J.; Tavernier, J. Leptin receptor signaling: Pathways to leptin resistance. Front. Biosci. (Landmark Ed.) 2011, 16, 2771–2793. [Google Scholar] [CrossRef]
- Deacon-Diaz, N.; Malhotra, A. Inherent vs. Induced Loop Gain Abnormalities in Obstructive Sleep Apnea. Front. Neurol. 2018, 9, 896. [Google Scholar] [CrossRef]
- Khoo, M.C. Determinants of ventilatory instability and variability. Respir. Physiol. 2000, 122, 167–182. [Google Scholar] [CrossRef]
- Younes, M.; Ostrowski, M.; Thompson, W.; Leslie, C.; Shewchuk, W. Chemical control stability in patients with obstructive sleep apnea. Am. J. Respir. Crit. Care Med. 2001, 163, 1181–1190. [Google Scholar] [CrossRef] [PubMed]
- Eckert, D.J.; Malhotra, A.; Jordan, A.S. Mechanisms of apnea. Prog. Cardiovasc. Dis. 2009, 51, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Younes, M. Role of respiratory control mechanisms in the pathogenesis of obstructive sleep disorders. J. Appl. Physiol. 2008, 105, 1389–1405. [Google Scholar] [CrossRef] [PubMed]
- Deacon-Diaz, N.L.; Sands, S.A.; McEvoy, R.D.; Catcheside, P.G. Daytime loop gain is elevated in obstructive sleep apnea but not reduced by CPAP treatment. J. Appl. Physiol. 2018, 125, 1490–1497. [Google Scholar] [CrossRef] [PubMed]
- Narkiewicz, K.; van de Borne, P.J.; Pesek, C.A.; Dyken, M.E.; Montano, N.; Somers, V.K. Selective potentiation of peripheral chemoreflex sensitivity in obstructive sleep apnea. Circulation 1999, 99, 1183–1189. [Google Scholar] [CrossRef]
- Peng, Y.; Kline, D.D.; Dick, T.E.; Prabhakar, N.R. Chronic intermittent hypoxia enhances carotid body chemoreceptor response to low oxygen. Adv. Exp. Med. Biol. 2001, 499, 33–38. [Google Scholar]
- Deacon, N.L.; Catcheside, P.G. The role of high loop gain induced by intermittent hypoxia in the pathophysiology of obstructive sleep apnoea. Sleep Med. Rev. 2015, 22, 3–14. [Google Scholar] [CrossRef]
- Bach, K.B.; Mitchell, G.S. Hypoxia-induced long-term facilitation of respiratory activity is serotonin dependent. Respir. Physiol. 1996, 104, 251–260. [Google Scholar] [CrossRef]
- Mateika, J.H.; Mendello, C.; Obeid, D.; Badr, M.S. Peripheral chemoreflex responsiveness is increased at elevated levels of carbon dioxide after episodic hypoxia in awake humans. J. Appl. Physiol. 2004, 96, 1197–1205. [Google Scholar] [CrossRef]
- Pialoux, V.; Hanly, P.J.; Foster, G.E.; Brugniaux, J.V.; Beaudin, A.E.; Hartmann, S.E.; Pun, M.; Duggan, C.T.; Poulin, M.J. Effects of exposure to intermittent hypoxia on oxidative stress and acute hypoxic ventilatory response in humans. Am. J. Respir. Crit. Care Med. 2009, 180, 1002–1009. [Google Scholar] [CrossRef]
- Chowdhuri, S.; Shanidze, I.; Pierchala, L.; Belen, D.; Mateika, J.H.; Badr, M.S. Effect of episodic hypoxia on the susceptibility to hypocapnic central apnea during NREM sleep. J. Appl. Physiol. 2010, 108, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.-J.; Overholt, J.L.; Kline, D.; Kumar, G.K.; Prabhakar, N.R. Induction of sensory long-term facilitation in the carotid body by intermittent hypoxia: Implications for recurrent apneas. Proc. Natl. Acad. Sci. USA 2003, 100, 10073–10078. [Google Scholar] [CrossRef] [PubMed]
- Kline, D.D.; Peng, Y.-J.; Manalo, D.J.; Semenza, G.L.; Prabhakar, N.R. Defective carotid body function and impaired ventilatory responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1 alpha. Proc. Natl. Acad. Sci. USA 2002, 99, 821–826. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.W.; Peskind, E.; Raskind, M.; Boyko, E.J.; Porte, D. Cerebrospinal fluid leptin levels: Relationship to plasma levels and to adiposity in humans. Nat. Med. 1996, 2, 589–593. [Google Scholar] [CrossRef]
- Berry, R.B.; Kouchi, K.; Bower, J.; Prosise, G.; Light, R.W. Triazolam in patients with obstructive sleep apnea. Am. J. Respir. Crit. Care Med. 1995, 151, 450–454. [Google Scholar] [CrossRef]
- Parrino, L.; Terzano, M.G. Polysomnographic effects of hypnotic drugs. A review. Psychopharmacology (Berl.) 1996, 126, 1–16. [Google Scholar] [CrossRef]
- Rosenberg, R.; Roach, J.M.; Scharf, M.; Amato, D.A. A pilot study evaluating acute use of eszopiclone in patients with mild to moderate obstructive sleep apnea syndrome. Sleep Med. 2007, 8, 464–470. [Google Scholar] [CrossRef]
- Heinzer, R.C.; White, D.P.; Jordan, A.S.; Lo, Y.L.; Dover, L.; Stevenson, K.; Malhotra, A. Trazodone increases arousal threshold in obstructive sleep apnoea. Eur. Respir. J. 2008, 31, 1308–1312. [Google Scholar] [CrossRef]
- Eckert, D.J.; Owens, R.L.; Kehlmann, G.B.; Wellman, A.; Rahangdale, S.; Yim-Yeh, S.; White, D.P.; Malhotra, A. Eszopiclone increases the respiratory arousal threshold and lowers the apnoea/hypopnoea index in obstructive sleep apnoea patients with a low arousal threshold. Clin. Sci. 2011, 120, 505–514. [Google Scholar] [CrossRef]
- Smales, E.T.; Edwards, B.A.; Deyoung, P.N.; McSharry, D.G.; Wellman, A.; Velasquez, A.; Owens, R.; Orr, J.E.; Malhotra, A. Trazodone Effects on Obstructive Sleep Apnea and Non-REM Arousal Threshold. Ann. Am. Thorac. Soc. 2015, 12, 758–764. [Google Scholar] [CrossRef]
- Carter, S.G.; Berger, M.S.; Carberry, J.C.; Bilston, L.E.; Butler, J.E.; Tong, B.K.Y.; Martins, R.T.; Fisher, L.P.; McKenzie, D.K.; Grunstein, R.R.; et al. Zopiclone Increases the Arousal Threshold without Impairing Genioglossus Activity in Obstructive Sleep Apnea. Sleep 2016, 39, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Sands, S.A.; Terrill, P.I.; Edwards, B.A.; Taranto Montemurro, L.; Azarbarzin, A.; Marques, M.; de Melo, C.M.; Loring, S.H.; Butler, J.P.; White, D.P.; et al. Quantifying the Arousal Threshold Using Polysomnography in Obstructive Sleep Apnea. Sleep 2018, 41, zsx183. [Google Scholar] [CrossRef] [PubMed]
- Younes, M.; Park, E.; Horner, R.L. Pentobarbital sedation increases genioglossus respiratory activity in sleeping rats. Sleep 2007, 30, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Pedersen, N.P.; Yokota, S.; Hur, E.E.; Fuller, P.M.; Lazarus, M.; Chamberlin, N.L.; Saper, C.B. Glutamatergic signaling from the parabrachial nucleus plays a critical role in hypercapnic arousal. J. Neurosci. 2013, 33, 7627–7640. [Google Scholar] [CrossRef]
- Yokota, S.; Kaur, S.; VanderHorst, V.G.; Saper, C.B.; Chamberlin, N.L. Respiratory-related outputs of glutamatergic, hypercapnia-responsive parabrachial neurons in mice. J. Comp. Neurol. 2015, 523, 907–920. [Google Scholar] [CrossRef]
- Kaur, S.; Saper, C.B. Neural Circuitry Underlying Waking Up to Hypercapnia. Front. Neurosci. 2019, 13, 401. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).