Relationship between HDL Cholesterol Efflux Capacity, Calcium Coronary Artery Content, and Antibodies against ApolipoproteinA-1 in Obese and Healthy Subjects

 , , ,
, , ,
Abstract
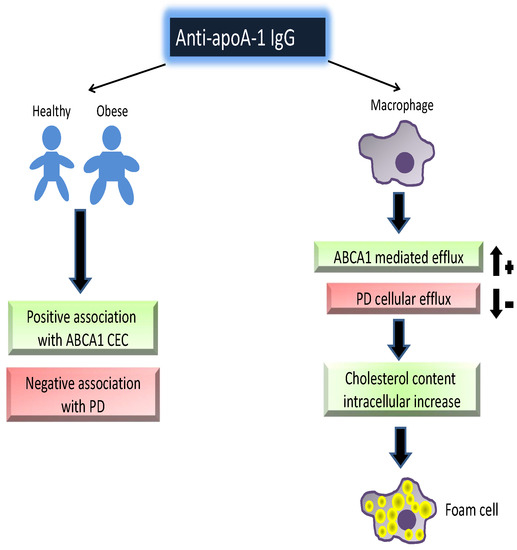
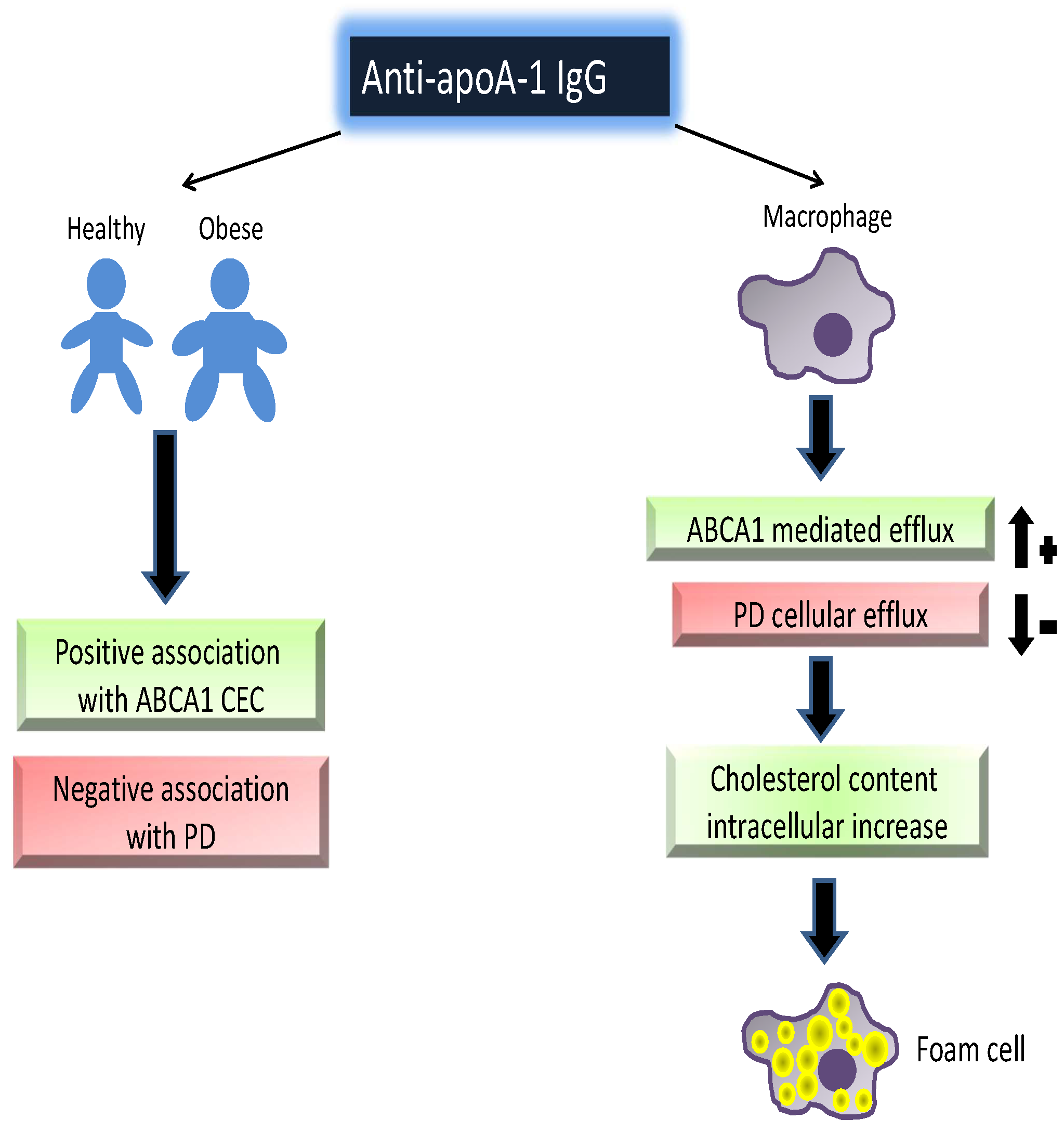
:
1. Introduction
2. Experimental Section
2.1. Study Population and Design
2.2. Study Endpoints
2.3. Assessment of Coronary Artery Calcification by Chest CTscan
2.4. Biochemical Analyses
2.5. Serum HDL Cholesterol Efflux Capacity (CEC)
2.6. Antibody Anti-apoA-1 Modulation of Cellular Cholesterol Efflux
2.7. Measurement of Intracellular Cholesterol Content
2.8. Antibody Anti-apoA-1 Modulation of Membrane Free Cholesterol: Assay of Cholesterol Oxidase
2.9. Anti-apoA-1 IgG Modulation of Cellular Cholesterol Esterification: ACAT Activity
2.10. Measurement of Free Cholesterol Content in Cell Supernatant
2.11. Statistical Analyses
3. Results
3.1. Study Characteristics
3.2. Associations between CEC Pathways, CAC Score, Framingham Risk Score, Pre-Beta-HDL, and hsCRP Levels
3.3. Associations between Anti-apoA-1 IgGs, Specific CEC Pathways, Pre-β-HDL, and hsCRP Levels
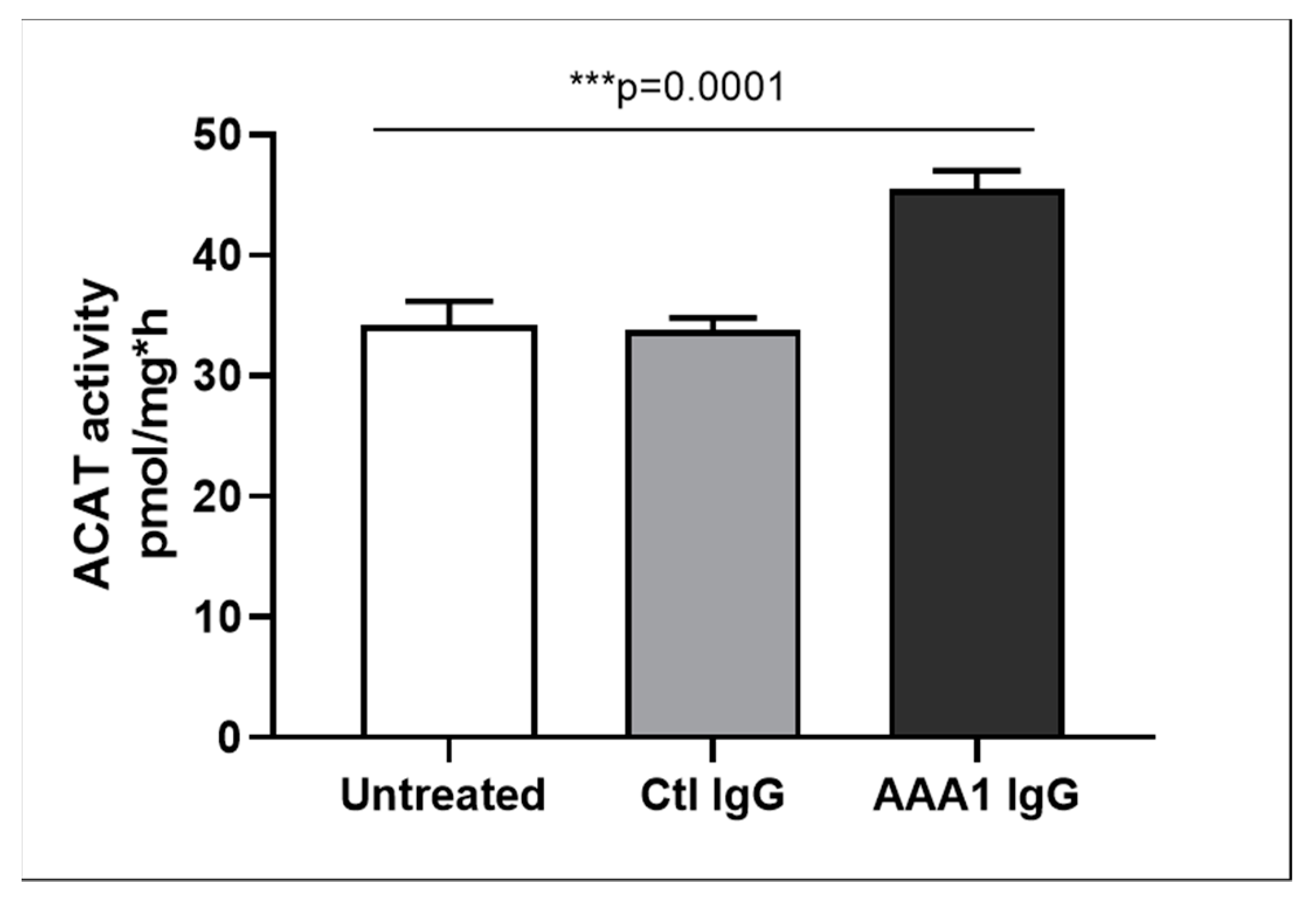
3.4. Anti-apoA-1 IgG-Mediated Modulation of Cellular Cholesterol Efflux
3.5. Modulation of Anti-apoA-1 IgGs on Intracellular and Membrane Cholesterol Content
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Khera, A.V.; Cuchel, M.; de la Llera-Moya, M.; Rodrigues, A.; Burke, M.F.; Jafri, K.; French, B.C.; Phillips, J.A.; Mucksavage, M.L.; Wilensky, R.L.; et al. Cholesterol Efflux Capacity, High-Density Lipoprotein Function, and Atherosclerosis. N. Engl. J. Med. 2011, 364, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Demler, O.V.; Adelman, S.J.; Collins, H.L.; Glynn, R.J.; Ridker, P.M.; Rader, D.J.; Mora, S. Cholesterol Efflux Capacity, High-Density Lipoprotein Particle Number, and Incident Cardiovascular Events: An Analysis from the Jupiter Trial (Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin). Circulation 2017, 135, 2494–2504. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, A.; Khera, A.; Berry, J.D.; Givens, E.G.; Ayers, C.R.; Wedin, K.E.; Neeland, I.J.; Yuhanna, I.S.; Rader, D.R.; de Lemos, J.A.; et al. Hdl Cholesterol Efflux Capacity and Incident Cardiovascular Events. N. Engl. J. Med. 2014, 371, 2383–2393. [Google Scholar] [CrossRef] [PubMed]
- Li, X.M.; Tang, W.H.; Mosior, M.K.; Huang, Y.; Wu, Y.; Matter, W.; Gao, V.; Schmitt, D.; Didonato, J.A.; Fisher, E.A.; et al. Paradoxical Association of Enhanced Cholesterol Efflux with Increased Incident Cardiovascular Risks. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1696–1705. [Google Scholar] [CrossRef] [PubMed]
- Lucero, D.; Sviridov, D.; Freeman, L.; Lopez, G.I.; Fassio, E.; Remaley, A.T.; Schreier, L. Increased Cholesterol Efflux Capacity in Metabolic Syndrome: Relation with Qualitative Alterations in Hdl and Lcat. Atherosclerosis 2015, 242, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Nestel, P.; Hoang, A.; Sviridov, D.; Straznicky, N. Cholesterol Efflux from Macrophages Is Influenced Differentially by Plasmas from Overweight Insulin-Sensitive and-Resistant Subjects. Int. J. Obes. 2012, 36, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Ormseth, M.J.; Yancey, P.G.; Yamamoto, S.; Oeser, A.M.; Gebretsadik, T.; Shintani, A.; Linton, M.F.; Fazio, S.; Davies, S.S.; Roberts, L.J., II; et al. Net Cholesterol Efflux Capacity of Hdl Enriched Serum and Coronary Atherosclerosis in Rheumatoid Arthritis. IJC Metab. Endocr. 2016, 13, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Antiochos, P.; Marques-Vidal, P.; Virzi, J.; Pagano, S.; Satta, N.; Bastardot, F.; Hartley, O.; Montecucco, F.; Mach, F.; Waeber, G.; et al. Association between Anti-Apolipoprotein a-1 Antibodies and Cardiovascular Disease in the General Population. Results from the Colaus Study. Thromb. Haemost. 2016, 116, 764–771. [Google Scholar] [CrossRef]
- Antiochos, P.; Marques-Vidal, P.; Virzi, J.; Pagano, S.; Satta, N.; Hartley, O.; Montecucco, F.; Mach, F.; Kutalik, Z.; Waeber, G.; et al. Anti-Apolipoprotein a-1 Igg Predict All-Cause Mortality and Are Associated with Fc Receptor-Like 3 Polymorphisms. Front. Immunol. 2017, 8, 437. [Google Scholar] [CrossRef]
- El-Lebedy, D.; Rasheed, E.; Kafoury, M.; Abd-El Haleem, D.; Awadallah, E.; Ashmawy, I. Anti-Apolipoprotein a-1 Autoantibodies as Risk Biomarker for Cardiovascular Diseases in Type 2 Diabetes Mellitus. J. Diabetes Complic. 2016, 30, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Vuilleumier, N.; Bas, S.; Pagano, S.; Montecucco, F.; Guerne, P.A.; Finckh, A.; Lovis, C.; Mach, F.; Hochstrasser, D.; Roux-Lombard, P.; et al. Anti-Apolipoprotein a-1 Igg Predicts Major Cardiovascular Events in Patients with Rheumatoid Arthritis. Arthr. Rheum. 2010, 62, 2640–2650. [Google Scholar] [CrossRef] [PubMed]
- Vuilleumier, N.; Montecucco, F.; Spinella, G.; Pagano, S.; Bertolotto, M.; Pane, B.; Pende, A.; Galan, K.; Roux-Lombard, P.; Combescure, C.; et al. Serum Levels of Anti-Apolipoprotein a-1 Auto-Antibodies and Myeloperoxidase as Predictors of Major Adverse Cardiovascular Events after Carotid Endarterectomy. Thromb. Haemost. 2013, 109, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Vuilleumier, N.; Rossier, M.F.; Pagano, S.; Python, M.; Charbonney, E.; Nkoulou, R.; James, R.; Reber, G.; Mach, F.; Roux-Lombard, P. Anti-Apolipoprotein a-1 Igg as an Independent Cardiovascular Prognostic Marker Affecting Basal Heart Rate in Myocardial Infarction. Eur. Heart J. 2010, 31, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Mannic, T.; Satta, N.; Pagano, S.; Python, M.; Virzi, J.; Montecucco, F.; Frias, M.A.; James, R.W.; Maturana, A.D.; Rossier, M.F.; et al. Cd14 as a Mediator of the Mineralocorticoid Receptor-Dependent Anti-Apolipoprotein a-1 Igg Chronotropic Effect on Cardiomyocytes. Endocrinology 2015, 156, 4707–4719. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, F.; Braunersreuther, V.; Burger, F.; Lenglet, S.; Pelli, G.; Carbone, F.; Fraga-Silva, R.; Stergiopulos, N.; Monaco, C.; Mueller, C.; et al. Anti-Apoa-1 Auto-Antibodies Increase Mouse Atherosclerotic Plaque Vulnerability, Myocardial Necrosis and Mortality Triggering Tlr2 and Tlr4. Thromb. Haemost. 2015, 114, 410–422. [Google Scholar] [CrossRef]
- Montecucco, F.; Vuilleumier, N.; Pagano, S.; Lenglet, S.; Bertolotto, M.; Braunersreuther, V.; Pelli, G.; Kovari, E.; Pane, B.; Spinella, G.; et al. Anti-Apolipoprotein a-1 Auto-Antibodies Are Active Mediators of Atherosclerotic Plaque Vulnerability. Eur. Heart J. 2011, 32, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Pagano, S.; Carbone, F.; Burger, F.; Roth, A.; Bertolotto, M.; Pane, B.; Spinella, G.; Palombo, D.; Pende, A.; Dallegri, F.; et al. Anti-Apolipoprotein a-1 Auto-Antibodies as Active Modulators of Atherothrombosis. Thromb. Haemost. 2016, 116, 554–564. [Google Scholar] [CrossRef]
- Pagano, S.; Satta, N.; Werling, D.; Offord, V.; de Moerloose, P.; Charbonney, E.; Hochstrasser, D.; Roux-Lombard, P.; Vuilleumier, N. Anti-Apolipoprotein a-1 Igg in Patients with Myocardial Infarction Promotes Inflammation through Tlr2/Cd14 Complex. J. Intern. Med. 2012, 272, 344–357. [Google Scholar] [CrossRef]
- Quercioli, A.; Montecucco, F.; Galan, K.; Ratib, O.; Roux-Lombard, P.; Pagano, S.; Mach, F.; Schindler, T.H.; Vuilleumier, N. Anti-Apolipoprotein a-1 Igg Levels Predict Coronary Artery Calcification in Obese but Otherwise Healthy Individuals. Mediat. Inflamm. 2012, 2012, 243158. [Google Scholar] [CrossRef]
- Ahmed, M.M.; Elserougy, E.M.; Al, G., II; Fikry, I.M.; Habib, D.F.; Younes, K.M.; Salem, N.A. Anti-Apolipoprotein a-I Antibodies and Paraoxonase 1 Activity in Systemic Lupus Erythematosus. EXCLI J. 2013, 12, 719–732. [Google Scholar]
- Ames, P.R.; Matsuura, E.; Batuca, J.R.; Ciampa, A.; Lopez, L.L.; Ferrara, F.; Iannaccone, L.; Alves, J.D. High-Density Lipoprotein Inversely Relates to Its Specific Autoantibody Favoring Oxidation in Thrombotic Primary Antiphospholipid Syndrome. Lupus 2010, 19, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Batuca, J.R.; Amaral, M.C.; Favas, C.; Paula, F.S.; Ames, P.R.J.; Papoila, A.L.; Delgado Alves, J. Extended-Release Niacin Increases Anti-Apolipoprotein a-I Antibodies That Block the Antioxidant Effect of High-Density Lipoprotein-Cholesterol: The Explore Clinical Trial. Br. J. Clin. Pharmacol. 2017, 83, 1002–1010. [Google Scholar] [CrossRef] [PubMed]
- Batuca, J.R.; Ames, P.R.; Amaral, M.; Favas, C.; Isenberg, D.A.; Delgado Alves, J. Anti-Atherogenic and Anti-Inflammatory Properties of High-Density Lipoprotein Are Affected by Specific Antibodies in Systemic Lupus Erythematosus. Rheumatology 2009, 48, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.; Yu, S.; Parks, B.W.; Black, L.L.; Kabarowski, J.H. Autoimmune-Mediated Reduction of High-Density Lipoprotein-Cholesterol and Paraoxonase 1 Activity in Systemic Lupus Erythematosus-Prone Gld Mice. Arthritis Rheum. 2011, 63, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Dullaart, R.P.F.; Pagano, S.; Perton, F.G.; Vuilleumier, N. Antibodies against the C-Terminus of Apoa-1 Are Inversely Associated with Cholesterol Efflux Capacity and Hdl Metabolism in Subjects with and without Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- Villines, T.C.; Hulten, E.A.; Shaw, L.J.; Goyal, M.; Dunning, A.; Achenbach, S.; Al-Mallah, M.; Berman, D.S.; Budoff, M.J.; Cademartiri, F.; et al. Prevalence and Severity of Coronary Artery Disease and Adverse Events among Symptomatic Patients with Coronary Artery Calcification Scores of Zero Undergoing Coronary Computed Tomography Angiography: Results from the Confirm (Coronary Ct Angiography Evaluation for Clinical Outcomes: An International Multicenter) Registry. J. Am. Coll. Cardiol. 2011, 58, 2533–2540. [Google Scholar] [CrossRef] [PubMed]
- Greenland, P.; Alpert, J.S.; Beller, G.A.; Benjamin, E.J.; Budoff, M.J.; Fayad, Z.A.; Foster, E.; Hlatky, M.A.; Hodgson, J.M.; Kushner, F.G.; et al. 2010 Accf/Aha Guideline for Assessment of Cardiovascular Risk in Asymptomatic Adults: A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2010, 56, e50–e103. [Google Scholar] [CrossRef]
- Miyazaki, O.; Kobayashi, J.; Fukamachi, I.; Miida, T.; Bujo, H.; Saito, Y. A New Sandwich Enzyme Immunoassay for Measurement of Plasma Pre-Beta1-Hdl Levels. J. Lipid Res. 2000, 41, 2083–2088. [Google Scholar]
- Vigna, G.B.; Satta, E.; Bernini, F.; Boarini, S.; Bosi, C.; Giusto, L.; Pinotti, E.; Tarugi, P.; Vanini, A.; Volpato, S.; et al. Flow-Mediated Dilation, Carotid Wall Thickness and Hdl Function in Subjects with Hyperalphalipoproteinemia. Nutr. Metab. Cardiovasc. Dis. NMCD 2014, 24, 777–783. [Google Scholar] [CrossRef]
- Favari, E.; Ronda, N.; Adorni, M.P.; Zimetti, F.; Salvi, P.; Manfredini, M.; Bernini, F.; Borghi, C.; Cicero, A.F. Abca1-Dependent Serum Cholesterol Efflux Capacity Inversely Correlates with Pulse Wave Velocity in Healthy Subjects. J. Lipid Res. 2013, 54, 238–243. [Google Scholar] [CrossRef]
- Ronda, N.; Favari, E.; Borghi, M.O.; Ingegnoli, F.; Gerosa, M.; Chighizola, C.; Zimetti, F.; Adorni, M.P.; Bernini, F.; Meroni, P.L. Impaired Serum Cholesterol Efflux Capacity in Rheumatoid Arthritis and Systemic Lupus Erythematosus. Ann. Rheum. Dis. 2014, 73, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Favari, E.; Calabresi, L.; Adorni, M.P.; Jessup, W.; Simonelli, S.; Franceschini, G.; Bernini, F. Small Discoidal Pre-Beta1 Hdl Particles Are Efficient Acceptors of Cell Cholesterol Via Abca1 and Abcg1. Biochemistry 2009, 48, 11067–11074. [Google Scholar] [CrossRef] [PubMed]
- Zimetti, F.; Adorni, M.P.; Ronda, N.; Gatti, R.; Bernini, F.; Favari, E. The Natural Compound Berberine Positively Affects Macrophage Functions Involved in Atherogenesis. Nutr. Metab. Cardiovasc. Dis. NMCD 2015, 25, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Markwell, M.A.; Haas, S.M.; Bieber, L.L.; Tolbert, N.E. A Modification of the Lowry Procedure to Simplify Protein Determination in Membrane and Lipoprotein Samples. Anal. Biochem. 1978, 87, 206–210. [Google Scholar] [CrossRef]
- Kellner-Weibel, G.; de La Llera-Moya, M.; Connelly, M.A.; Stoudt, G.; Christian, A.E.; Haynes, M.P.; Williams, D.L.; Rothblat, G.H. Expression of Scavenger Receptor Bi in Cos-7 Cells Alters Cholesterol Content and Distribution. Biochemistry 2000, 39, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Ho, Y.K.; Goldstein, J.L. The Cholesteryl Ester Cycle in Macrophage Foam Cells. Continual Hydrolysis and Re-Esterification of Cytoplasmic Cholesteryl Esters. J. Biol. Chem. 1980, 255, 9344–9352. [Google Scholar] [PubMed]
- Wilson, P.W.; D’Agostino, R.B.; Levy, D.; Belanger, A.M.; Silbershatz, H.; Kannel, W.B. Prediction of Coronary Heart Disease Using Risk Factor Categories. Circulation 1998, 97, 1837–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adorni, M.P.; Zimetti, F.; Billheimer, J.T.; Wang, N.; Rader, D.J.; Phillips, M.C.; Rothblat, G.H. The Roles of Different Pathways in the Release of Cholesterol from Macrophages. J. Lipid Res. 2007, 48, 2453–2462. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.C. Molecular Mechanisms of Cellular Cholesterol Efflux. J. Biolog. Chem. 2014, 289, 24020–24029. [Google Scholar] [CrossRef] [Green Version]
- Phillips, M.C.; Gillotte, K.L.; Haynes, M.P.; Johnson, W.J.; Lund-Katz, S.; Rothblat, G.H. Mechanisms of High Density Lipoprotein-Mediated Efflux of Cholesterol from Cell Plasma Membranes. Atherosclerosis 1998, 137, S13–S17. [Google Scholar] [CrossRef]
- Tomita, K.; Teratani, T.; Suzuki, T.; Shimizu, M.; Sato, H.; Narimatsu, K.; Usui, S.; Furuhashi, H.; Kimura, A.; Nishiyama, K.; et al. Acyl-Coa:Cholesterol Acyltransferase 1 Mediates Liver Fibrosis by Regulating Free Cholesterol Accumulation in Hepatic Stellate Cells. J. Hepatol. 2014, 61, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.Y.; Chang, C.C.; Cheng, D. Acyl-Coenzyme A:Cholesterol Acyltransferase. Annu. Rev. Biochem. 1997, 66, 613–638. [Google Scholar] [CrossRef] [PubMed]
- Gillotte, K.L.; Zaiou, M.; Lund-Katz, S.; Anantharamaiah, G.M.; Holvoet, P.; Dhoest, A.; Palgunachari, M.N.; Segrest, J.P.; Weisgraber, K.H.; Rothblat, G.H.; et al. Apolipoprotein-Mediated Plasma Membrane Microsolubilization. Role of Lipid Affinity and Membrane Penetration in the Efflux of Cellular Cholesterol and Phospholipid. J. Biolog. Chem. 1999, 274, 2021–2028. [Google Scholar] [CrossRef] [PubMed]
- Pagano, S.; Gaertner, H.; Cerini, F.; Mannic, T.; Satta, N.; Teixeira, P.C.; Cutler, P.; Mach, F.; Vuilleumier, N.; Hartley, O. The Human Autoantibody Response to Apolipoprotein a-I Is Focused on the C-Terminal Helix: A New Rationale for Diagnosis and Treatment of Cardiovascular Disease? PLoS ONE 2015, 10, e0132780. [Google Scholar] [CrossRef] [PubMed]
- Zannis, V.I.; Chroni, A.; Krieger, M. Role of Apoa-I, Abca1, Lcat, and Sr-Bi in the Biogenesis of Hdl. J. Mol. Med. 2006, 84, 276–294. [Google Scholar] [CrossRef] [PubMed]
- Frisdal, E.; Lesnik, P.; Olivier, M.; Robillard, P.; Chapman, M.J.; Huby, T.; Guerin, M.; Le Goff, W. Interleukin-6 Protects Human Macrophages from Cellular Cholesterol Accumulation and Attenuates the Proinflammatory Response. J. Biolog. Chem. 2011, 286, 30926–30936. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Overall (n = 69) | Obese (n = 35) | Non-Obese (n = 34) | * p Value | |
|---|---|---|---|---|
| Age, mean (+/−SD) | 44 (11.4) | 44.6 (12.0) | 43.5 (11.0) | 0.61 |
| Male Gender; n (%) | 42 (60.8) | 23 (65.7) | 19 (55.8) | 0.46 |
| Weight in Kg, mean (+/−SD) | 97.5 (26.37) | 116 (19.3) | 74.4 (12.5) | <0.001 |
| Height in cm, mean (+/−SD) | 74 (9.3) | 173 (9.4) | 172.2 (9.5) | 0.56 |
| BMI in kg/m2, mean (+/−SD) | 32.2 (8.8) | 39.0 (7.1) | 25.1 (3.0) | <0.001 |
| Framingham risk score; median (IQR; range) | 1 (0.9–4; 0.9–16) | 1 (0.9–6; 0.9–16) | 1 (0.9–3; 0.9–15) | 0.13 |
| FRS > 10%; n (%) | 7 (10.1) | 6 (17) | 1 (2.9) | 0.10 |
| Chest CT: | ||||
| Total CAC score, mean (+/−SD) | 4.9 (15.8) | 4.65 (14.8) | 5 (17) | 0.75 |
| Number of CAC lesions, mean (+/−SD) | 0.28 (0.75) | 0.28 (0.71) | 0.26 (0.79) | 0.74 |
| Presence of any CAC lesion; n (%) | 10 (14.4) | 6 (17.1) | 4 (11.7) | 0.73 |
| Lipid profile: | ||||
| Total cholesterol in mg/dL, mean (+/−SD) | 198.9 (36.4) | 198.5 (41.4) | 202.1 (31.5) | 0.69 |
| LDL cholesterol in mg/dL mean (+/−SD) | 127.9 (32.2) | 126.9 (36.9) | 133.9 (26.7) | 0.42 |
| HDL cholesterol in mg/dL, mean (+/−SD) | 47.1 (13.0) | 42.1 (11.5) | 50.4 (13.9) | 0.04 |
| Triglycerides in mg/dl, mean (+/−SD) | 97.5 (73.2) | 122.8 (70.8) | 83.0 (67.0) | <0.001 |
| Pre-β-HDL in μg/mL | 42.6 (16.62) | 49.2 (17.26) | 35.6 (12.8) | <0.001 |
| Cholesterol Efflux Capacity: | ||||
| ABCG1-mediated, mean (+/−SD) | 4.04 (1.2) | 4.04 (0.89) | 3.85 (1.49) | 0.96 |
| ABCA1-mediated, mean (+/−SD) | 3.98 (1.49) | 4.18 (1.34) | 3.67 (1.61) | 0.18 |
| Total, mean (+/-SD) | 13.73 (1.55) | 13.5 (1.57) | 13.80 (1.57) | 0.64 |
| Passive diffusion, mean (+/−SD) | 9.75 (2.05) | 9.5 (1.9) | 9.70 (2.16) | 0.19 |
| Anti-apoA-1 IgG OD; median (IQR; range) | 0.31 (0.18–0.43; 0–1.3) | 0.33 (0.2-0.48; 0.1–1.3) | 0.26 (0.16–0.38; 0–0.56) | 0.05 |
| Anti-apoA-1 positivity, n (%) hsCRP, median (IQR; range) | 6 (8.7) 2.9 (0.9–6; 0.9–26.1) | 6 (17.1) 5 (2.5–8.7; 0.9–26.1) | 0 (0) 1 (0.9–3; 0.32–11) | 0.02 < 0.0001 |
| Overall Subjects (n = 69) | Obese Subjects (n = 35) | Non-Obese Subjects (n = 34) | ||||
|---|---|---|---|---|---|---|
| Correlations | R Value (Spearman) | p Value | R Value (Spearman) | p Value | R Value (Spearman) | p Value |
| ABCG1-mediated CEC vs.: | ||||||
| CAC score | 0.02 | 0.84 | −0.03 | 0.82 | 0.10 | 0.57 |
| Nr of CAC lesions | 0.02 | 0.86 | −0.04 | 0.81 | 0.09 | 0.61 |
| Framingham RS | −0.01 | 0.90 | 0.06 | 0.70 | −0.09 | 0.59 |
| Pre-β-HDL | −0.22 | 0.09 | −0.19 | 0.27 | −0.23 | 0.18 |
| hsCRP | 0.007 | 0.94 | 0.04 | 0.77 | 0.22 | 0.20 |
| ABCA1-mediated CEC vs.: | ||||||
| CAC score | 0.26 | 0.02 | 0.47 | 0.004 | 0.05 | 0.76 |
| Nr of CAC lesions | 0.26 | 0.02 | 0.47 | 0.004 | 0.04 | 0.80 |
| Framingham RS | 0.30 | 0.01 | 0.18 | 0.29 | 0.40 | 0.01 |
| Pre-β-HDL | −0.08 | 0.48 | −0.15 | 0.38 | −0.25 | 0.15 |
| hsCRP | 0.05 | 0.44 | 0.04 | 0.79 | −0.12 | 0.47 |
| Passive Diffusion vs.: | ||||||
| CAC score | −0.30 | 0.01 | −0.46 | 0.006 | −0.10 | 0.53 |
| Nr of CAC lesions | −0.29 | 0.01 | −0.45 | 0.006 | −0.09 | 0.60 |
| Framingham RS | −0.33 | 0.006 | −0.40 | 0.01 | −0.22 | 0.21 |
| Pre-β-HDL | 0.15 | 0.20 | 0.39 | 0.02 | 0.06 | 0.72 |
| hsCRP | −0.01 | 0.90 | 0.06 | 0.63 | 0.17 | 0.35 |
| Correlations after excluding anti-apoA-1 IgG seropositive individuals in obese participants (n = 29) | ||||||
| Correlations | R Value (Spearman) | p Value | ||||
| ABCG1-mediated CEC vs.: | ||||||
| CAC score | 0.01 | 0.72 | ||||
| Nr of CAC lesions | 0.01 | 0.97 | ||||
| Framingham RS | 0.06 | 0.72 | ||||
| Pre-β HDL | −0.13 | 0.49 | ||||
| ABCA1-mediated CEC vs.: | ||||||
| CAC score | 0.39 | 0.03 | ||||
| Nr of CAC lesions | 0.39 | 0.03 | ||||
| Framingham RS | 0.24 | 0.20 | ||||
| Pre-β HDL | −0.14 | 0.48 | ||||
| Passive diffusion vs.: | ||||||
| CAC score | −0.39 | 0.04 | ||||
| Nr of CAC lesions | −0.39 | 0.04 | ||||
| Framingham RS | −0.45 | 0.009 | ||||
| Pre-β-HDL | 0.41 | 0.03 | ||||
| Univariate Analyses | Multivariate Analyses | |||||
|---|---|---|---|---|---|---|
| Continuous Predictors | Odds Ratio | 95% CI | p | Odds Ratio | 95% CI | p |
| ABCG1 CEC | 1.10 | 0.64–1.91 | 0.72 | 1.02 * | 0.51–2.05 | 0.94 |
| ABCA1 CEC | 1.17 | 1.17–3.13 | 0.009 | 1.38 ** | 0.67–2.86 | 0.38 |
| Passive diffusion | 0.56 | 0.32–0.97 | 0.03 | 0.78 *** | 0.37–1.54 | 0.44 |
| Framingham RS | 1.31 | 1.13 | 0.004 | 1.27 **** | 1.09–1.49 | 0.002 |
| Overall Subjects (n = 69) | Obese Subjects (n = 35) | Non-Obese Subjects (n = 34) | ||||
|---|---|---|---|---|---|---|
| Correlations | R value | p value | R value | p value | R value | p value |
| Anti-apoA-1 IgG (OD) vs.: | ||||||
| CAC score | 0.21 | 0.09 | 0.37 | 0.03 | 0.01 | 0.91 |
| Nr of CAC lesions | 0.21 | 0.09 | 0.37 | 0.03 | 0.02 | 0.88 |
| Framingham Risk Score | −0.03 | 0.81 | −0.03 | 0.82 | −0.11 | 0.51 |
| ABCG1-mediated CEC | −0.16 | 0.17 | −0.23 | 0.18 | −0.10 | 0.54 |
| ABCA1-mediated CEC | 0.48 | 0.00002 | 0.49 | 0.002 | 0.44 | 0.007 |
| Passive diffusion | −0.33 | 0.004 | −0.29 | 0.09 | −0.36 | 0.03 |
| Pre-β-HDL | 0.17 | 0.89 | −0.04 | 0.81 | −0.06 | 0.9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vuilleumier, N.; Pagano, S.; Montecucco, F.; Quercioli, A.; Schindler, T.H.; Mach, F.; Cipollari, E.; Ronda, N.; Favari, E. Relationship between HDL Cholesterol Efflux Capacity, Calcium Coronary Artery Content, and Antibodies against ApolipoproteinA-1 in Obese and Healthy Subjects. J. Clin. Med. 2019, 8, 1225. https://doi.org/10.3390/jcm8081225
Vuilleumier N, Pagano S, Montecucco F, Quercioli A, Schindler TH, Mach F, Cipollari E, Ronda N, Favari E. Relationship between HDL Cholesterol Efflux Capacity, Calcium Coronary Artery Content, and Antibodies against ApolipoproteinA-1 in Obese and Healthy Subjects. Journal of Clinical Medicine. 2019; 8(8):1225. https://doi.org/10.3390/jcm8081225
Chicago/Turabian StyleVuilleumier, Nicolas, Sabrina Pagano, Fabrizio Montecucco, Alessandra Quercioli, Thomas H. Schindler, François Mach, Eleonora Cipollari, Nicoletta Ronda, and Elda Favari. 2019. "Relationship between HDL Cholesterol Efflux Capacity, Calcium Coronary Artery Content, and Antibodies against ApolipoproteinA-1 in Obese and Healthy Subjects" Journal of Clinical Medicine 8, no. 8: 1225. https://doi.org/10.3390/jcm8081225