Advances and Remaining Challenges in the Study of Influenza and Anthrax Infection in Lung Cell Culture

Chemical and Biological Signature Sciences Group, Pacific Northwest National Laboratory, Richland, WA 99352, USA
*
Author to whom correspondence should be addressed.
Challenges 2018, 9(1), 2; https://doi.org/10.3390/challe9010002
Submission received: 16 December 2017
/
Revised: 12 January 2018
/
Accepted: 15 January 2018
/
Published: 17 January 2018
Abstract
:For over 30 years, immortalized lung cells have enabled researchers to elucidate lung-pathogen molecular interactions. However, over the last five years, numerous commercial companies are now providing affordable, ready-to-use primary lung cells for use in research laboratories. Despite advances in primary cell culture, studies using immortalized lung cells still dominate the recent scientific literature. In this review, we highlight recent influenza and anthrax studies using in vitro primary lung tissue models and how these models are providing better predictive outcomes for when extrapolated to in vivo observations. By focusing on one virus (influenza) and one bacterium (Bacillus anthracis), it is the intent that these primary lung cell culture observations may translate into more useful studies for other related viral and bacterial lung pathogens of interest.
1. Introduction
Immortalized cells, created through genetic techniques or isolated from cancerous tumors, have been instrumental in our understanding of molecular biology. Additionally, immortalized cells have been invaluable in helping biotechnology companies produce a variety of cell-derived vaccines on a global scale [1,2,3]. While convenient, a drawback of using immortalized cells to answer basic research biological questions is that these cells have rapid and uncharacteristic proliferation that is not typical of a normal cell state. In prior decades, immortalized cell lines were often the only option for use by cell biologists to understand host cellular responses to pathogens in vitro. At the time, extraction and culturing protocols for use of more physiologically relevant primary cells were not well established and often resulted in limited cell numbers from a harvested organ. Additionally, for some primary cells, conditions were not yet optimized, and therefore the cells did not proliferate after extraction. This meant extracting more tissue to have sufficient cell numbers for a given study. Due to the difficulty in using primary cells until recently, it is only in the last decade that we are starting to understand the finer details of certain aspects of molecular host-pathogen interactions.
In vitro studies using both immortalized and primary cell lines present challenges in terms of host physiological relevance. For immortalized cell lines, under the right growth conditions, a single cell can give rise to >100 progenitor cells in one week [4]. While these fast-growing cells still serve an important purpose in vaccine production and in some screening studies, this type of cell proliferation for immortalized cells can be argued to be in no way reflective of the healthy human body. The dysregulation of cell cycling and excessive growth of an immortalized cell is often more proliferative in tissue culture than even the most aggressive cancers in the human body. If cellular studies take into consideration this excessive proliferation by using serum-free medium that reduces proliferation, one could still argue these cells are still not metabolically normal. Conversely, with every passage of primary cells, the sub-population of cells that proliferate more rapidly than other population sub-types could potentially change the host response profile due to selection of the outcompeting rapidly proliferating cells. Even though primary cells such as human lung epithelial cells can survive in culture for 40–60 days and be passaged approximately 10–15 times [5], care should be taken that these cells be used at the lowest passage possible, so that they are more relevant to the organ in which they were isolated from.
In the last five years, numerous primary cell types derived from a range of diseased and non-diseased tissues can now be ordered commercially in ample quantities. Companies have streamlined and optimized primary tissue processing, making it a competitive option for in vitro studies. Additionally, primary cells can be purchased from a range of mammalian species and a variety of tissue/organ sites. For example, and within the scope of this review, it is now possible to purchase epithelial cells from the nasal, tracheal or bronchial lung region of a human donor. Epithelial cells from cystic fibrosis patients and Chronic Obstructive Pulmonary Disease (COPD) patients are also commercially available from Lonza (Basel, Switzerland), as well as lung fibroblasts from patients that suffered from pulmonary fibrosis. A few select companies, such as MatTek Corporation (Ashland, MA, USA) and Epithelix (Archamps, France), provide primary lung monocultures and co-culture lung cells in pre-defined well formats that are ready to use upon arrival. Lastly, commercial companies that provide primary cells often make available ready-to-use optimized medium for culturing their respective tissue type. Table 1 summarizes some of the various commercial options that make use of immortalized and primary cells from the human lung environment.
Considering both the increasing convenience and affordability for using primary cells, one could make a compelling argument that immortalized cells should not be used when there is a primary cell counterpart commercially available. At the very least, key findings identified in immortalized cell screening should be validated in a primary cell counterpart for relevancy. A key question that has been minimally addressed is how do the molecular responses at the RNA and protein level for an immortalized cell compare to that of its primary healthy cell counterpart after infection with pathogens? As we try to understand molecular outcomes for host cell responses to pathogenic infection (i.e., protein and RNA biomarkers), a “primary only” approach in low passage primary cells could provide better predictive outcomes. In the following two sections, recent experimental studies utilizing primary lung models to study both a viral respiratory pathogen (influenza) and a bacterial pathogen (Bacillus anthracis—anthrax) are discussed. Some of the major differences in experimental outcomes and interpretations that can occur from using immortalized versus primary cells are also highlighted.
2. Influenza A Virus (IAV)
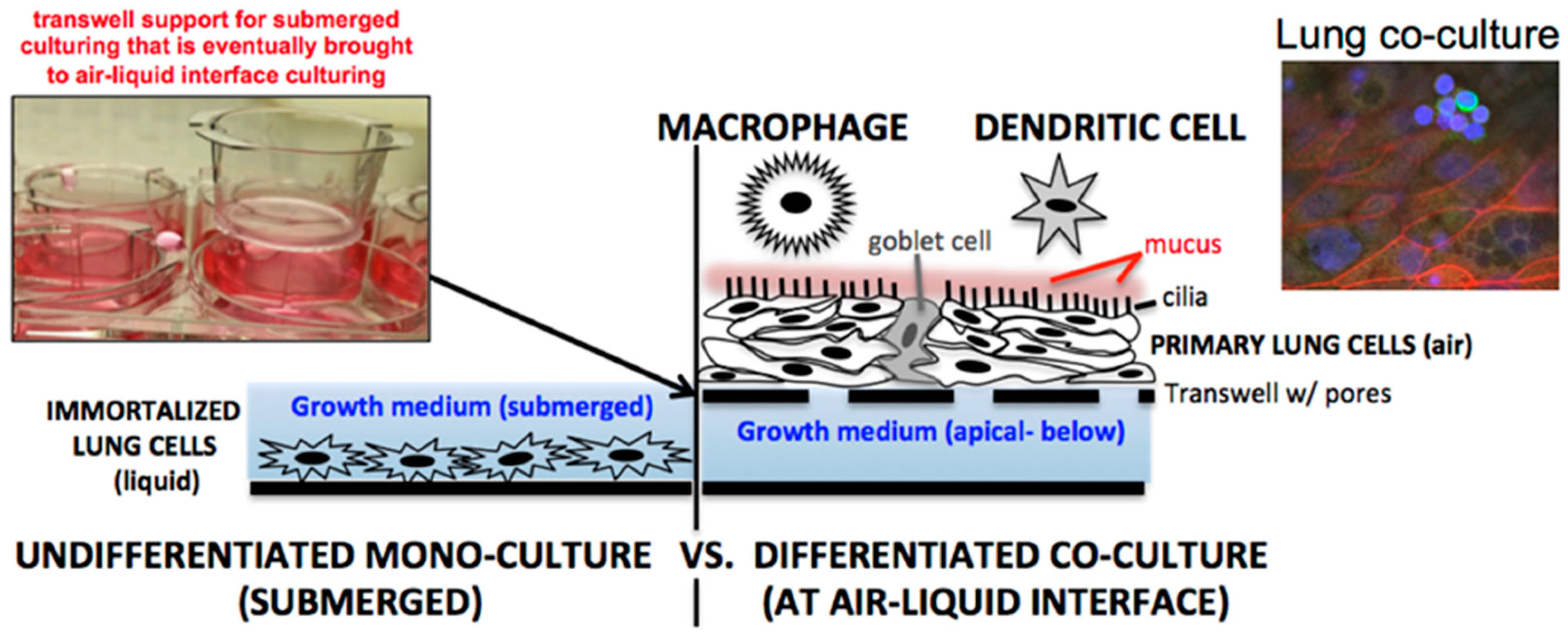
In the last decade, numerous influenza A virus (IAV) respiratory studies have used primary human lung epithelial cells to study host cell responses (see review [6]). Primary lung epithelial cells are often cultured at air-liquid-interface (ALI), undergo polarization, and produce mucus (Figure 1, right) [7]. Primary lung cells cultured at ALI therefore represent a more physiologically relevant system for viral infection than the submerged culturing conditions traditionally used for A549, Calu-3, and BEAS-2B immortalized lung cells (Figure 1, left). In our own laboratory we have found that NHBE cells (Lonza, CC-2540) should be used within the first six passages. However, multiple vials of high titer NHBEs can be cryo-preserved by the user at the end of passage two for later use. This allows an affordable option for long-term studies to be performed at either passage four or five.
Primary lung cell influenza A virus (IAV) studies have been published characterizing host lung responses to both human and avian influenza strains [8,9,10,11]. What makes primary lung cells highly relevant for IAV study is the cell’s ability to naturally secrete a class of proteases that cleave influenza hemagglutinin (HA) for viral cell entry [12]. Prior to the development of lung cell culturing at ALI, submerged immortalized cells (Figure 1, left) were cultured in medium supplemented with exogenous trypsin (~1 µg/mL) serving as an artificial way to cleave the viral HA through proteolysis to initiate infection [13]. The kinetics of viral infection for artificial trypsin supplementation versus cell-based secreted protease for cleaving of viral HA could represent differing IAV infection outcomes in vitro. For example, recent results supplementing pig primary lung cells at ALI with and without exogenous trypsin revealed higher viral titers for IAV in samples for the latter (6). One can conclude from this study that the use of trypsin speeds up the infection process and therefore could lead to alterations in the kinetics of host cell antiviral responses. In addition to differences in protease secretion in primary versus immortalized cell models, the presence of mucus also impacts how influenza spreads in cell culture. A mucus layer does not form on submerged cells covered in liquid, but at ALI, mucus readily forms about a week after exposure to air. Mucus can shield the virus from escaping apically into a liquid medium, forcing the virus to travel laterally to the neighboring cell, much like a natural infection. Interestingly, a mucus layer at ALI in human cells can also serve as a natural barrier against avian IAV strains such as H5N1 [8]. The production of mucus at ALI induces up-regulation of sialic acid alpha-2-6 expression used by human influenza viruses but minimal alpha-2-3 expression that is preferred for entry by avian influenza viruses. This feature of lung cells at ALI partially explains why avian H5N1 but to a lesser extent human IAV strains are highly attenuated in well-differentiated NHBE cells versus the undifferentiated state [9]. It can be concluded from these studies related to mucus and proteases that there are key differences in the study of IAV using two-dimensional immortalized cells versus the more physiologically relevant three-dimensional ALI primary lung cell platforms. These differences warrant further investigation.
A recent intriguing influenza-primary lung cell finding appears to show that both human and rat differentiated primary lung epithelial cells have antiviral activity toward influenza through the production of the oxidase hypothiocyanite [14]. The extracellular oxidation and inactivation of influenza as a mechanism to combat influenza infection is intriguing as it may indicate a certain low-level antiviral protection potential in the human lung. Results from this study showing inactivation of IAV by host secretions (present in the supernatant) infers that performing influenza studies with primary lung cells may alter infection outcomes and viral titers under particular culturing conditions. How hypothiocyanite is diluted or washed away during medium exchange in submerged lung cell versus ALI culturing is a question that has not been explored. In our own unpublished studies, we have noted that NHBE cells at ALI for some IAV strains seem to be refractory to infection when low level of virus are added (MOI below 0.01). The range of secreted host cellular enzymes that can inactivate influenza infection outcomes has been largely unexplored. Further studies exploring the natural low-level antiviral and antibacterial properties of primary lung cells will continue to be insightful and improve interpretations using these cell platforms.
Swine primary lung epithelial cells have also been used to characterize innate immune responses to IAV infection [15] and the reassortment tendencies of IAV in co-infected cells [16]. Previous results have shown that primary swine lung epithelial cells compared to the immortalized porcine kidney epithelial cell line (PK-1) give rise to different reassortment outcomes after co-infection when introducing human pandemic H1N1 and swine H3N2 [16]. Considering that pigs are the mixing vessel for IAV reassortment, understanding how lung cells support novel influenza variants after a cell is co-infected with two IAV strains is an active research area. Besides pigs, primary ferret tracheal-lung cells have also been infected with IAV in vitro [17]. While these ferret studies were proof-of-principle, an extensively implemented primary ferret lung cell platform would nicely complement animal ferret studies that are often used to study at IAV aerosol transmission. Of note, the company Cell Biologics Inc (Chicago, IL, USA) now sells bronchial/alveolar primary lung epithelial cells derived from numerous species that include pig, rat, rabbit, hamster and monkey. However, a commercial option for ferret primary lung cells to our knowledge, is not yet available.
Interestingly, the Pekosz lab has established a system for isolating and differentiating primary human nasal epithelial cell (hNECs) as an in vitro model. These cells allow for the in vitro study of live attenuated influenza vaccine (LAIV) strains that are delivered to the nasal cavity [18,19]. When the authors subjected hNECs to LAIV, they found a much greater degree of attenuation in hNEC cultures compared to other cell culture systems (MDCK) suggesting there are nasal epithelial cell specific factors contributing to attenuated replication [19]. Follow up studies showed that hNEC cells infected with LAIV elicits robust antiviral responses, particularly chemokines and type III interferons which may explain the restricted viral replication [18]. Furthermore, certain mutations in LAIV strains show greater attenuation in hNECs, but are not attenuated in immortalized MDCK cells [20]. Also using hNECs, Peretz and co-workers [21] have shown that seasonal IAV is more attenuated in hNECs isolated from female versus male donors that are subjected to estrogen compounds. Further studies with primary nasal and bronchial cells should prove enlightening as it relates to improving LAIV nasal delivery-based in vitro studies.
Lastly, a 2012 study has shown that differentiated NHBE cells exhibited greater restriction for H1N1, H3N2, and B vaccine viruses compared to immortalized MDCK cells, in agreement with human clinical trial data [22]. The authors note that NHBE cultures should be considered for use as a screening step for evaluating the restricted replication of influenza vaccine candidates. While this would be a nice use of the NHBE platform, the issue of donor to donor variation in influenza outcomes in lung epithelial cells has been minimally explored. Moving forward, it will be informative and insightful to characterize influenza fate across differing NHBE cells isolated from a diverse donor population to determine if these cells are suitable for screening purposes. A challenge that will remain, particularly for larger multi-year studies looking at genetic differences, is that there is a finite amount of a donor’s primary cells that will be available.
3. Inhalation Anthrax/Bacillus anthracis (BA)
B. anthracis (BA) is the etiological agent of anthrax. Spores of BA pose a significant health and bioterrorism risk to humans through inhalation. It is widely considered that inhaling BA spores leads to higher mortality rates versus either cutaneous or ingestion routes of spore exposure [23]. Once BA spores are inhaled and migrate to the more favorable environment of the lymphatic system, amplification (replication) of BA and the production of toxin ultimately kills the host. Although there is detailed knowledge regarding the structural and molecular aspects of the BA toxin components, it is only in the last decade that we are beginning to understand BA infection events in the lung at earlier time points of exposure. The physical interactions of BA with the lung that allows migration to the lymph node is an active research area (for review, see [23,24]). The mechanism of transport of BA from the lung to the lymph node is still controversial [25]. Whether the mechanism relies on phagocytic immune cells [26] or occurs through passive transport [27] is up for debate. Furthermore, the production of lethal toxin (LT) by the bacterium has been shown to compromise cytoskeletal integrity in NHBE cells, potentially facilitating a means for BA to travel to the lymph node [28].
There have been some insightful studies using both primary and immortalized lung cell lines to understand the relationship between the lung epithelium and BA [27,29,30,31]. Primary airway lung cells are susceptible to anthrax lethal toxin effects, whereas the immortalized A549 cell line is more resistant [29]. Recently, our laboratory has shown that BA proliferation, even after 48 h of culture, does not seem to harm cell integrity in vitro for submerged primary and immortalized cells, but does begin to compromise the ALI barrier by eight hours post-infection [30]. Additionally, these same studies show that proliferation of BA was slower in the primary versus immortalized (A549, Calu-3) cells. Lastly, these studies also showed that BA germination and proliferation occurred in the apical exterior cell environment outside the cell in both differentiated primary lung cells and submerged immortalized lung cells. These results differ from two earlier studies using the same cell types that appear to show that human lung epithelial cells internalize spores [31,32]. However, these earlier studies have been met with some debate [33]. Taken together, the aforementioned BA studies indicate in vitro model differences between primary and immortalized cells, and even differences in interpretation between labs. Lastly, as tissue culture supplements (serum, ultra-glutamine, etc.) can induce BA germination and proliferation [30,34], additional work is needed to increase the relevancy of these cell culture platforms so they are used appropriately.
As depicted in Figure 1, one aspect to consider when studying BA spores in lung cell cultures is that when lung cells are grown at air-liquid interface (ALI), they produce a mucus layer. This sticky substance traps BA (both spores and germinate bacteria) closely to the cell surface [30]. When cells are submerged in a defined volume of medium, such as for immortalized cells or when primary cells are not at ALI, mucus is minimally present, and the BA germinated bacterium tend to “float” within the liquid medium. One could conclude that BA toxin is more heavily concentrated in ALI systems compared to being diluted out in liquid medium. How secreted lung factors, and in turn how BA secreted toxins would dictate outcomes in submerged versus ALI lung cell culture systems, warrants further study. How different anthrax wild isolates versus the laboratory strain (Ames) impact lung cell host responses warrants further study. The use of lung cells to screen for more virulent versus avirulent isolates of BA may be possible, but additional testing is needed.
Using immune cells, in combination with lung epithelial cells in co-culture, can increase the relevancy of in vitro anthrax studies (discussed in Section 4 below). Immortalized mouse macrophage cell lines (either RAW 264.7 or J774A.1) [35,36,37] and two immortalized human monocyte cell lines (THP-1 and U937) [38,39,40] that are often differentiated into a macrophage-like cells have been used extensively for BA spore/bacterium phagocytosis studies. Primary mouse immune cells and human primary peripheral blood mononuclear cells (PBMCs) that can be differentiated into either macrophage-like or dendritic-like cell types have also have been used to study BA immune clearance [41,42]. Comparatively profiling immortalized versus primary phagocytic cell clearing of BA spores side-by-side has not been investigated. Because immortalized PBMC cell lines proliferate, whereas primary PBMCs do not, it would be expected that there could be substantial differences between these model cell systems for studying BA infection in vitro. Lastly, while immortalized PBMCs (~$500) cost less in the long run, as they can be expanded and stored for multiple experiments, the price of primary PBMC is also reasonable. For instance, primary PBMCs available are for less than $500 for >40 million cells from Lonza (CC-2702). Lonza also provides peripheral blood dendritic cells (CC-2701) that have already been differentiated from monocytes and are verified to be CD11c+ for a cost of $549.00 for >3 million cells, leading to further savings and convenience.
4. Future Directions and Considerations
A natural progression in primary cell culturing would be to establish co-culture of two or more primary cell types to try to more closely mimic the multi-cellular aspects of the in vivo environment. For example, incorporating immune cells such as dendritic cells and macrophages that are placed in the presence of lung epithelial cells could better allow for the study of lung innate immune responses (Figure 1, right). Australian researchers have already developed a pilot platform of immune-lung co-cultures for the study of Aspergillus fumigatus infections in vitro [43]. A triple co-culture system using lung cells, dendritic cells, and macrophages has also been constructed by Swiss researchers [44]. Yonker et al. [45] have recently created an inflamed airway mucosa co-culture model that is comprised of neutrophils and primary lung cells that were studied in the context of Pseudomonas aeruginosa infection. As already mentioned, there are lung co-cultures commercially available through Epithelix (MucilAir™-HF) or MatTek (EpiAirwayFT) (Table 1). These cells do not have an immune component but do incorporate a basal human airway fibroblast layer embedded below the lung epithelium. Both companies also provide customers with ready-to-infect convenient lung cells at ALI in a range of options so that researchers do not have to culture their own time-intensive lung cells.
In our own laboratory, we have further developed lung cell culturing by growing primary lung cells in a rotating rotisserie format (Figure 2) to dictate the three-dimensional architecture of the overall cell population. Optimizing this type of growth could pave the way in creating three-dimensional lung platforms on plastic scaffolds that mimic the branched architecture of the lung. Of note, a group at Columbia University [46] along with Japanese scientists [47] have recently culture 3D lung organoids from human-induced pluripotent stem cells (hiPSCs). While these platforms are primarily used to look at lung development and disease, there may be unique opportunities for use with pathogens. Of note, for further reading in the use of lung organoid systems, Nadkami at co-authors provide an informative review [48]. As additional technologies, such as the popularized lung-on-a-chip [49,50], are being established, and even the recent report of closed circulatory perfusion systems that includes a liver-heart-lung cell platform [51], scientists are now being enticed with next-generation platforms to study respiratory pathogens. Nevertheless, before scientists incorporate recent lung cell platform advances, there are still plenty of unknowns (i.e., lot-to-lot variability and optimized culture conditions) that need to be addressed in simplified lung tissue systems as it relates to bacterial and viral pathogens.
Acknowledgments
Pacific Northwest National Laboratory (PNNL) is a multi-program national laboratory operated by Battelle for the DOE under Contract DE-AC05-76RLO 1830.
Author Contributions
J.D.P. conceived and generated the manuscript; T.S.B. provided critical comments, feedback and technical expertise.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Milian, E.; Kamen, A.A. Current and emerging cell culture manufacturing technologies for influenza vaccines. BioMed Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Perdue, M.L.; Arnold, F.; Li, S.; Donabedian, A.; Cioce, V.; Warf, T.; Huebner, R. The future of cell culture-based influenza vaccine production. Expert Rev. Vaccines 2011, 10, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Barrett, P.N.; Mundt, W.; Kistner, O.; Howard, M.K. Vero cell platform in vaccine production: Moving towards cell culture-based viral vaccines. Expert Rev. Vaccines 2009, 8, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Masters, J.R.; Stacey, G.N. Changing medium and passaging cell lines. Nat. Protoc. 2007, 2, 2276–2284. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, A.S.; Randell, S.H.; Stewart, S.A.; Elenbaas, B.; Hartwell, K.A.; Brooks, M.W.; Fleming, M.D.; Olsen, J.C.; Miller, S.W.; Weinberg, R.A.; et al. Immortalization and transformation of primary human airway epithelial cells by gene transfer. Oncogene 2002, 21, 4577–4586. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.D.; Waters, K.M. Influenza-omics and the host response: Recent advances and future prospects. Pathogens 2017, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.E.; Torr, E.E.; Mohd Jamili, N.H.; Bosquillon, C.; Sayers, I. Evaluation of differentiated human bronchial epithelial cell culture systems for asthma research. J. Allergy 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, H.; Levin, D.; Forrest, S.; Beauchemin, C.A.; Tipper, J.; Knight, J.; Donart, N.; Layton, R.C.; Pyles, J.; Gao, P.; et al. Higher level of replication efficiency of 2009 (H1N1) pandemic influenza virus than those of seasonal and avian strains: Kinetics from epithelial cell culture and computational modeling. J. Virol. 2011, 85, 1125–1135. [Google Scholar] [CrossRef] [PubMed]
- Chan, R.W.; Yuen, K.M.; Yu, W.C.; Ho, C.C.; Nicholls, J.M.; Peiris, J.S.; Chan, M.C. Influenza H5N1 and H1N1 virus replication and innate immune responses in bronchial epithelial cells are influenced by the state of differentiation. PLoS ONE 2010, 5, e8713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerlach, R.L.; Camp, J.V.; Chu, Y.K.; Jonsson, C.B. Early host responses of seasonal and pandemic influenza a viruses in primary well-differentiated human lung epithelial cells. PLoS ONE 2013, 8, e78912. [Google Scholar] [CrossRef] [PubMed]
- Oshansky, C.M.; Pickens, J.A.; Bradley, K.C.; Jones, L.P.; Saavedra-Ebner, G.M.; Barber, J.P.; Crabtree, J.M.; Steinhauer, D.A.; Tompkins, S.M.; Tripp, R.A. Avian influenza viruses infect primary human bronchial epithelial cells unconstrained by sialic acid α2,3 residues. PLoS ONE 2011, 6, e21183. [Google Scholar] [CrossRef] [PubMed]
- Bottcher-Friebertshauser, E.; Klenk, H.D.; Garten, W. Activation of influenza viruses by proteases from host cells and bacteria in the human airway epithelium. Pathog. Dis. 2013, 69, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Klenk, H.D.; Rott, R.; Orlich, M.; Blodorn, J. Activation of influenza a viruses by trypsin treatment. Virology 1975, 68, 426–439. [Google Scholar] [CrossRef]
- Gingerich, A.; Pang, L.; Hanson, J.; Dlugolenski, D.; Streich, R.; Lafontaine, E.R.; Nagy, T.; Tripp, R.A.; Rada, B. Hypothiocyanite produced by human and rat respiratory epithelial cells inactivates extracellular H1N2 influenza a virus. Inflamm. Res. 2016, 65, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Hauser, M.J.; Dlugolenski, D.; Culhane, M.R.; Wentworth, D.E.; Tompkins, S.M.; Tripp, R.A. Antiviral responses by swine primary bronchoepithelial cells are limited compared to human bronchoepithelial cells following influenza virus infection. PLoS ONE 2013, 8, e70251. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.D.; Dlugolenski, D.; Nagy, T.; Gabbard, J.; Lee, C.; Tompkins, S.M.; Tripp, R.A. Polymerase discordance in novel swine influenza H3N2v constellations is tolerated in swine but not human respiratory epithelial cells. PLoS ONE 2014, 9, e110264. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Goldsmith, C.S.; Maines, T.R.; Belser, J.A.; Gustin, K.M.; Pekosz, A.; Zaki, S.R.; Katz, J.M.; Tumpey, T.M. Tropism and infectivity of influenza virus, including highly pathogenic avian H5N1 virus, in ferret tracheal differentiated primary epithelial cell cultures. J. Virol. 2013, 87, 2597–2607. [Google Scholar] [CrossRef] [PubMed]
- Forero, A.; Fenstermacher, K.; Wohlgemuth, N.; Nishida, A.; Carter, V.; Smith, E.A.; Peng, X.; Hayes, M.; Francis, D.; Treanor, J.; et al. Evaluation of the innate immune responses to influenza and live-attenuated influenza vaccine infection in primary differentiated human nasal epithelial cells. Vaccine 2017, 35, 6112–6121. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W.A., II; King, L.S.; Lane, A.P.; Pekosz, A. Restricted replication of the live attenuated influenza a virus vaccine during infection of primary differentiated human nasal epithelial cells. Vaccine 2015, 33, 4495–4504. [Google Scholar] [CrossRef] [PubMed]
- Wohlgemuth, N.; Ye, Y.; Fenstermacher, K.J.; Liu, H.; Lane, A.P.; Pekosz, A. The M2 protein of live, attenuated influenza vaccine encodes a mutation that reduces replication in human nasal epithelial cells. Vaccine 2017, 35, 6691–6699. [Google Scholar] [CrossRef] [PubMed]
- Peretz, J.; Pekosz, A.; Lane, A.P.; Klein, S.L. Estrogenic compounds reduce influenza a virus replication in primary human nasal epithelial cells derived from female, but not male, donors. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L415–L425. [Google Scholar] [CrossRef] [PubMed]
- Ilyushina, N.A.; Ikizler, M.R.; Kawaoka, Y.; Rudenko, L.G.; Treanor, J.J.; Subbarao, K.; Wright, P.F. Comparative study of influenza virus replication in MDCK cells and in primary cells derived from adenoids and airway epithelium. J. Virol. 2012, 86, 11725–11734. [Google Scholar] [CrossRef] [PubMed]
- Beierlein, J.M.; Anderson, A.C. New developments in vaccines, inhibitors of anthrax toxins, and antibiotic therapeutics for Bacillus anthracis. Curr. Med. Chem. 2011, 18, 5083–5094. [Google Scholar] [CrossRef] [PubMed]
- Goossens, P.L.; Tournier, J.N. Crossing of the epithelial barriers by Bacillus anthracis: The known and the unknown. Front. Microbiol. 2015, 6, 1122. [Google Scholar] [CrossRef] [PubMed]
- Weiner, Z.P.; Glomski, I.J. Updating perspectives on the initiation of Bacillus anthracis growth and dissemination through its host. Infect. Immun. 2012, 80, 1626–1633. [Google Scholar] [CrossRef] [PubMed]
- Guidi-Rontani, C. The alveolar macrophage: The Trojan horse of Bacillus anthracis. Trends Microbiol. 2002, 10, 405–409. [Google Scholar] [CrossRef]
- Booth, J.L.; Duggan, E.S.; Patel, V.I.; Langer, M.; Wu, W.; Braun, A.; Coggeshall, K.M.; Metcalf, J.P. Bacillus anthracis spore movement does not require a carrier cell and is not affected by lethal toxin in human lung models. Microbes Infect. 2016, 18, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.; Noack, D.; Wood, M.; Perego, M.; Knaus, U.G. Lung epithelial injury by B. Anthracis lethal toxin is caused by MKK-dependent loss of cytoskeletal integrity. PLoS ONE 2009, 4, e4755. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.Y.; Kang, C.I.; Hur, G.H.; Yang, J.M.; Shin, S. Bacillus anthracis lethal toxin induces cell-type-specific cytotoxicity in human lung cell lines. J. Appl. Microbiol. 2014, 116, 1334–1343. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.D.; Hutchison, J.R.; Hess, B.M.; Straub, T.M. Bacillus anthracis spores germinate extracellularly at air-liquid interface in an in vitro lung model under serum-free conditions. J. Appl. Microbiol. 2015, 119, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Russell, B.H.; Vasan, R.; Keene, D.R.; Koehler, T.M.; Xu, Y. Potential dissemination of Bacillus anthracis utilizing human lung epithelial cells. Cell. Microbiol. 2008, 10, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Russell, B.H.; Liu, Q.; Jenkins, S.A.; Tuvim, M.J.; Dickey, B.F.; Xu, Y. In vivo demonstration and quantification of intracellular Bacillus anthracis in lung epithelial cells. Infect. Immun. 2008, 76, 3975–3983. [Google Scholar] [CrossRef] [PubMed]
- Tournier, J.N.; Cleret, A.; Quesnel-Hellmann, A. What is the relevance of lung epithelial cells during the dissemination of spores in inhalational anthrax? Infect. Immun. 2009, 77, 565–566. [Google Scholar] [CrossRef] [PubMed]
- Bensman, M.D.; Mackie, R.S.; Minter, Z.A.; Gutting, B.W. Effect of animal sera on Bacillus anthracis sterne spore germination and vegetative cell growth. J. Appl. Microbiol. 2012, 113, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Bergman, N.H.; Passalacqua, K.D.; Gaspard, R.; Shetron-Rama, L.M.; Quackenbush, J.; Hanna, P.C. Murine macrophage transcriptional responses to Bacillus anthracis infection and intoxication. Infect. Immun. 2005, 73, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.; Cho, M.; Chun, J.H.; Cho, M.H.; Park, J.; Oh, H.B.; Yoo, C.K.; Rhie, G.E. The poly-gamma-d-glutamic acid capsule of Bacillus anthracis enhances lethal toxin activity. Infect. Immun. 2011, 79, 3846–3854. [Google Scholar] [CrossRef] [PubMed]
- Bergman, N.H.; Anderson, E.C.; Swenson, E.E.; Janes, B.K.; Fisher, N.; Niemeyer, M.M.; Miyoshi, A.D.; Hanna, P.C. Transcriptional profiling of Bacillus anthracis during infection of host macrophages. Infect. Immun. 2007, 75, 3434–3444. [Google Scholar] [CrossRef] [PubMed]
- Bradburne, C.; Chung, M.C.; Zong, Q.; Schlauch, K.; Liu, D.; Popova, T.; Popova, A.; Bailey, C.; Soppet, D.; Popov, S. Transcriptional and apoptotic responses of thp-1 cells to challenge with toxigenic, and non-toxigenic Bacillus anthracis. BMC Immunol. 2008, 9, 67. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, N.; Li, J.; Ferreira, C.S.; Little, S.F.; Friedlander, A.M.; Spitalny, G.L.; Casey, L.S. Enhancement of anthrax lethal toxin cytotoxicity: A subset of monoclonal antibodies against protective antigen increases lethal toxin-mediated killing of murine macrophages. Infect. Immun. 2004, 72, 3276–3283. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.H.; Lee, H.R.; Cho, M.H.; Park, O.K.; Park, J.; Rhie, G.E. The poly-γ-d-glutamic acid capsule surrogate of the Bacillus anthracis capsule is a novel toll-like receptor 2 agonist. Infect. Immun. 2015, 83, 3847–3856. [Google Scholar] [CrossRef] [PubMed]
- Brittingham, K.C.; Ruthel, G.; Panchal, R.G.; Fuller, C.L.; Ribot, W.J.; Hoover, T.A.; Young, H.A.; Anderson, A.O.; Bavari, S. Dendritic cells endocytose Bacillus anthracis spores: Implications for anthrax pathogenesis. J. Immunol. 2005, 174, 5545–5552. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.M.; Ramachandran, G.; Basu, S.; Rollins, S.; Mann, D.; Cross, A.S. The il-23/th17 axis is involved in the adaptive immune response to Bacillus anthracis in humans. Eur. J. Immunol. 2014, 44, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Chandorkar, P.; Posch, W.; Zaderer, V.; Blatzer, M.; Steger, M.; Ammann, C.G.; Binder, U.; Hermann, M.; Hortnagl, P.; Lass-Florl, C.; et al. Fast-track development of an in vitro 3D lung/immune cell model to study Aspergillus infections. Sci. Rep. 2017, 7, 11644. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, A.D.; Daum, N.; Bur, M.; Lehr, C.M.; Gehr, P.; Rothen-Rutishauser, B.M. An in vitro triple cell co-culture model with primary cells mimicking the human alveolar epithelial barrier. Eur. J. Pharm. Biopharm. 2011, 77, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Yonker, L.M.; Mou, H.; Chu, K.K.; Pazos, M.A.; Leung, H.; Cui, D.; Ryu, J.; Hibbler, R.M.; Eaton, A.D.; Ford, T.N.; et al. Development of a primary human co-culture model of inflamed airway mucosa. Sci. Rep. 2017, 7, 8182. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Huang, S.X.; de Carvalho, A.; Ho, S.H.; Islam, M.N.; Volpi, S.; Notarangelo, L.D.; Ciancanelli, M.; Casanova, J.L.; Bhattacharya, J.; et al. A three-dimensional model of human lung development and disease from pluripotent stem cells. Nat. Cell Biol. 2017, 19, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Gotoh, S.; Korogi, Y.; Seki, M.; Konishi, S.; Ikeo, S.; Sone, N.; Nagasaki, T.; Matsumoto, H.; Muro, S.; et al. Long-term expansion of alveolar stem cells derived from human iPS cells in organoids. Nat. Methods 2017, 14, 1097–1106. [Google Scholar] [CrossRef] [PubMed]
- Nadkarni, R.R.; Abed, S.; Draper, J.S. Organoids as a model system for studying human lung development and disease. Biochem. Biophys. Res. Commun. 2016, 473, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Esch, E.W.; Bahinski, A.; Huh, D. Organs-on-chips at the frontiers of drug discovery. Nat. Rev. Drug Discov. 2015, 14, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Matthews, B.D.; Mammoto, A.; Montoya-Zavala, M.; Hsin, H.Y.; Ingber, D.E. Reconstituting organ-level lung functions on a chip. Science 2010, 328, 1662–1668. [Google Scholar] [CrossRef] [PubMed]
- Skardal, A.; Murphy, S.V.; Devarasetty, M.; Mead, I.; Kang, H.W.; Seol, Y.J.; Shrike Zhang, Y.; Shin, S.R.; Zhao, L.; Aleman, J.; et al. Multi-tissue interactions in an integrated three-tissue organ-on-a-chip platform. Sci. Rep. 2017, 7, 8837. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Lung cell culturing through either (left) submerging cells in liquid culture medium or (right) culturing cells at air-liquid-interface on transwell supports. Additional complexity can be integrated by co-culturing immune cells on the apical lung cell surface. Shown on the right is a cluster of monocyte-derived macrophages (green—CD14+) placed above lung epithelial cells (red—actin).
Figure 1.
Lung cell culturing through either (left) submerging cells in liquid culture medium or (right) culturing cells at air-liquid-interface on transwell supports. Additional complexity can be integrated by co-culturing immune cells on the apical lung cell surface. Shown on the right is a cluster of monocyte-derived macrophages (green—CD14+) placed above lung epithelial cells (red—actin).
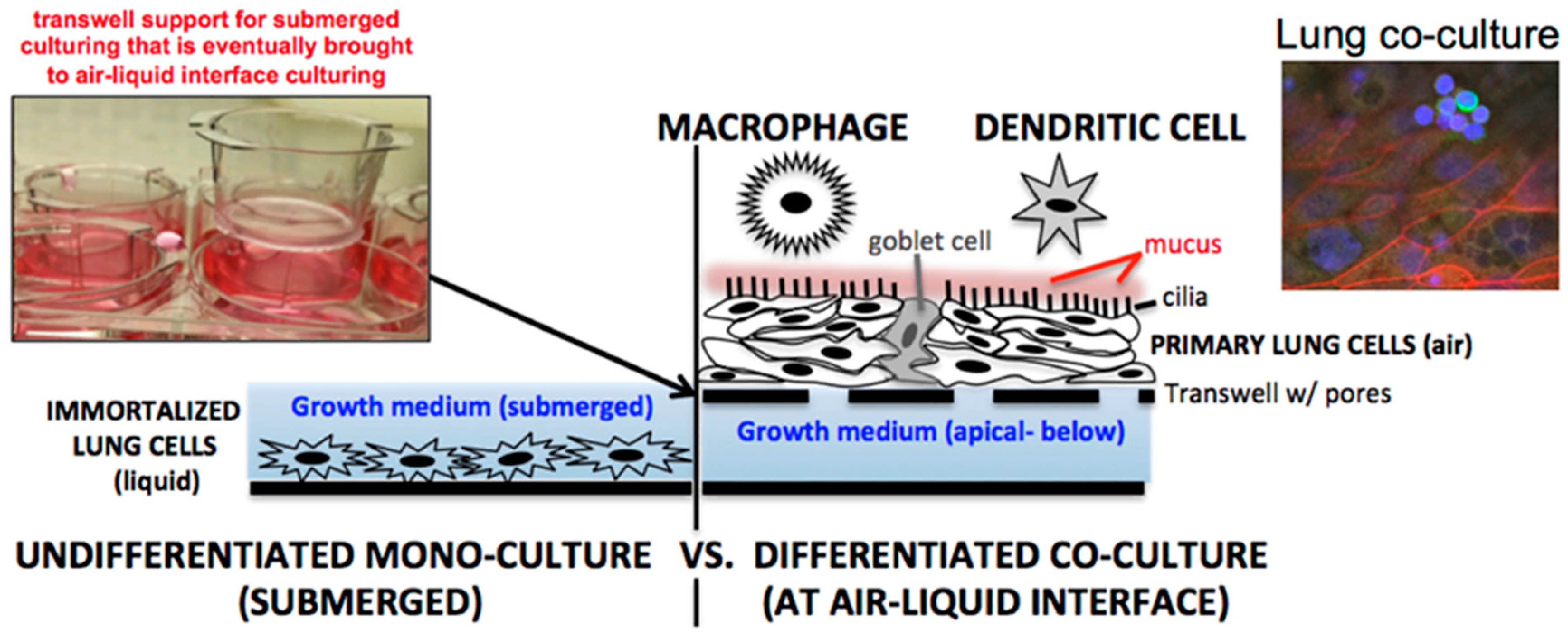
Figure 2.
A rotating vessel rotisserie format for the culturing of primary lung epithelial cells of three-dimensional tubular surfaces. By adjusting the angle of the rotation and rotation speed, the cells can be cultured and hydrated but also induced into secreting mucus as they are in contact with liquid only periodically.
Figure 2.
A rotating vessel rotisserie format for the culturing of primary lung epithelial cells of three-dimensional tubular surfaces. By adjusting the angle of the rotation and rotation speed, the cells can be cultured and hydrated but also induced into secreting mucus as they are in contact with liquid only periodically.


{kind=link}
{kind=link}
Table 1.
Commercially relevant primary lung monocultures and co-culture lung cell platforms.
| Cell Platform | Company, Product ID | Lung Cell Type |
|---|---|---|
| A549 | ATCC, CCL-185 | immortalized epithelial: monoculture |
| Calu-3 | ATCC, HTB-55 | immortalized epithelial: monoculture |
| BEAS-2B | ATCC, CRL-9609 | immortalized epithelial: monoculture |
| HBTEC 1 | Lifeline, FC-0035 | primary epithelial: monoculture |
| NHBE 2 | Lonza, CC-2540 3 | primary epithelial: monoculture |
| NHBE 2 COPD | Lonza, 195275 3 | primary epithelial: monoculture |
| NHBE 2 asthma | Lonza, 194911 3 | primary epithelial: monoculture |
| NHBE 2 cystic fibrosis | Lonza, 196979 3 | primary epithelial: monoculture |
| NHLF 4,5 | Lonza, CC-2512 | primary fibroblast: monoculture |
| Diseased HLF (iPF) 6 | Lonza, CC-7231 | primary fibroblast: monoculture |
| HLMEC 7 | Lifeline, FC-0058 | primary endothelial: monoculture |
| NHLSMC 8 | Lifeline, FC-0046 | primary smooth muscle: monoculture |
| EpiAirway™ | MatTek, (various) | primary epithelial: monoculture, ready-to-use at air-liquid-interface |
| EpiAirway™-FT | MatTek, (various) | primary epithelial + stromal fibroblasts: co-culture, ready-to-use at air-liquid-interface |
| MucilAir™ | Epithelix, (various) | primary epithelial: monoculture, ready-to-use at air-liquid-interface |
| MucilAir™-HF | Epithelix, (various) | primary epithelial + airway fibroblasts: co-culture, ready-to-use at air-liquid-interface |
| OncoCilAir™ | Epithelix, (various) | primary epithelial + airway fibroblasts + lung adenocarcinoma cells: triple-culture, ready-to-use at air-liquid-interface |
1 HBTEC—Human Bronchial Tracheal Epithelial Cells; 2 NHBE—Normal Human Bronchial Epithelial; 3 available in air-liquid interface options; 4 NHLF—Normal human lung fibroblasts; 5 available in diseased states (COPD, asthma); 6 iPF—Idiopathic Pulmonary Fibrosis; 7 HMLEC—Human Lung Microvascular Endothelial Cells; 8 NHLSMC—Normal Human Lung Smooth Muscles Cells.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Powell, J.D.; Straub, T.M. Advances and Remaining Challenges in the Study of Influenza and Anthrax Infection in Lung Cell Culture. Challenges 2018, 9, 2. https://doi.org/10.3390/challe9010002
AMA Style
Powell JD, Straub TM. Advances and Remaining Challenges in the Study of Influenza and Anthrax Infection in Lung Cell Culture. Challenges. 2018; 9(1):2. https://doi.org/10.3390/challe9010002
Chicago/Turabian StylePowell, Joshua D., and Timothy M. Straub. 2018. "Advances and Remaining Challenges in the Study of Influenza and Anthrax Infection in Lung Cell Culture" Challenges 9, no. 1: 2. https://doi.org/10.3390/challe9010002
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.