
Influence of the Localization of Ge Atoms within the Si(001)(4 × 2) Surface Layer on Semicore One-Electron States

Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
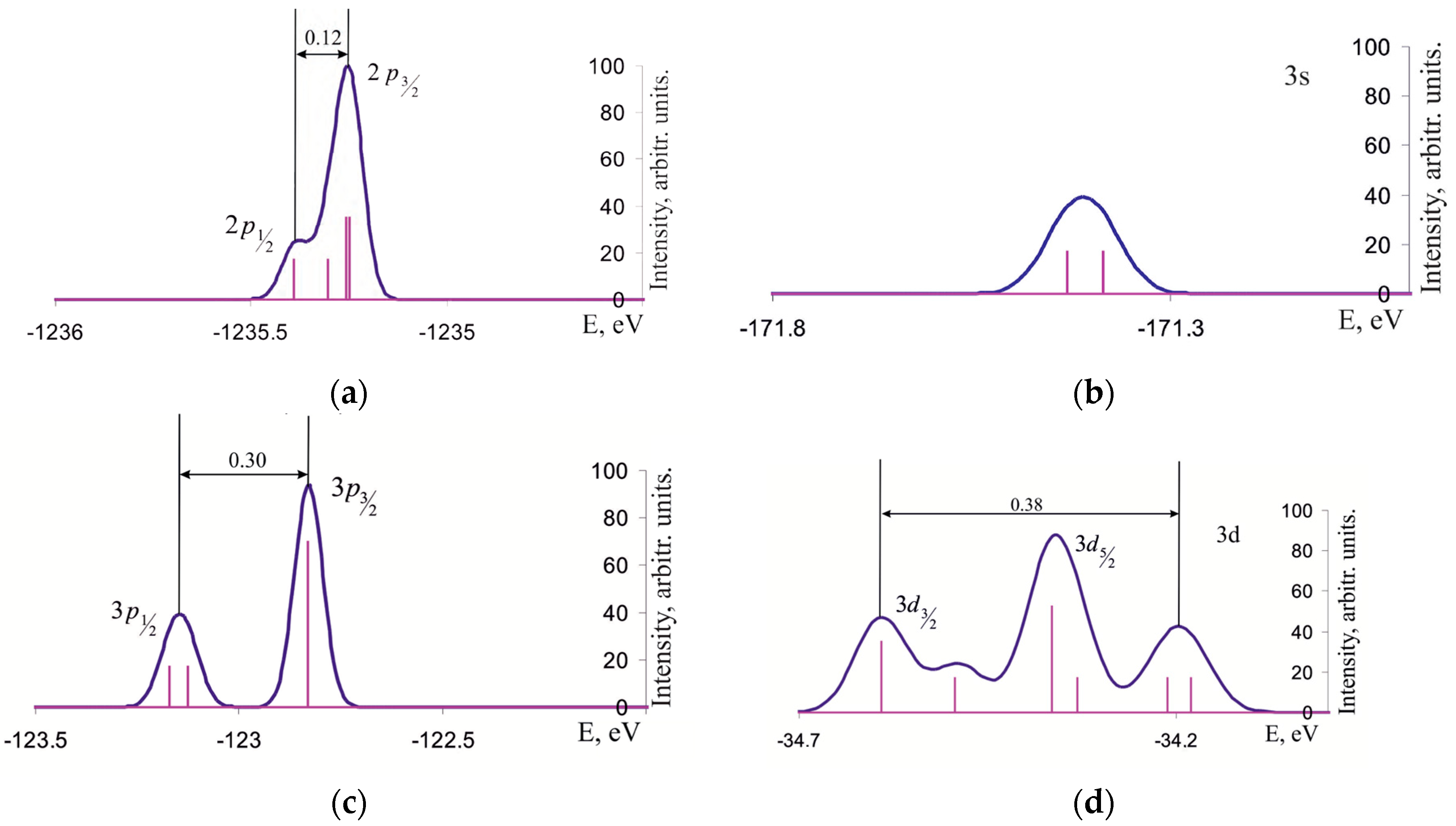
3.1. Density of One-Electron States of Ge2 Molecule
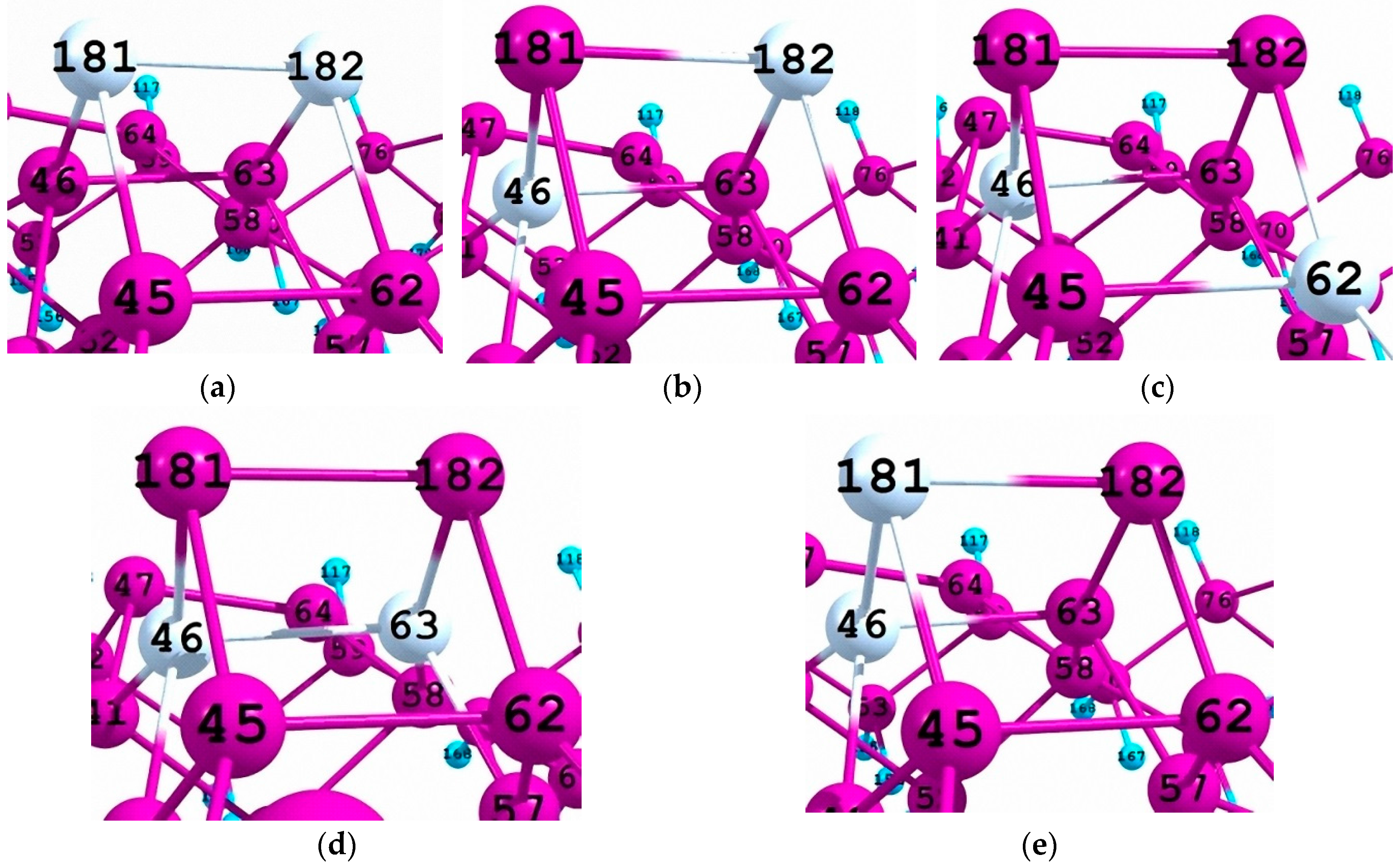
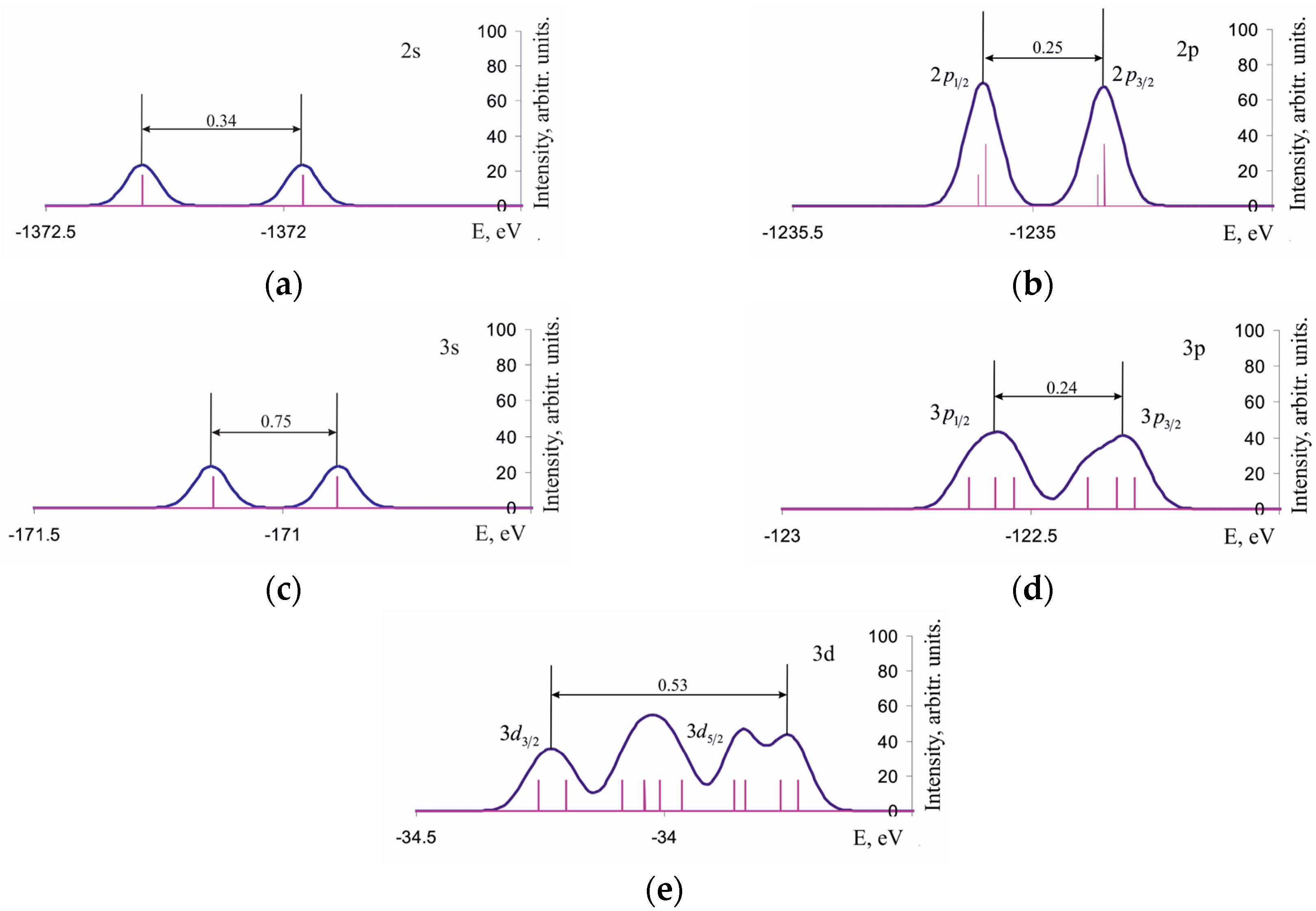
3.2. Density of One-Electron States of Si96H84Ge2 Cluster
3.3. Chemical Shifts of the Spin-Orbit Component inClusters A1–A4
4. Conclusions
- (i)
- Atomic orbitals from the closed d shell of germanium atom contribute to internal molecular orbitals that are responsible for a high binding energy of the =Ge-Ge= surface dimer.
- (ii)
- For Si96Ge2Н84 clusters containing only one germanium atom embedded in a crystalline silicon substrate, a chemical shift of the Ge(3d) line is positive (i.e., the binding energy of the corresponding electrons is higher comparing to that in the cluster containing =Ge-Ge= surface dimer). For clusters with both germanium atoms embedded in a substrate, such a chemical shift is negative.
Author Contributions
Conflicts of Interest
References
- Lozovoy, K.A.; Voytsekhovskiy, A.V.; Kokhanenko, A.P.; Satdarov, V.G.; Pchelyakov, O.P.; Nikiforov, A.I. Heterostructures with self-organized quantum dots of Ge on Si for optoelectronic devices. Opto-Electron. Rev. 2014, 22, 171–177. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, T.; Li, L.; Zuo, Y.; He, C.; Li, C.; Xue, C.; Cheng, B.; Wang, Q. Ge/Si quantum dots thin film solar cells. Appl. Phys. Lett. 2013, 103, 082101. [Google Scholar] [CrossRef]
- Chang, H.-T.; Wang, S.-Y.; Lee, S.-W. Designer Ge/Si composite quantum dots with enhanced thermoelectric properties. Nanoscale 2014, 6, 3593–3598. [Google Scholar] [CrossRef] [PubMed]
- Gomoyunova, M.G.; Pronin, I.I. Photoelectron spectroscopy of atomic core-shell levels at silicon surface. Zhurnal Tekhnicheskoi Fiziki 2004, 74, 1–34. (In Russian) [Google Scholar]
- Shklyaev, A.A.; Ichikawa, M. Creation of germanium and silicon nanostructures using scanning tunneling microscope. Uspekhi Fizicheskikh Nauk 2006, 176, 913–930. (In Russian) [Google Scholar] [CrossRef]
- Cotton, F.A.; Wilkinson, G. Advanced Inorganic Chemistry, 3rd ed.; Wiley: New York, NY, USA, 1972. [Google Scholar]
- Ratto, F.; Locatelli, A.; Fontana, S.; Kharrazi, S.; Ashtaputre, S.; Kulkarni, S.K.; Heun, S.; Rosei, F. Diffusion Dynamics during the Nucleation and Grown Ge/Si Nanoctructures on Si(111). Phys. Rev. Lett. 2006, 96, 096103. [Google Scholar] [CrossRef] [PubMed]
- Simonsen, A.C.; Schleberger, M.; Tougaard, S.; Hansen, J.L.; Larsen, A.N. Nanoctructure of Ge deposited on Si(111): A study by XPS peak shape analysis and AFM. Thin Solid Films 1999, 338, 165–171. [Google Scholar] [CrossRef]
- Siegbahn, K.; Nordling, C.; Fahlman, A.; Nerdberg, R.; Hamrin, K.; Hedman, J.; Johansson, G.; Bergmark, T.; Karlsson, S.-E.; Lindgrem, I.; et al. ESCA Atomic, Molecular and Solid State Structure Studied by Means of Electron Spectroscopy; Almqvist & Wiksells Boktryckeri AB: Uppsala, Sweden, 1967. [Google Scholar]
- Briggs, D.; Seah, M.P. Practical Surface Analysis. Auger and X-ray Photoelectron Spectroscopy, 2nd ed.; Wiley: New York, NY, USA, 1990. [Google Scholar]
- Teterin, Y.A.; Gagarin, S.G. Internal valence molecular orbitals of compounds and structure of photoelectron spectra. Uspekhi Khimii 1996, 65, 895–919. (In Russian) [Google Scholar]
- Ku, W.; Eguiluz, A.G. Band-gap problem in semiconductors revisited: Effects of core states and many-body self-consistency. Phys. Rev. Lett. 2002, 89, 126401. [Google Scholar] [CrossRef] [PubMed]
- Messmer, R.P.; Watkins, G.D. Molecular-orbital treatment for deep levels in semiconductors: Substitutional nitrogen and the lattice vacancy in diamond. Phys. Rev. B 1973, 7, 2568–2590. [Google Scholar] [CrossRef]
- Posternac, M.; Krakauer, H.; Freeman, A.J.; Koelling, D.D. Self-consistent electronic structure of surface: Surface states and surface resonance on W(001). Phys. Rev. B 1980, 21, 5601–5312. [Google Scholar] [CrossRef]
- Salahub, D.R.; Messmer, R.P. Molecular-orbital study of aluminum clusters containing up to 43 atoms. Phys. Rev. B 1977, 16, 2526–2536. [Google Scholar] [CrossRef]
- Abergel, D.S.L.; Apalkov, V.; Berashevich, J.; Zielger, K.; Chakraborty, T. Properties of graphene: A theoretical perspective. Adv. Phys. 2010, 59, 261–482. [Google Scholar] [CrossRef]
- Barnarda, A.S.; Snook, I.K. Size- and shape-dependence of the graphene transformation in the absence of hydrogen. J. Mater. Chem. 2010, 20, 10459–10464. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation energy density functionals of Becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Konecny, R.; Doren, D.J. Adsorption of water on Si(100)-(2 × 1): A study with density functional theory. J. Chem. Phys. 1997, 106, 2426–2435. [Google Scholar] [CrossRef]
- Yang, C.; Kang, H.C. Geometry of dimer reconstruction on the C(100), Si(100), and Ge(100) surfaces. J. Chem. Phys. 1999, 110, 11029–11037. [Google Scholar] [CrossRef]
- Penev, E.; Kratzer, P.; Scheffler, M. Effect of the cluster size in modeling the H2 desorption and dissociative adsorption on Si(001). J. Chem. Phys. 1999, 110, 3986–3994. [Google Scholar] [CrossRef]
- Gay, S.C.A.; Srivastava, G.P. Dimer length variation for different reconstructions of Si, Ge, and mixed Si-Ge dimers on Si(001) and Ge(001) substrates. Phys. Rev. B 1999, 60, 1488–1492. [Google Scholar] [CrossRef]
- Hess, B.A.; Marian, C.M.; Wahlgren, U.; Gropen, O. A mean-field spin-orbit method applicable to correlated wavefunctions. Chem. Phys. Lett. 1996, 251, 365–371. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic-structure system. Review. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Tkachuk, O.I.; Terebinska, M.I.; Lobanov, V.V. Structure of the dimeric adsorption complex Ge on the face of Si(001). Surface 2013, 5, 26–33. (In Ukrainian) [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Cluster | A1 | A2 | A3 | A4 |
|---|---|---|---|---|
| Relative energy, kcal/mol | 1.83 | 5.15 | 2.72 | 5.11 |
| Chemical shift, eV | +0.12 | −0.08 | −0.07 | +0.10 |
| Number of an Atom in the Cluster | Chemical Symbol of an Atom | Atomic Charge | Sum of Charges on Neighboring Atoms |
|---|---|---|---|
| Cluster А | |||
| 181 | Ge | −0.068 | 0.042 |
| 182 | Ge | −0.070 | 0.040 |
| 45 | Si | 0.068 | −0.179 |
| 62 | Si | 0.039 | −0.103 |
| 63 | Si | 0.071 | −0.098 |
| 46 | Si | 0.044 | −0.112 |
| Cluster А1 | |||
| 181 | Si | 0.018 | −0.095 |
| 182 | Ge | −0.065 | 0.118 |
| 45 | Si | 0.051 | −0.108 |
| 62 | Si | 0.008 | −0.094 |
| 63 | Si | 0.092 | −0.216 |
| 46 | Ge | −0.081 | 0.074 |
| Cluster А2 | |||
| 181 | Si | 0.014 | −0.021 |
| 182 | Si | 0.027 | −0.050 |
| 45 | Si | 0.075 | −0.224 |
| 62 | Ge | −0.010 | 0.095 |
| 63 | Si | 0.081 | −0.165 |
| 46 | Ge | −0.023 | 0.029 |
| Cluster А3 | |||
| 181 | Si | 0.020 | −0.037 |
| 182 | Si | 0.032 | −0.001 |
| 45 | Si | −0.004 | −0.107 |
| 62 | Si | −0.015 | −0.043 |
| 63 | Ge | −0.006 | −0.030 |
| 46 | Ge | −0.065 | −0.023 |
| Cluster А4 | |||
| 181 | Ge | −0.118 | 0.096 |
| 182 | Si | −0.014 | 0.125 |
| 45 | Si | 0.014 | −0.055 |
| 62 | Si | 0.032 | −0.151 |
| 63 | Si | 0.111 | −0.250 |
| 46 | Ge | −0.096 | 0.116 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tkachuk, O.I.; Terebinskaya, M.I.; Lobanov, V.V.; Arbuznikov, A.V. Influence of the Localization of Ge Atoms within the Si(001)(4 × 2) Surface Layer on Semicore One-Electron States. Computation 2016, 4, 14. https://doi.org/10.3390/computation4010014
Tkachuk OI, Terebinskaya MI, Lobanov VV, Arbuznikov AV. Influence of the Localization of Ge Atoms within the Si(001)(4 × 2) Surface Layer on Semicore One-Electron States. Computation. 2016; 4(1):14. https://doi.org/10.3390/computation4010014
Chicago/Turabian StyleTkachuk, Olha I., Maria I. Terebinskaya, Victor V. Lobanov, and Alexei V. Arbuznikov. 2016. "Influence of the Localization of Ge Atoms within the Si(001)(4 × 2) Surface Layer on Semicore One-Electron States" Computation 4, no. 1: 14. https://doi.org/10.3390/computation4010014