
Exploiting Polyelectrolyte Complexation for the Development of Adhesive and Bioactive Membranes Envisaging Guided Tissue Regeneration

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of the Chitosan–Catechol Conjugate (CHI–Cat)
2.3. Synthesis of the Hyaluronic Acid–Catechol Conjugate (HA–Cat)
2.4. Production of the Ternary Bioactive Glass Nanoparticles (BGNPs)
2.5. Ultraviolet-Visible Spectrophotometry Characterization
2.6. FTIR Spectroscopy Analysis
2.7. Zeta Potential (ζ) Measurements
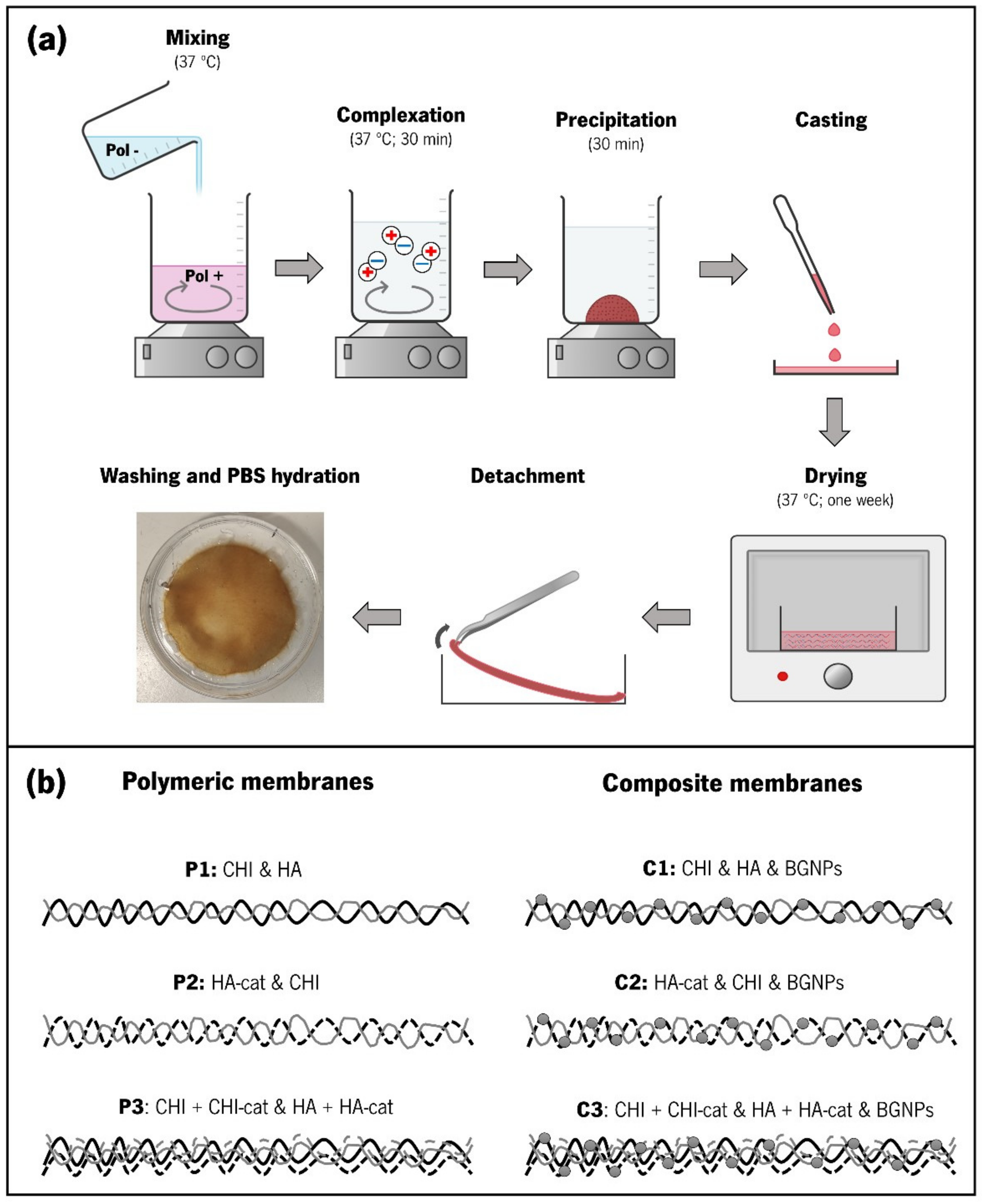
2.8. Production of Membranes through CoPEC Methodology
2.9. Morphological and Topographic Characterization of the CoPEC Membranes
2.10. Wettability and Surface Energy Analysis
2.11. Water Uptake (WU) and Weight Loss (WL)
2.12. Mechanical Characterization
2.13. In Vitro Bioactivity Studies
2.14. Cellular Assays
2.15. Statistical Analysis
3. Results and Discussion
3.1. UV-Vis Analysis of Catechol-Modified Polymers
3.2. Zeta (ζ)-Potential Measurements
3.3. Production of the CoPEC Membranes
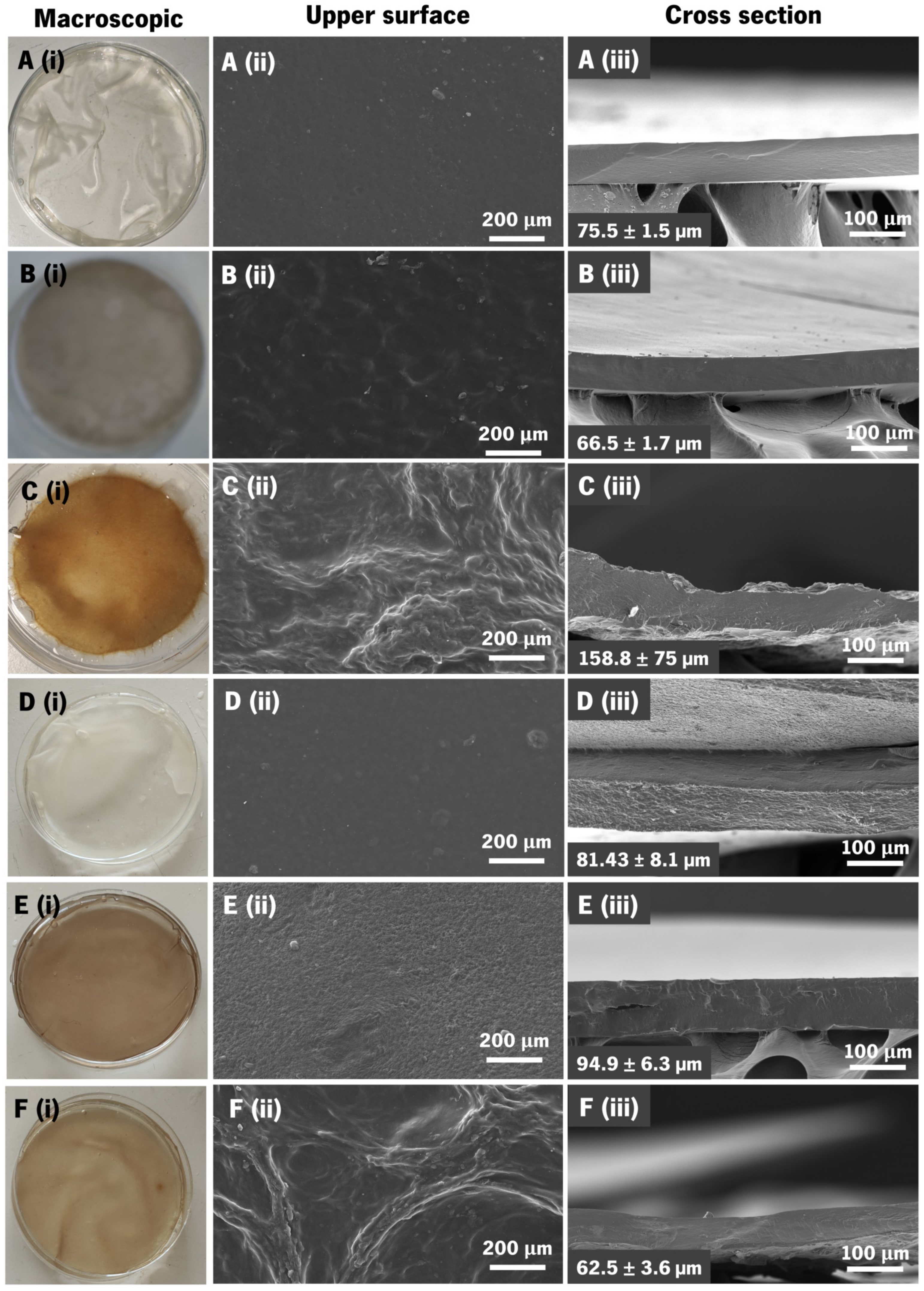
3.4. Membranes’ Morphological and Topographic Characterization
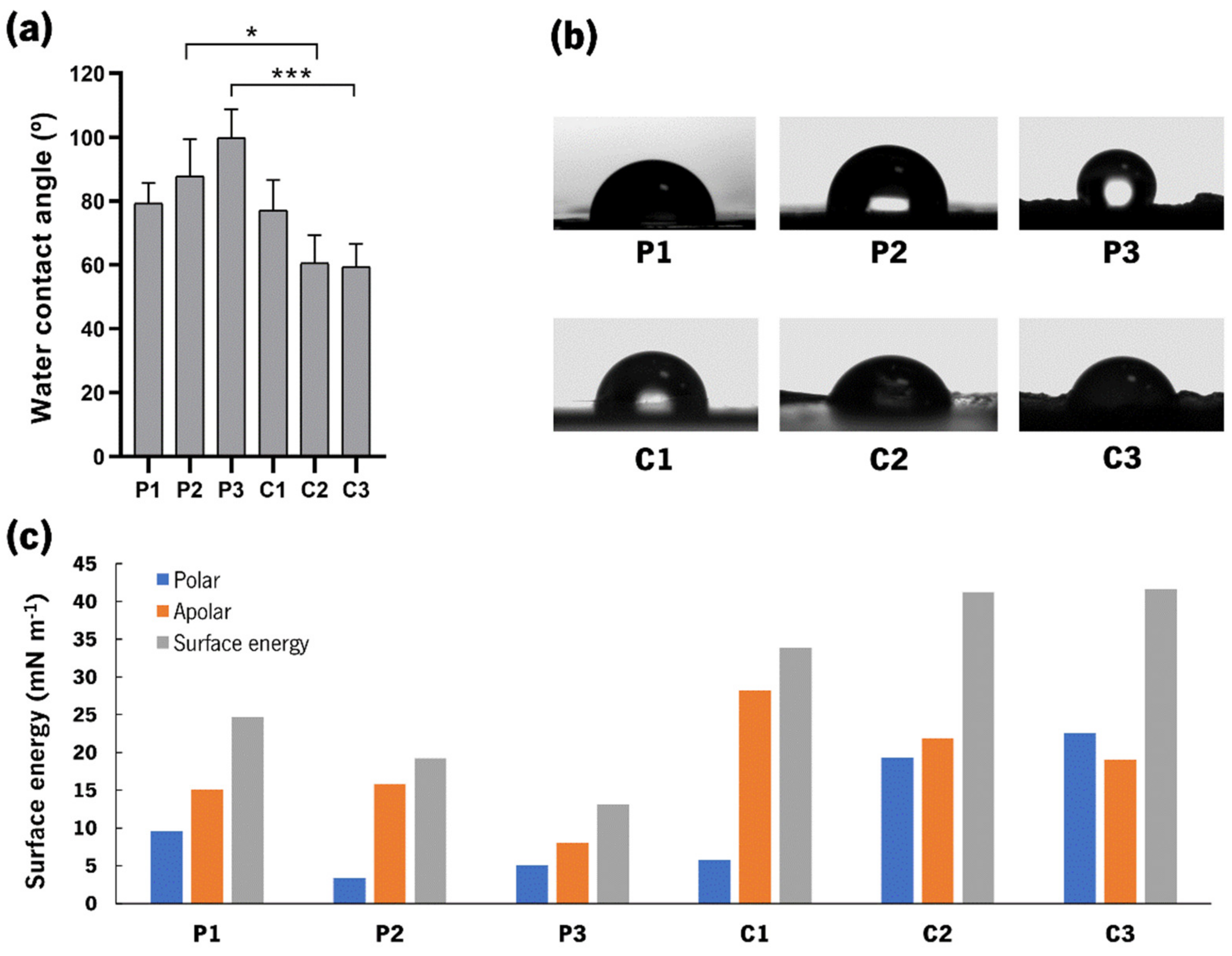
3.5. Water Contact Angle Measurements
3.6. Swelling and Degradation Studies
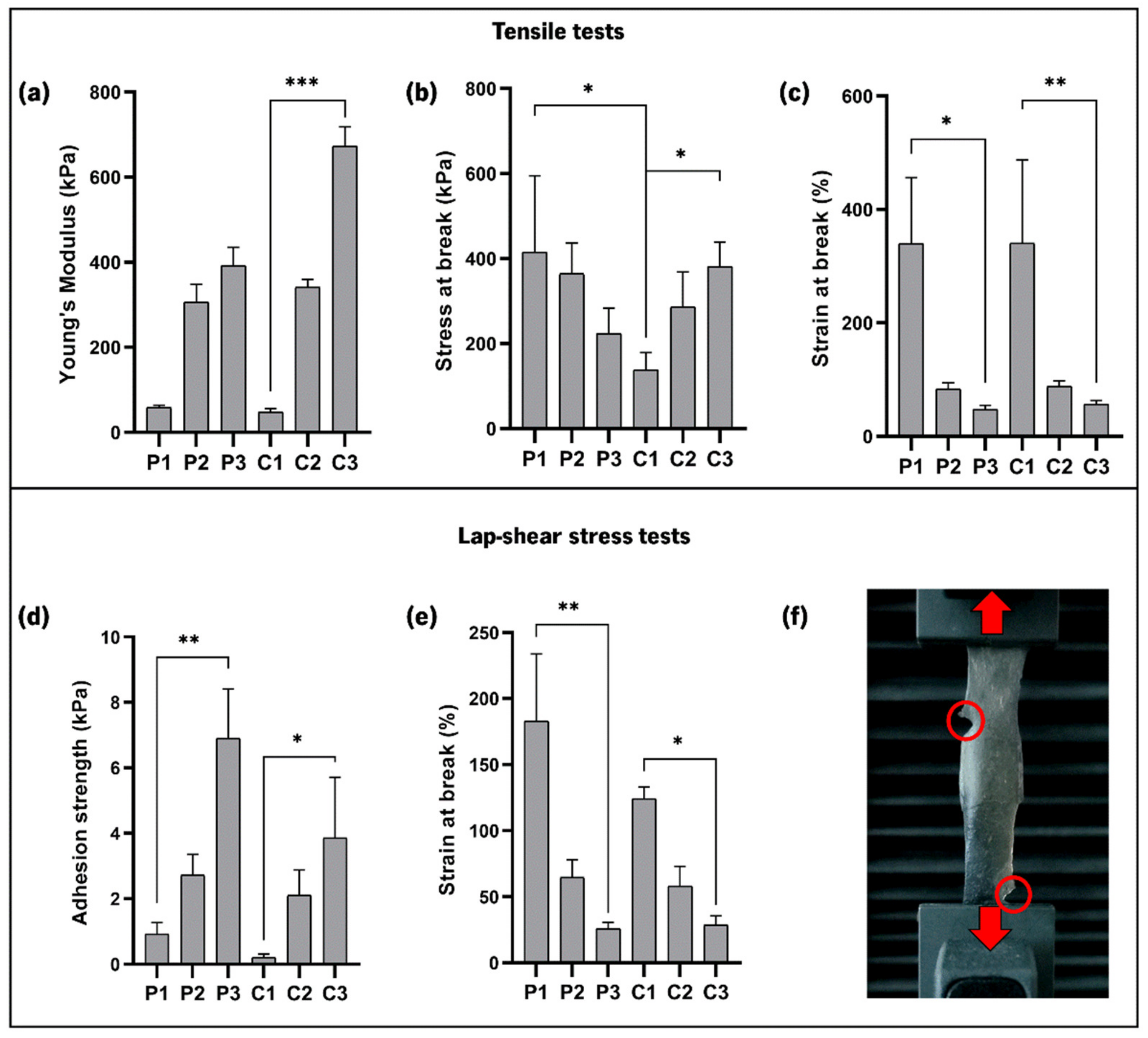
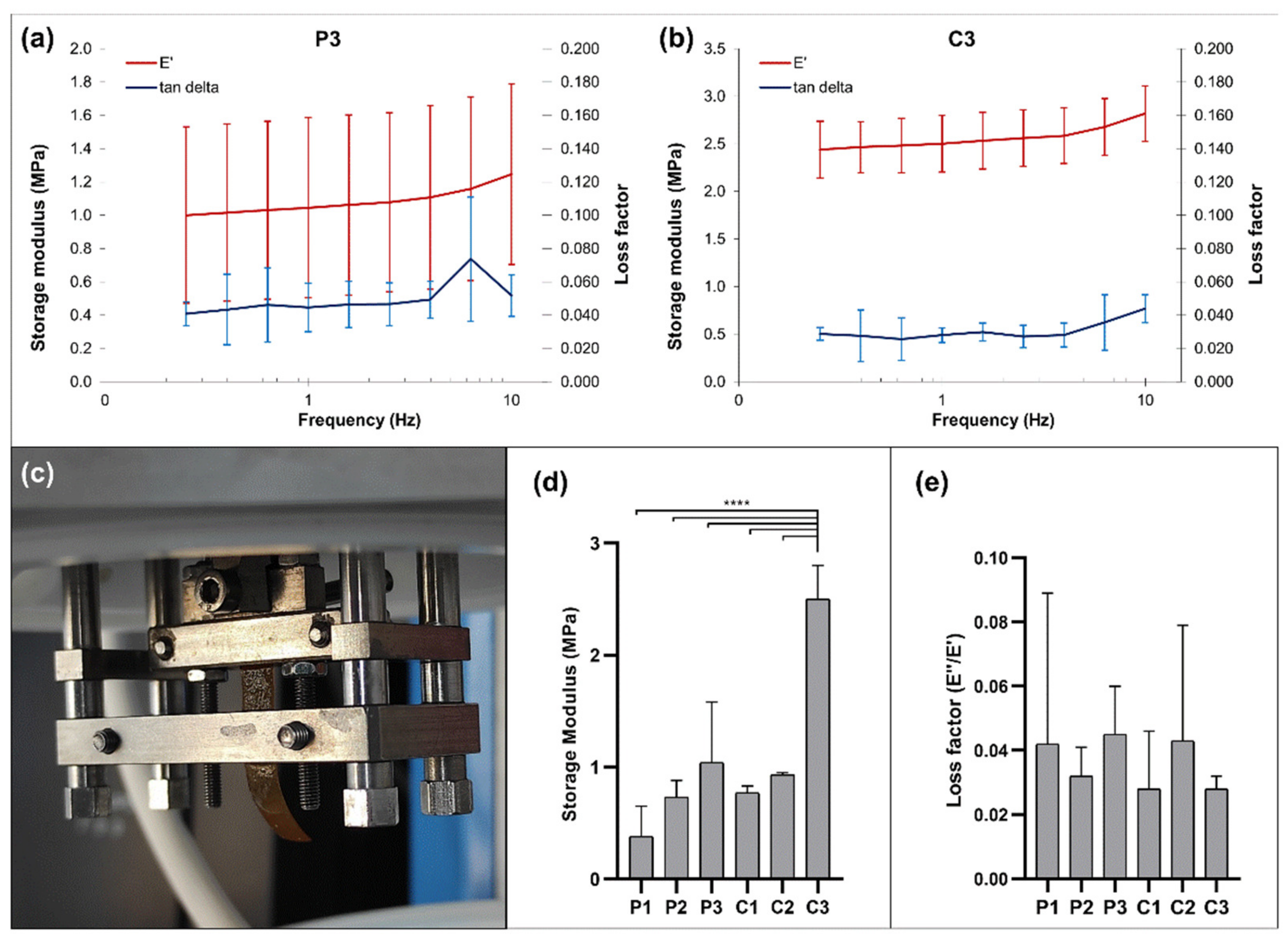
3.7. Mechanical Properties
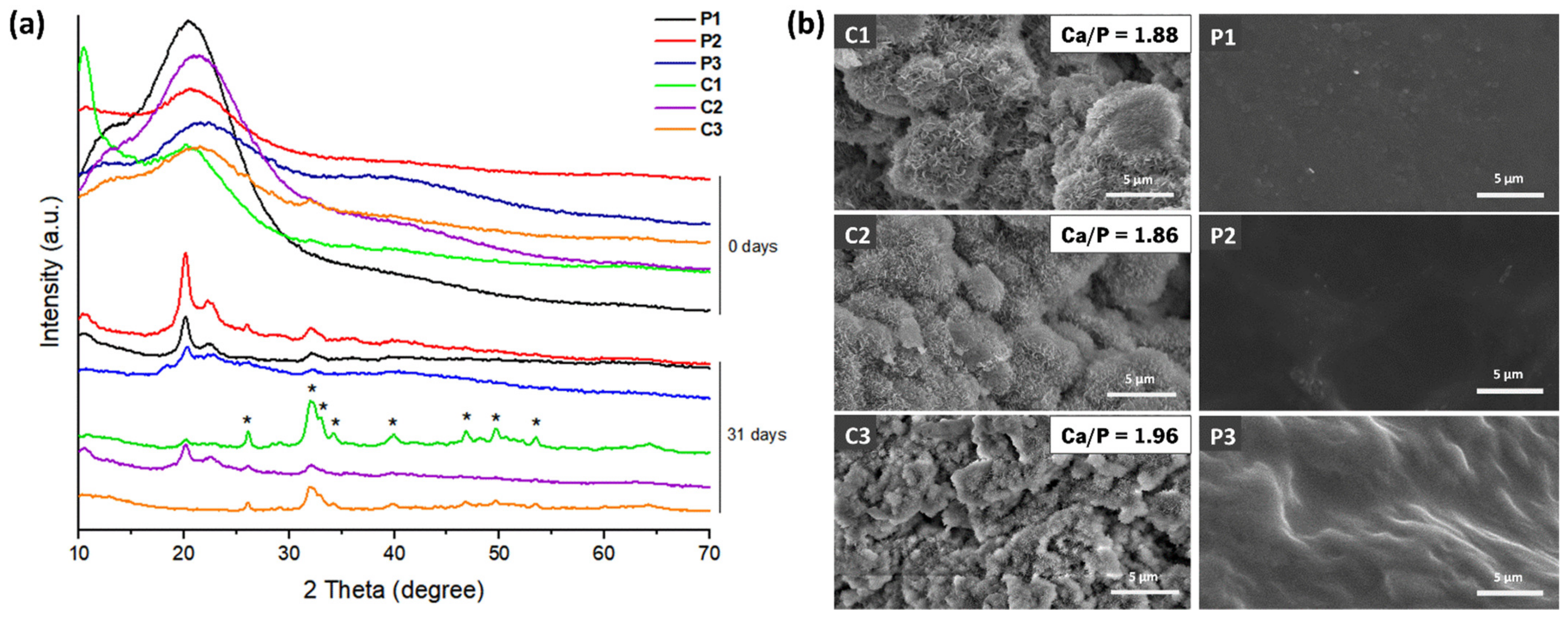
3.8. In Vitro Mineralization Studies
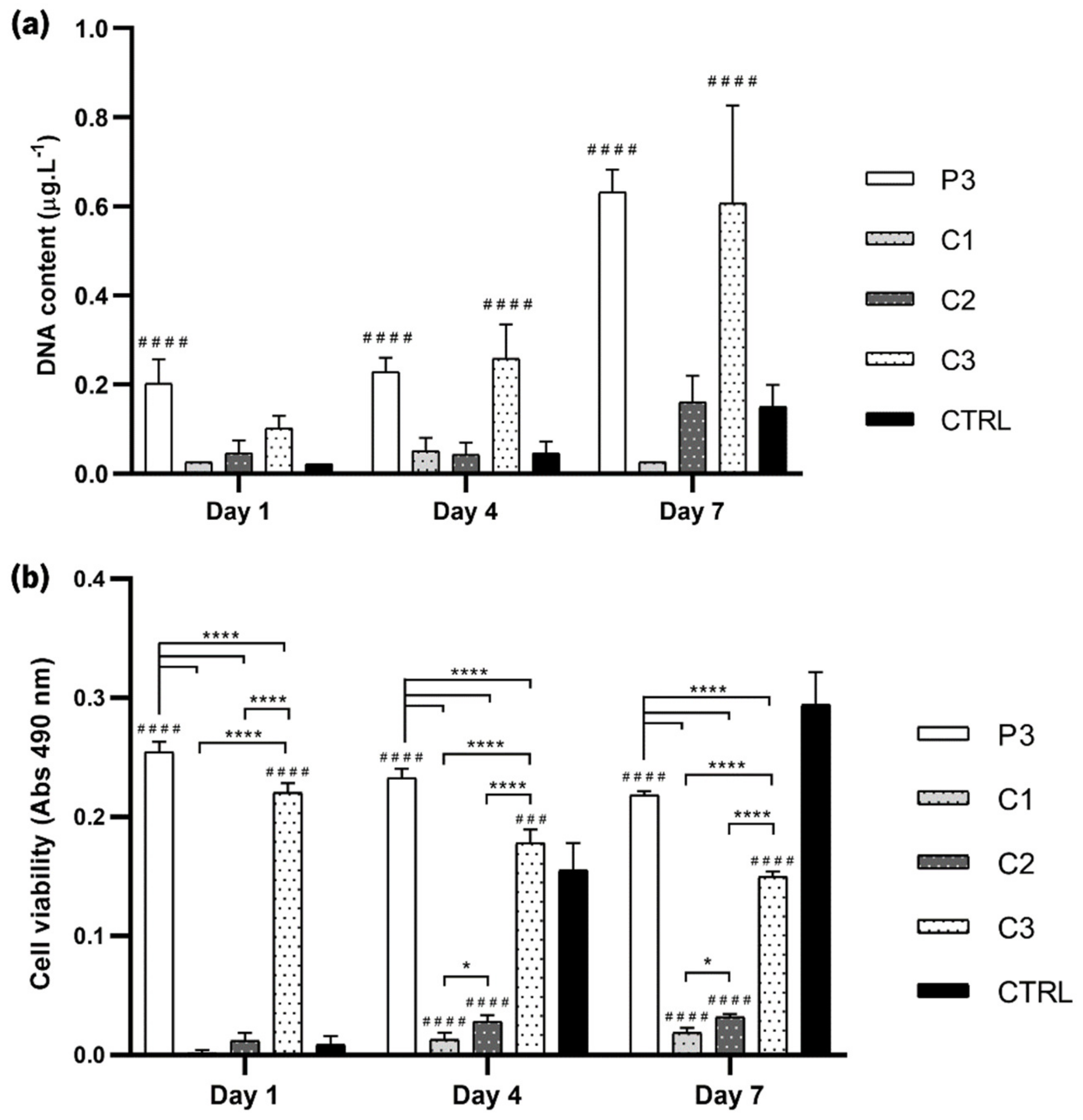
3.9. Cellular Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abdelrasoul, A.; Westphalen, H.; Saadati, S.; Shoker, A. Hemodialysis biocompatibility mathematical models to predict the inflammatory biomarkers released in dialysis patients based on hemodialysis membrane characteristics and clinical practices. Sci. Rep. 2021, 11, 23080. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, C.; Xie, Y.; Yang, Y.; Zheng, Y.; Meng, H.; He, W.; Qiao, K. Highly transparent, highly flexible composite membrane with multiple antimicrobial effects used for promoting wound healing. Carbohydr. Polym. 2019, 222, 114985. [Google Scholar] [CrossRef] [PubMed]
- Mirbagheri, M.; Adibnia, V.; Hughes, B.R.; Waldman, S.D.; Banquy, X.; Hwang, D.K. Advanced cell culture platforms: A growing quest for emulating natural tissues. Mater. Horiz. 2019, 6, 45–71. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, J.; Wan, Y.; Romeis, S.; Esper, J.D.; Peukert, W.; Zheng, K.; Boccaccini, A.R. Bioactive Glass Flakes as Innovative Fillers in Chitosan Membranes for Guided Bone Regeneration. Adv. Eng. Mater. 2022, 24, 2101042. [Google Scholar] [CrossRef]
- Pazarçeviren, A.E.; Evis, Z.; Keskin, D.; Tezcaner, A. Resorbable PCEC/gelatin-bismuth doped bioglass-graphene oxide bilayer membranes for guided bone regeneration. Biomed. Mater. 2019, 14, 035018. [Google Scholar] [CrossRef]
- Castillo Dalí, G.; Torres Lagares, D. Chapter 1—Nanobiomaterials in hard tissue engineering. In Nanobiomaterials in Hard Tissue Engineering; Grumezescu, A.M., Ed.; William Andrew Publishing: Norwich, NY, USA, 2016; pp. 1–31. [Google Scholar]
- Borges, J.; Mano, J.F. Molecular Interactions Driving the Layer-by-Layer Assembly of Multilayers. Chem. Rev. 2014, 114, 8883–8942. [Google Scholar] [CrossRef]
- Costa, R.R.; Mano, J.F. Polyelectrolyte multilayered assemblies in biomedical technologies. Chem. Soc. Rev. 2014, 43, 3453–3479. [Google Scholar] [CrossRef]
- Richardson, J.J.; Cui, J.; Björnmalm, M.; Braunger, J.A.; Ejima, H.; Caruso, F. Innovation in Layer-by-Layer Assembly. Chem. Rev. 2016, 116, 14828–14867. [Google Scholar] [CrossRef] [Green Version]
- Costa, R.R.; Costa, A.M.S.; Caridade, S.G.; Mano, J.F. Compact Saloplastic Membranes of Natural Polysaccharides for Soft Tissue Engineering. Chem. Mater. 2015, 27, 7490–7502. [Google Scholar] [CrossRef]
- Rodrigues, M.N.; Oliveira, M.B.; Costa, R.R.; Mano, J.F. Chitosan/Chondroitin Sulfate Membranes Produced by Polyelectrolyte Complexation for Cartilage Engineering. Biomacromolecules 2016, 17, 2178–2188. [Google Scholar] [CrossRef]
- Costa, R.R.; Reis, R.L.; Pashkuleva, I. Glycosaminoglycans as polyelectrolytes: Implications in bioactivity and assembly of biomedical devices. Int. Mater. Rev. 2022, 67, 765–795. [Google Scholar] [CrossRef]
- Tao, F.; Cheng, Y.; Shi, X.; Zheng, H.; Du, Y.; Xiang, W.; Deng, H. Applications of chitin and chitosan nanofibers in bone regenerative engineering. Carbohydr. Polym. 2020, 230, 115658. [Google Scholar] [CrossRef]
- Vale, A.C.; Pereira, P.R.; Alves, N.M. Polymeric biomaterials inspired by marine mussel adhesive proteins. React. Funct. Polym. 2021, 159, 104802. [Google Scholar] [CrossRef]
- Vale, A.C.; Pereira, P.; Barbosa, A.M.; Torrado, E.; Mano, J.F.; Alves, N.M. Antibacterial free-standing polysaccharide composite films inspired by the sea. Int. J. Biol. Macromol. 2019, 133, 933–944. [Google Scholar] [CrossRef]
- Li, Y.; Cheng, J.; Delparastan, P.; Wang, H.; Sigg, S.J.; DeFrates, K.G.; Cao, Y.; Messersmith, P.B. Molecular design principles of Lysine-DOPA wet adhesion. Nat. Commun. 2020, 11, 3895. [Google Scholar] [CrossRef]
- Almeida, A.C.; Vale, A.C.; Reis, R.L.; Alves, N.M. Bioactive and adhesive properties of multilayered coatings based on catechol-functionalized chitosan/hyaluronic acid and bioactive glass nanoparticles. Int. J. Biol. Macromol. 2020, 157, 119–134. [Google Scholar] [CrossRef]
- Moreira, J.; Vale, A.C.; Pires, R.A.; Botelho, G.; Reis, R.L.; Alves, N.M. Spin-Coated Polysaccharide-Based Multilayered Freestanding Films with Adhesive and Bioactive Moieties. Molecules 2020, 25, 840. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Kim, K.; Ryu, J.H.; Lee, H. Chitosan-catechol: A polymer with long-lasting mucoadhesive properties. Biomaterials 2015, 52, 161–170. [Google Scholar] [CrossRef]
- Xu, J.; Strandman, S.; Zhu, J.X.X.; Barralet, J.; Cerruti, M. Genipin-crosslinked catechol-chitosan mucoadhesive hydrogels for buccal drug delivery. Biomaterials 2015, 37, 395–404. [Google Scholar] [CrossRef]
- Ghadban, A.; Ahmed, A.S.; Ping, Y.; Ramos, R.; Arfin, N.; Cantaert, B.; Ramanujan, R.V.; Miserez, A. Bioinspired pH and magnetic responsive catechol-functionalized chitosan hydrogels with tunable elastic properties. Chem. Commun. 2016, 52, 697–700. [Google Scholar] [CrossRef]
- Lee, Y.; Chung, H.J.; Yeo, S.; Ahn, C.-H.; Lee, H.; Messersmith, P.B.; Park, T.G. Thermo-sensitive, injectable, and tissue adhesive sol–gel transition hyaluronic acid/pluronic composite hydrogels prepared from bio-inspired catechol-thiol reaction. Soft Matter 2010, 6, 977–983. [Google Scholar] [CrossRef]
- Kokubo, T.; Takadama, H. How useful is SBF in predicting in vivo bone bioactivity? Biomaterials 2006, 27, 2907–2915. [Google Scholar] [CrossRef] [PubMed]
- Forsey, R.W.; Chaudhuri, J.B. Validity of DNA analysis to determine cell numbers in tissue engineering scaffolds. Biotechnol. Lett. 2009, 31, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Rego, S.J.; Vale, A.C.; Luz, G.M.; Mano, J.F.; Alves, N.M. Adhesive Bioactive Coatings Inspired by Sea Life. Langmuir 2016, 32, 560–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barroso, N.; Guaresti, O.; Pérez-Álvarez, L.; Ruiz-Rubio, L.; Gabilondo, N.; Vilas-Vilela, J.L. Self-healable hyaluronic acid/chitosan polyelectrolyte complex hydrogels and multilayers. Eur. Polym. J. 2019, 120, 109268. [Google Scholar] [CrossRef]
- Doostmohammadi, A.; Monshi, A.; Salehi, R.; Fathi, M.H.; Golniya, Z.; Daniels, A.U. Bioactive glass nanoparticles with negative zeta potential. Ceram. Int. 2011, 37, 2311–2316. [Google Scholar] [CrossRef]
- Reisch, A.; Tirado, P.; Roger, E.; Boulmedais, F.; Collin, D.; Voegel, J.-C.; Frisch, B.; Schaaf, P.; Schlenoff, J.B. Compact Saloplastic Poly(Acrylic Acid)/Poly(Allylamine) Complexes: Kinetic Control Over Composition, Microstructure, and Mechanical Properties. Adv. Funct. Mater. 2013, 23, 673–682. [Google Scholar] [CrossRef] [Green Version]
- Costa, R.R.; Soares da Costa, D.; Reis, R.L.; Pashkuleva, I. Bioinspired baroplastic glycosaminoglycan sealants for soft tissues. Acta Biomater. 2019, 87, 108–117. [Google Scholar] [CrossRef]
- Nosonovsky, M.; Bhushan, B. Biologically Inspired Surfaces: Broadening the Scope of Roughness**. Adv. Funct. Mater. 2008, 18, 843–855. [Google Scholar] [CrossRef]
- Rodrigues, J.R.; Alves, N.M.; Mano, J.F. Biomimetic polysaccharide/bioactive glass nanoparticles multilayer membranes for guided tissue regeneration. RSC Adv. 2016, 6, 75988–75999. [Google Scholar] [CrossRef]
- Caridade, S.G.; Merino, E.G.; Alves, N.M.; Bermudez, V.d.Z.; Boccaccini, A.R.; Mano, J.F. Chitosan membranes containing micro or nano-size bioactive glass particles: Evolution of biomineralization followed by in situ dynamic mechanical analysis. J. Mech. Behav. Biomed. Mater. 2013, 20, 173–183. [Google Scholar] [CrossRef] [Green Version]
- Wenzel, R.N. Resistance of solid surfaces to wetting by water. Ind. Eng. Chem. 1936, 28, 988–994. [Google Scholar] [CrossRef]
- Harnett, E.M.; Alderman, J.; Wood, T. The surface energy of various biomaterials coated with adhesion molecules used in cell culture. Colloids Surf. B Biointerfaces 2007, 55, 90–97. [Google Scholar] [CrossRef]
- Lee, C.; Shin, J.; Lee, J.S.; Byun, E.; Ryu, J.H.; Um, S.H.; Kim, D.-I.; Lee, H.; Cho, S.-W. Bioinspired, Calcium-Free Alginate Hydrogels with Tunable Physical and Mechanical Properties and Improved Biocompatibility. Biomacromolecules 2013, 14, 2004–2013. [Google Scholar] [CrossRef]
- Zhu, W.; Peck, Y.; Iqbal, J.; Wang, D.A. A novel DOPA-albumin based tissue adhesive for internal medical applications. Biomaterials 2017, 147, 99–115. [Google Scholar] [CrossRef]
- Secrist, K.E.; Nolte, A.J. Humidity Swelling/Deswelling Hysteresis in a Polyelectrolyte Multilayer Film. Macromolecules 2011, 44, 2859–2865. [Google Scholar] [CrossRef]
- Banerjee, A.; Chatterjee, K.; Madras, G. Enzymatic degradation of polymers: A brief review. Mater. Sci. Technol. 2014, 30, 567–573. [Google Scholar] [CrossRef]
- Azevedo, H.S.; Santos, T.C.; Reis, R.L. 4—Controlling the degradation of natural polymers for biomedical applications. In Natural-Based Polymers for Biomedical Applications; Reis, R.L., Neves, N.M., Mano, J.F., Gomes, M.E., Marques, A.P., Azevedo, H.S., Eds.; Woodhead Publishing: Sawston, UK, 2008; pp. 106–128. [Google Scholar]
- Gentile, P.; Chiono, V.; Tonda-Turo, C.; Ferreira, A.M.; Ciardelli, G. Polymeric membranes for guided bone regeneration. Biotechnol. J. 2011, 6, 1187–1197. [Google Scholar] [CrossRef]
- Bottino, M.C.; Thomas, V.; Schmidt, G.; Vohra, Y.K.; Chu, T.-M.G.; Kowolik, M.J.; Janowski, G.M. Recent advances in the development of GTR/GBR membranes for periodontal regeneration—A materials perspective. Dent. Mater. 2012, 28, 703–721. [Google Scholar] [CrossRef]
- Lungu, R.; Anisiei, A.; Rosca, I.; Sandu, A.-I.; Ailincai, D.; Marin, L. Double functionalization of chitosan based nanofibers towards biomaterials for wound healing. React. Funct. Polym. 2021, 167, 105028. [Google Scholar] [CrossRef]
- Ren, D.; Yi, H.; Wang, W.; Ma, X. The enzymatic degradation and swelling properties of chitosan matrices with different degrees of N-acetylation. Carbohydr. Res. 2005, 340, 2403–2410. [Google Scholar] [CrossRef] [PubMed]
- Van den Mooter, G.; Samyn, C.; Kinget, R. The relation between swelling properties and enzymatic degradation of azo polymers designed for colon-specific drug delivery. Pharm. Res. 1994, 11, 1737–1741. [Google Scholar] [CrossRef] [PubMed]
- Nowell, C.S.; Odermatt, P.D.; Azzolin, L.; Hohnel, S.; Wagner, E.F.; Fantner, G.E.; Lutolf, M.P.; Barrandon, Y.; Piccolo, S.; Radtke, F. Chronic inflammation imposes aberrant cell fate in regenerating epithelia through mechanotransduction. Nat. Cell Biol. 2016, 18, 168–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mota, J.; Yu, N.; Caridade, S.G.; Luz, G.M.; Gomes, M.E.; Reis, R.L.; Jansen, J.A.; Walboomers, X.F.; Mano, J.F. Chitosan/bioactive glass nanoparticle composite membranes for periodontal regeneration. Acta Biomater. 2012, 8, 4173–4180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Liu, F.; Zheng, X.; Sun, J. Water-Enabled Self-Healing of Polyelectrolyte Multilayer Coatings. Angew. Chem. Int. Ed. 2011, 50, 11378–11381. [Google Scholar] [CrossRef] [PubMed]
- Hui, C.Y.; Lin, Y.Y.; Baney, J.M. The mechanics of tack: Viscoelastic contact on a rough surface. J. Polym. Sci. Part B Polym. Phys. 2000, 38, 1485–1495. [Google Scholar] [CrossRef]
- Creton, C.; Leibler, L. How does tack depend on time of contact and contact pressure? J. Polym. Sci. Part B Polym. Phys. 1996, 34, 545–554. [Google Scholar] [CrossRef]
- Haiat, G.; Barthel, E. An Approximate Model for the Adhesive Contact of Rough Viscoelastic Surfaces. Langmuir 2007, 23, 11643–11650. [Google Scholar] [CrossRef] [Green Version]
- Lauto, A.; Mawad, D.; Foster, L.J.R. Adhesive biomaterials for tissue reconstruction. J. Chem. Technol. Biotechnol. 2008, 83, 464–472. [Google Scholar] [CrossRef]
- Chartoff, R.P.; Menczel, J.D.; Dillman, S.H. Dynamic Mechanical Analysis (DMA). In Thermal Analysis of Polymers, Fundamentals and Applications; Menczel, J.D., Prime, R.B., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar]
- Loram, I.D.; Gawthrop, P.J.; Lakie, M. The frequency of human, manual adjustments in balancing an inverted pendulum is constrained by intrinsic physiological factors. J. Physiol. 2006, 577, 417–432. [Google Scholar] [CrossRef]
- Luz, G.M.; Mano, J.F. Preparation and characterization of bioactive glass nanoparticles prepared by sol–gel for biomedical applications. Nanotechnology 2011, 22, 494014. [Google Scholar] [CrossRef]
- Yun, J.; Holmes, B.; Fok, A.; Wang, Y. A Kinetic Model for Hydroxyapatite Precipitation in Mineralizing Solutions. Crystal Growth Design 2018, 18, 2717–2725. [Google Scholar] [CrossRef]
- Ergun, C.; Liu, H.; Webster, T.J.; Olcay, E.; Yilmaz, S.; Sahin, F.C. Increased osteoblast adhesion on nanoparticulate calcium phosphates with higher Ca/P ratios. J. Biomed. Mater. Res. Part A 2008, 85, 236–241. [Google Scholar] [CrossRef]
- Scognamiglio, F.; Travan, A.; Borgogna, M.; Donati, I.; Marsich, E.; Bosmans, J.W.A.M.; Perge, L.; Foulc, M.P.; Bouvy, N.D.; Paoletti, S. Enhanced bioadhesivity of dopamine-functionalized polysaccharidic membranes for general surgery applications. Acta Biomater. 2016, 44, 232–242. [Google Scholar] [CrossRef]
- Schweigert, N.; Zehnder, A.J.B.; Eggen, R.I.L. Chemical properties of catechols and their molecular modes of toxic action in cells, from microorganisms to mammals. Environ. Microbiol. 2001, 3, 81–91. [Google Scholar] [CrossRef]
- Cui, L.; Jia, J.; Guo, Y.; Liu, Y.; Zhu, P. Preparation and characterization of IPN hydrogels composed of chitosan and gelatin cross-linked by genipin. Carbohydr. Polym. 2014, 99, 31–38. [Google Scholar] [CrossRef]
- Silva, J.M.; Caridade, S.G.; Oliveira, N.M.; Reis, R.L.; Mano, J.F. Chitosan–alginate multilayered films with gradients of physicochemical cues. J. Mater. Chem. B 2015, 3, 4555–4568. [Google Scholar] [CrossRef]
- Caridade, S.G.; Monge, C.; Gilde, F.; Boudou, T.; Mano, J.F.; Picart, C. Free-Standing Polyelectrolyte Membranes Made of Chitosan and Alginate. Biomacromolecules 2013, 14, 1653–1660. [Google Scholar] [CrossRef]
- Gorgieva, S.; Kokol, V. Preparation, characterization, and in vitro enzymatic degradation of chitosan-gelatine hydrogel scaffolds as potential biomaterials. J. Biomed. Mater. Res. Part A 2012, 100A, 1655–1667. [Google Scholar] [CrossRef]
- Duarte, M.L.; Ferreira, M.C.; Marvão, M.R.; Rocha, J. An optimised method to determine the degree of acetylation of chitin and chitosan by FTIR spectroscopy. Int. J. Biol. Macromol. 2002, 31, 1–8. [Google Scholar] [CrossRef]
- Manna, U.; Bharani, S.; Patil, S. Layer-by-Layer Self-Assembly of Modified Hyaluronic Acid/Chitosan Based on Hydrogen Bonding. Biomacromolecules 2009, 10, 2632–2639. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Yang, K.; Kang, B.; Lee, C.; Song, I.T.; Byun, E.; Park, K.I.; Cho, S.-W.; Lee, H. Hyaluronic Acid Catechol: A Biopolymer Exhibiting a pH-Dependent Adhesive or Cohesive Property for Human Neural Stem Cell Engineering. Adv. Funct. Mater. 2013, 23, 1774–1780. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Polycation | Polyanion | Ceramic |
|---|---|---|---|
| P1 | CHI (50 %) | HA (50 %) | - |
| P2 | CHI (50 %) | HA–cat (50 %) | - |
| P3 | CHI (25 %) and CHI–cat (25 %) | HA (25 %) and HA–cat (25 %) | - |
| C1 | CHI (35 %) | HA (50 %) | BGNPs (15 %) |
| C2 | CHI (35 %) | HA–cat (50 %) | BGNPs (15 %) |
| C3 | CHI (17.5 %) and CHI–cat (17.5 %) | HA (25 %) and HA–cat (25 %) | BGNPs (15 %) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fonseca, M.C.; Vale, A.C.; Costa, R.R.; Reis, R.L.; Alves, N.M. Exploiting Polyelectrolyte Complexation for the Development of Adhesive and Bioactive Membranes Envisaging Guided Tissue Regeneration. J. Funct. Biomater. 2023, 14, 3. https://doi.org/10.3390/jfb14010003
Fonseca MC, Vale AC, Costa RR, Reis RL, Alves NM. Exploiting Polyelectrolyte Complexation for the Development of Adhesive and Bioactive Membranes Envisaging Guided Tissue Regeneration. Journal of Functional Biomaterials. 2023; 14(1):3. https://doi.org/10.3390/jfb14010003
Chicago/Turabian StyleFonseca, Mário C., Ana Catarina Vale, Rui R. Costa, Rui L. Reis, and Natália M. Alves. 2023. "Exploiting Polyelectrolyte Complexation for the Development of Adhesive and Bioactive Membranes Envisaging Guided Tissue Regeneration" Journal of Functional Biomaterials 14, no. 1: 3. https://doi.org/10.3390/jfb14010003