Calcium Phosphate Growth at Electropolished Titanium Surfaces


Abstract
:
1. Introduction
2. Results and Discussion
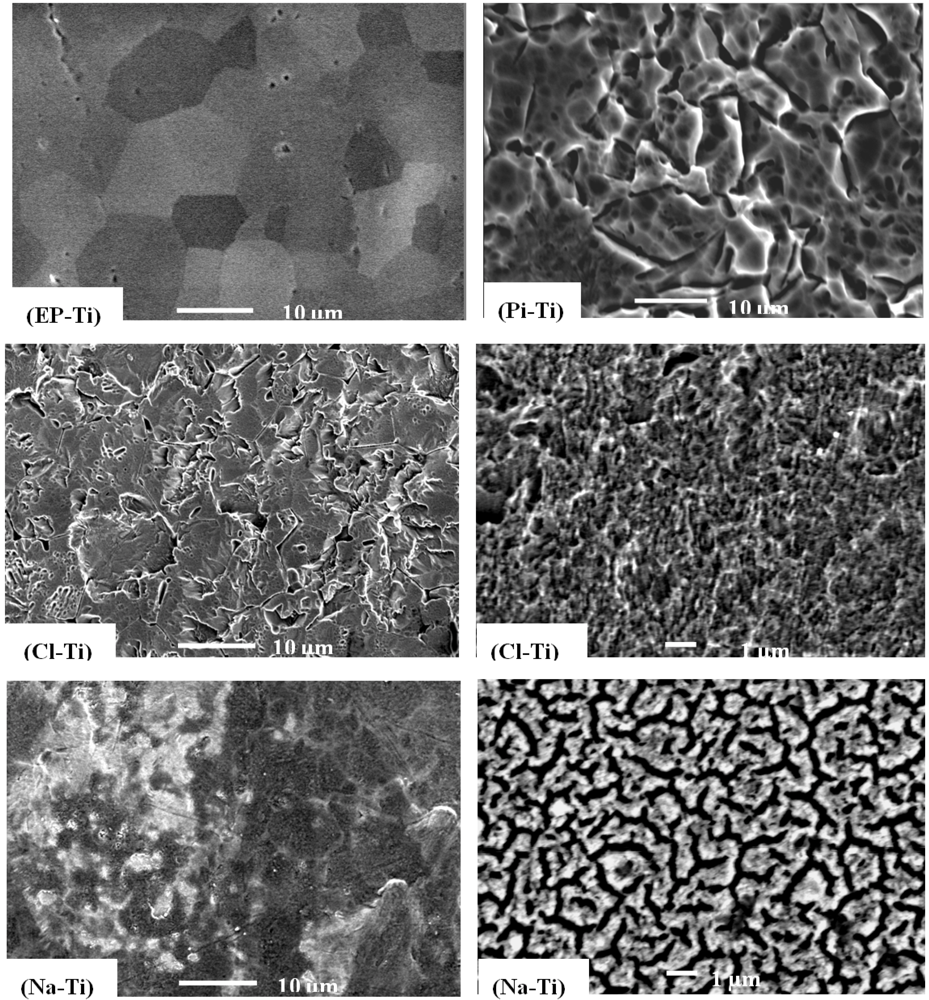
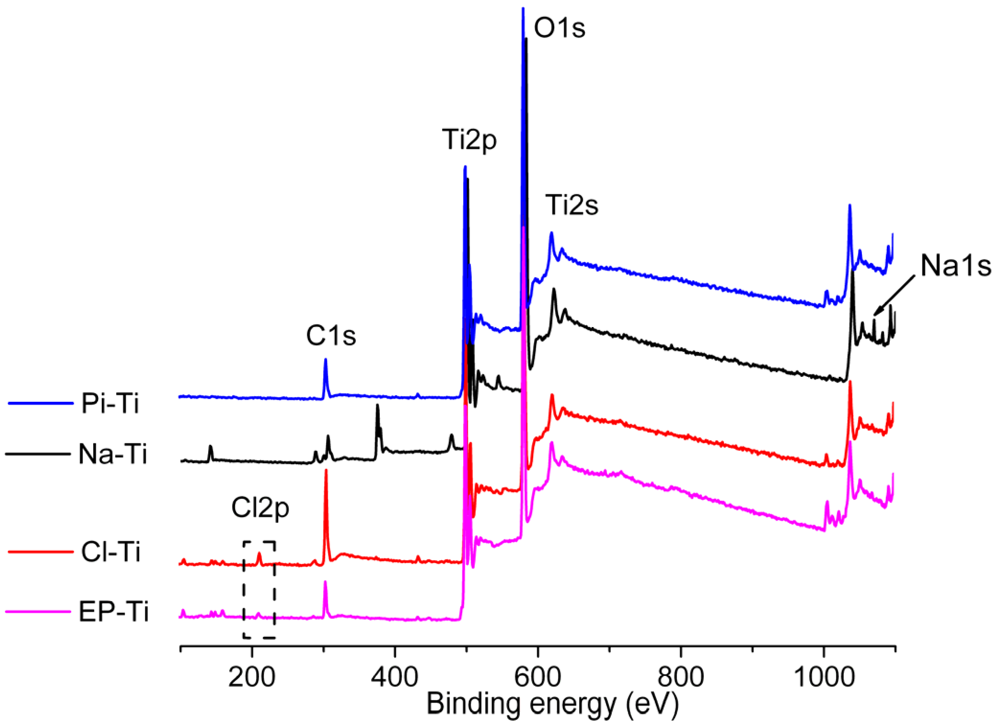
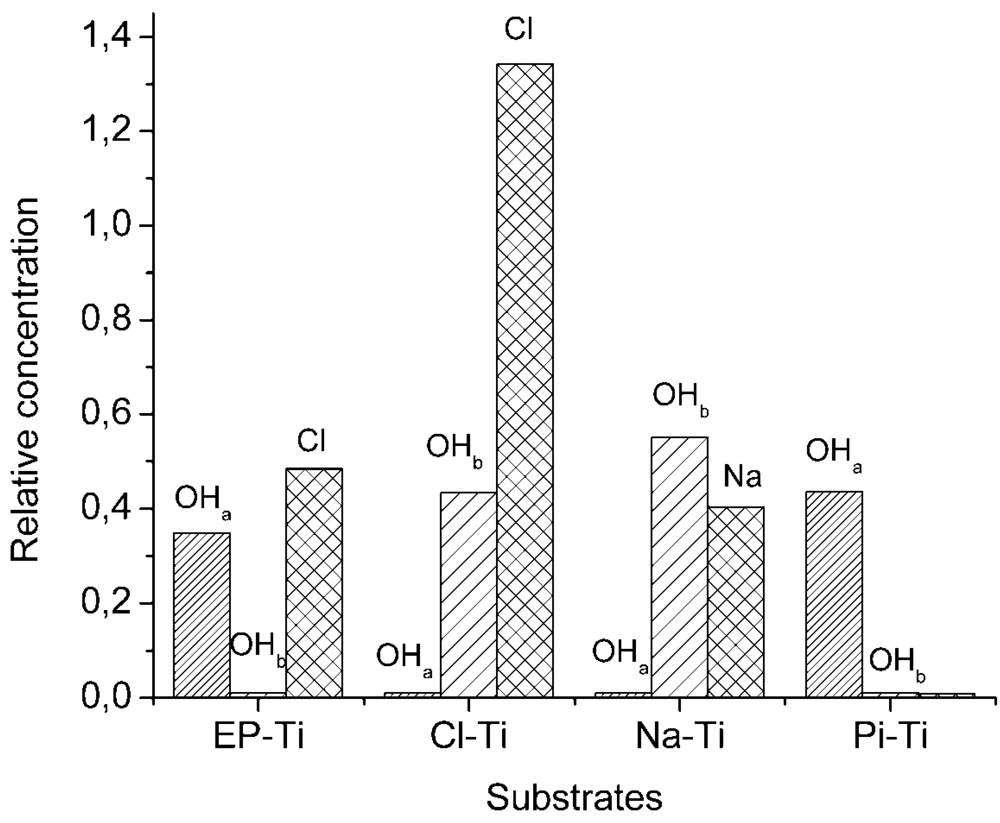
2.1. Surface Properties of the Pre-Treated Ti Substrates

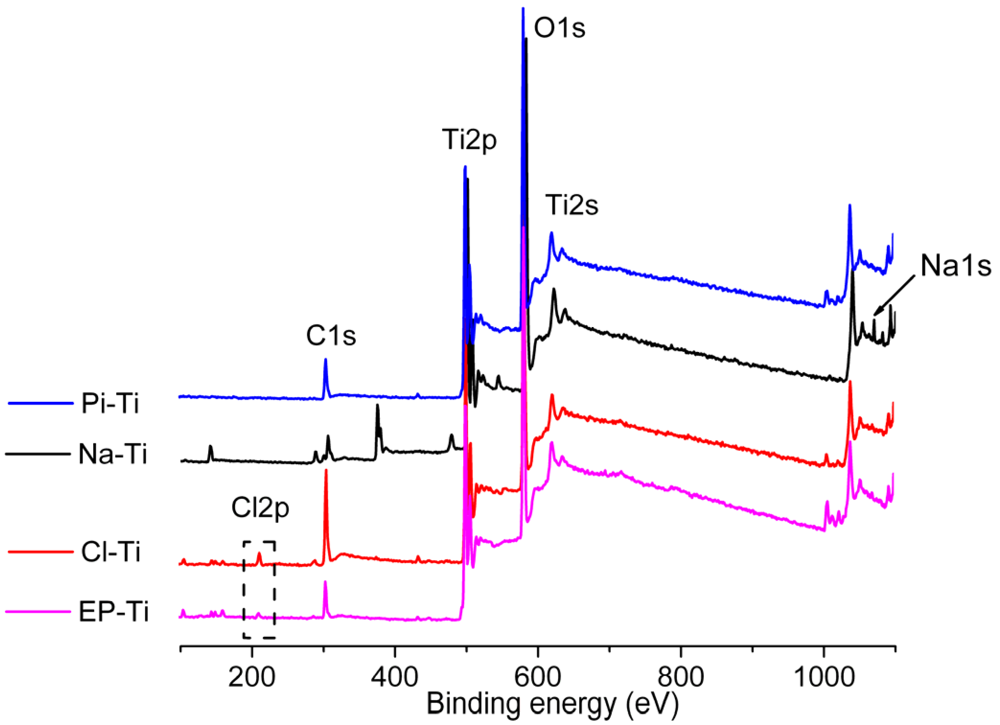
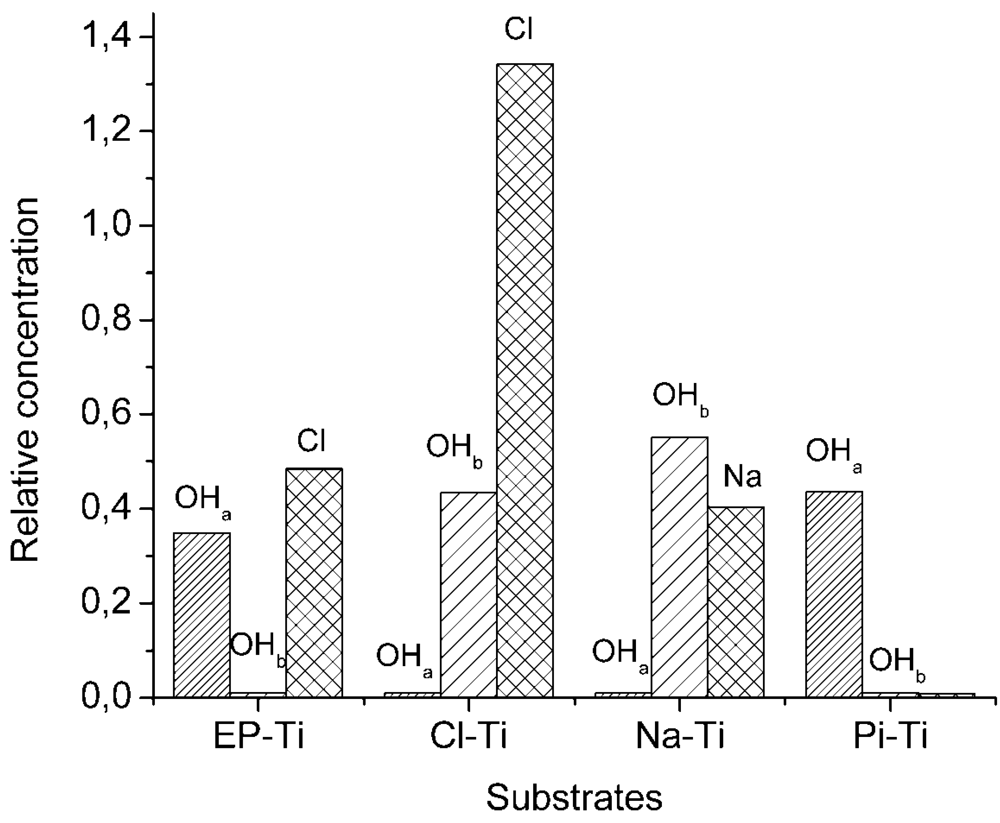
2.2. Ti Substrates upon CaP Coating
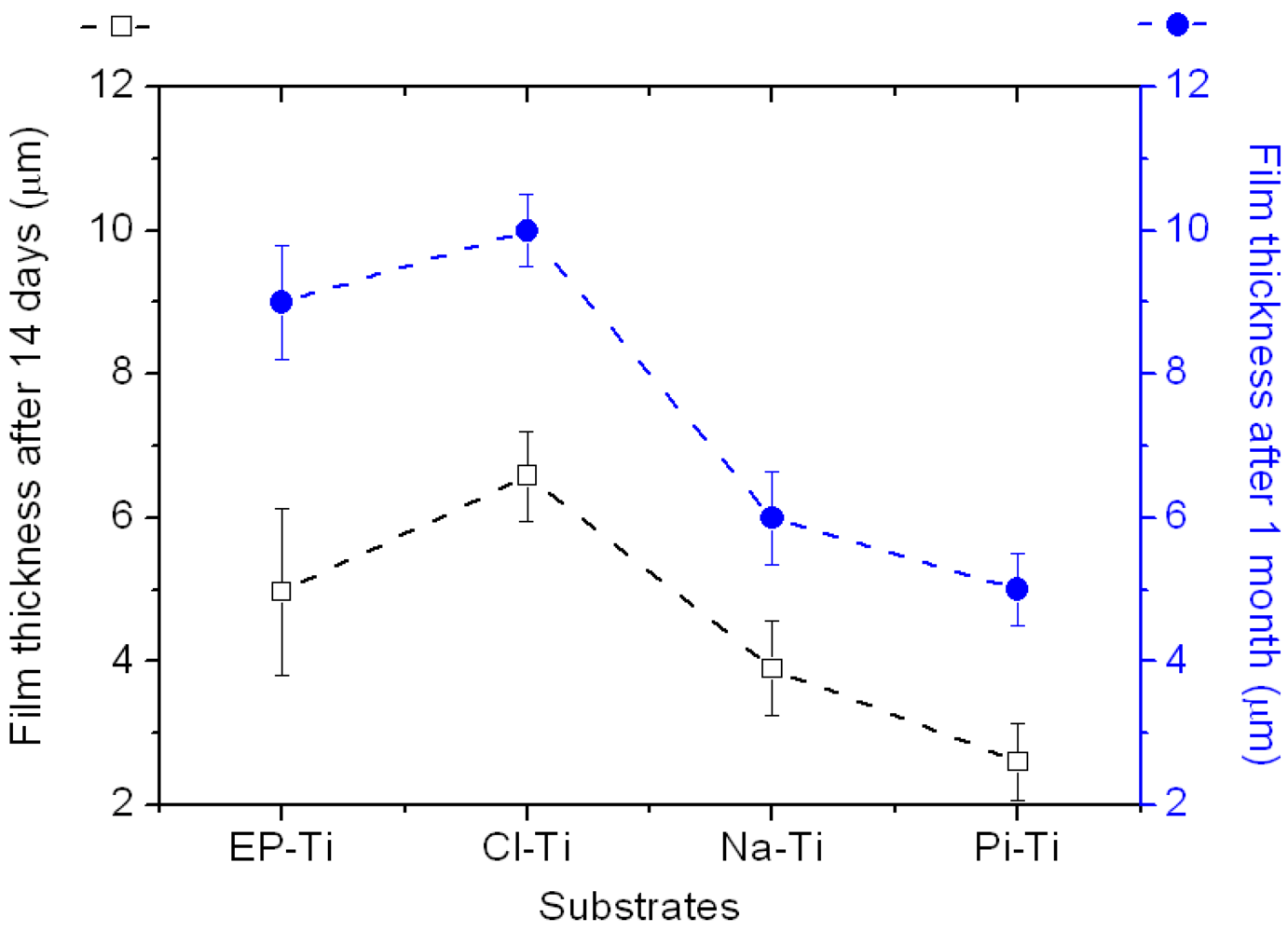
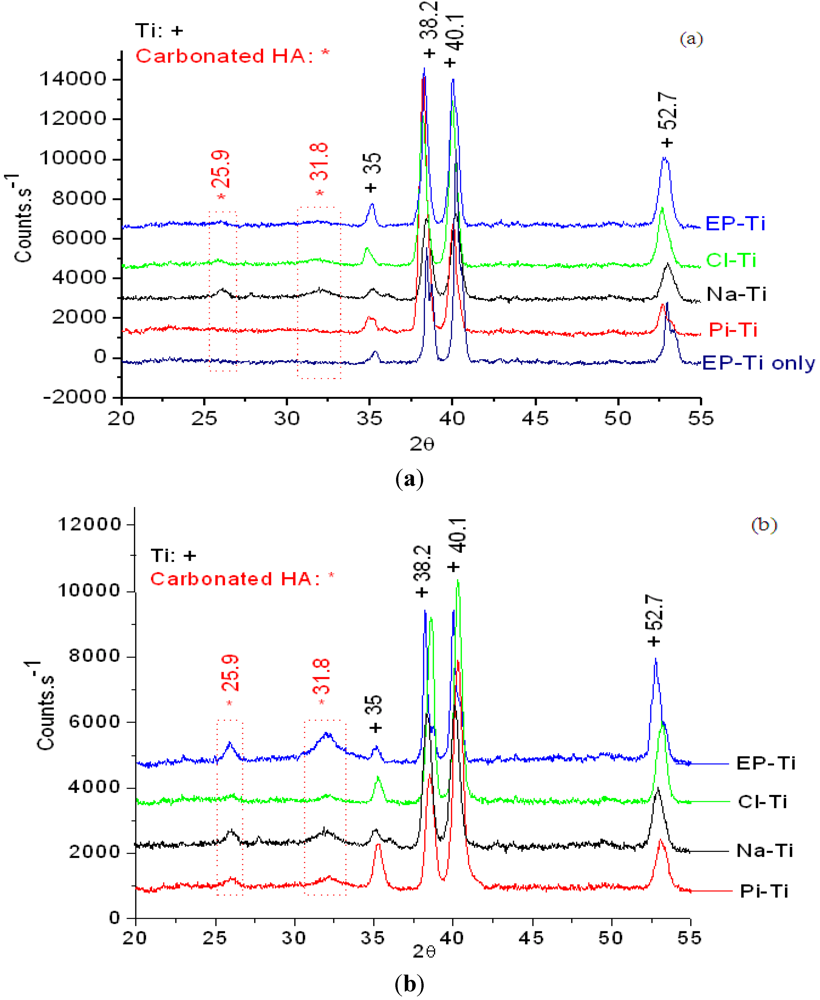
2.2.1. Physical Properties of the CaP Coating

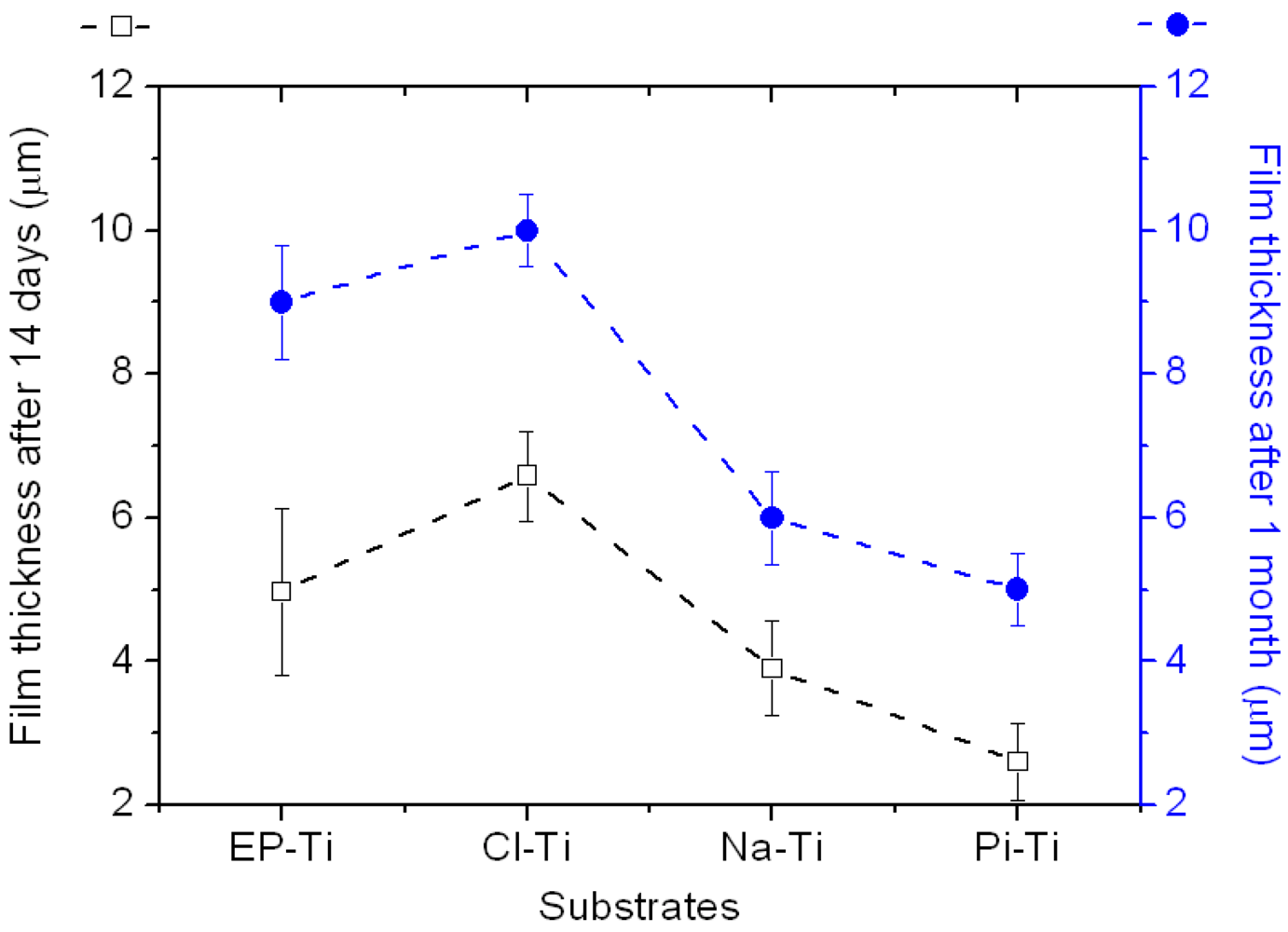
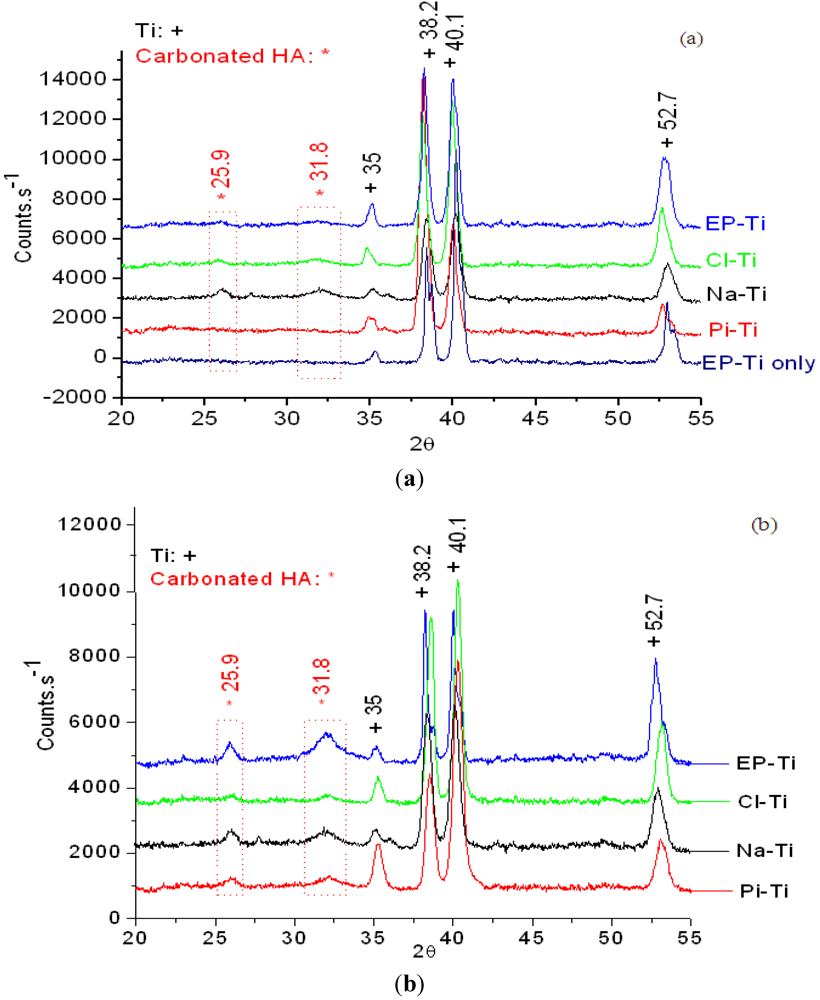

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HA crystallite size (±1.0 nm) | Na-Ti | EP-Ti | Pi-Ti | Cl-Ti |
|---|---|---|---|---|
| after 14 days | 5.5 | 3.5 | - | 4.4 |
| after 30 days | 6.0 | 6.4 | 5.2 | 5.2 |
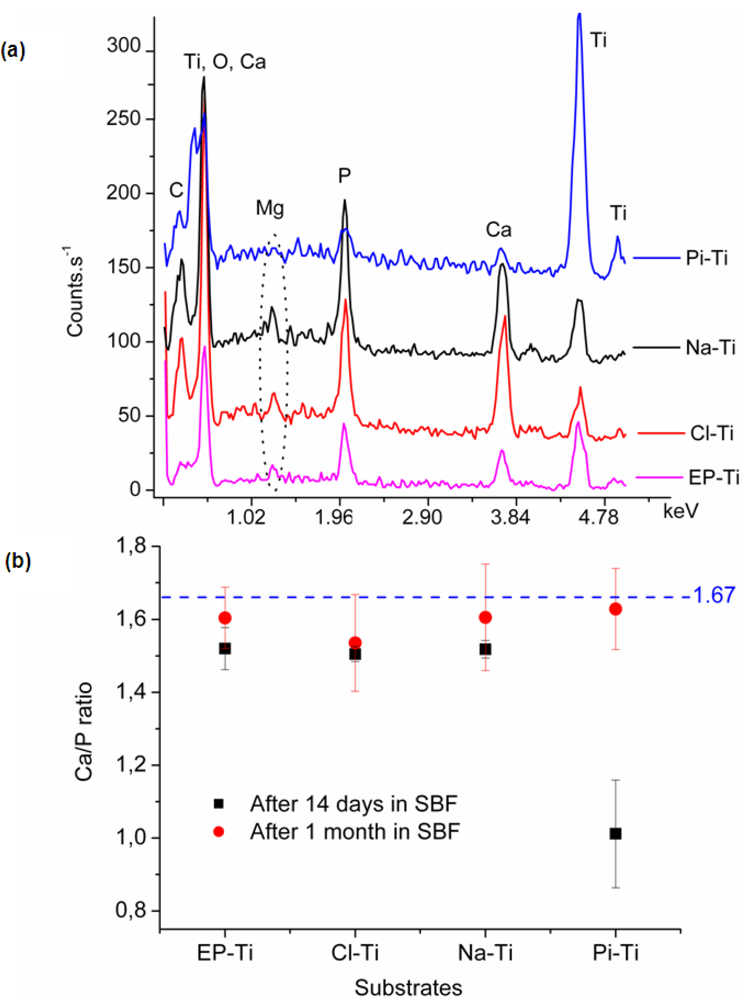
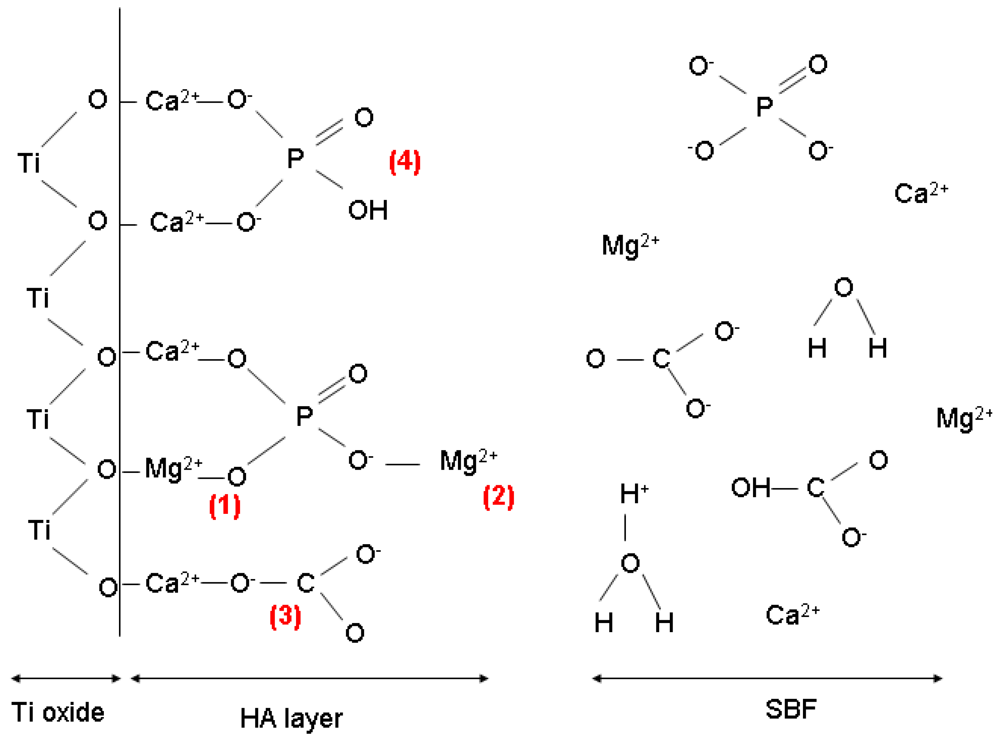
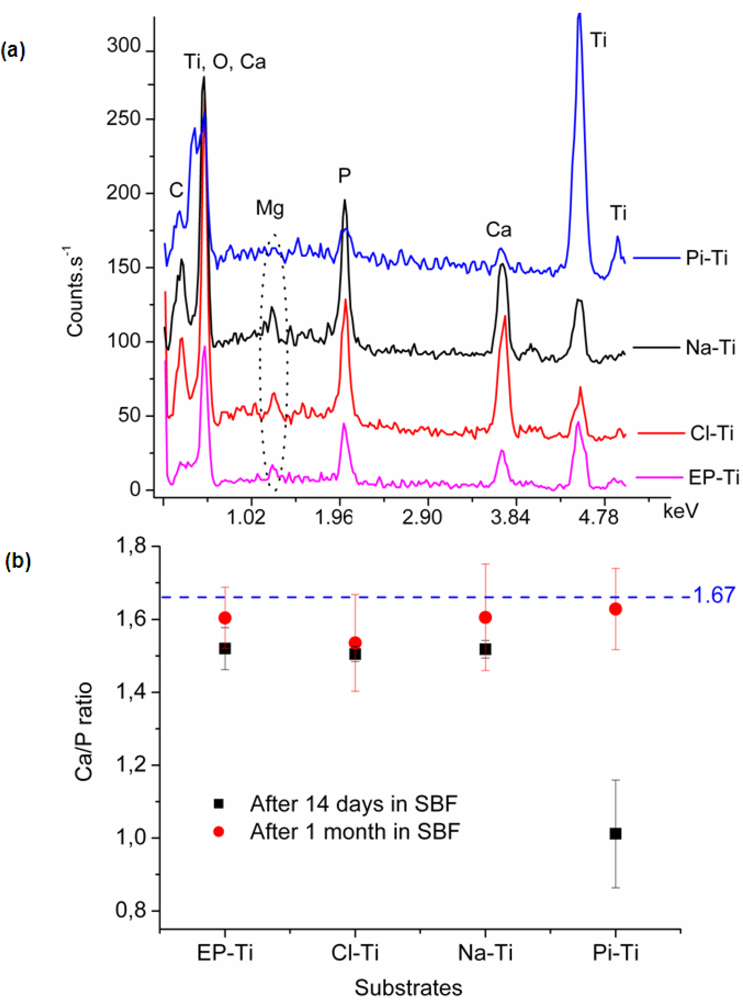
2.2.2. Chemical Composition of the Coating


| FTIR peaks related to PO43− | Wavenumber (cm−1) |
|---|---|
| ν4: O-P-O bending | Doublet peaks at 571 and 604 |
| ν1: P-O symmetric stretching | 960 |
| ν3: P-O asymmetric stretching | doublet peaks between 1,015–1,120 |
| FTIR peaks related to CO32− | Wavenumber (cm−1) |
| ν2: O-C-O out-of-plane bending | 872 |
| ν3: C-O asymmetric stretching | doublet peaks between 1,415–1,490 |
| ν3: O-C-O bending | Between 1,640–1,650 |
3. Experimental Section
| # | Chemicals | Amount (g) for 1.5 SBF in 1L of water |
|---|---|---|
| 1 | NaCl | 10.806 |
| 2 | NaHCO3 | 1.472 |
| 3 | Na2CO3 | 4.072 |
| 4 | KCl | 0.45 |
| 5 | K2HPO4.3H2O | 0.476 |
| 6 | MgCl2.H2O | 0.622 |
| 7 | HEPES | 23.856 |
| 8 | CaCl2 | 0.586 |
| 9 | Na2SO4 | 0.144 |
| 10 | 1M-NaOH | 3 mL |
3.1. Electropolishing
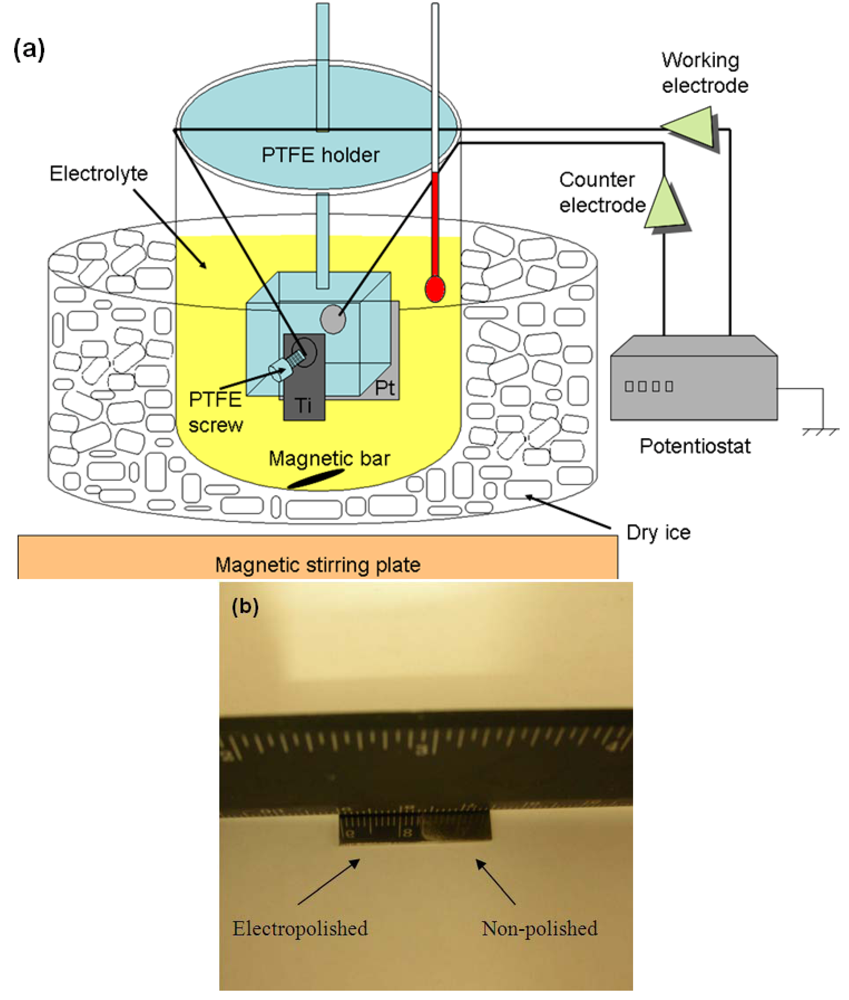
3.2. Conventional Surface Treatments
3.3. CaP Coating
3.4. Characterization Methods
4. Conclusions
Supplementary Files
Acknowledgments
References
- Liu, X.; Chu, P.K.; Ding, C. Surface modification of titanium, titanium alloys, and related materials for biomedical applications. Mater. Sci. Eng. A 2004, 47, 49–121. [Google Scholar] [CrossRef]
- Brunnette, D.M.; Tengvall, P.; Textor, M.; Thomsen, P. Titanium in Medicine. Materials Science, Surface Science, Engineering, Biological Responses and Medical Applications; Springer: Berlin, Germany, 2001. [Google Scholar]
- Gross, K.A.; Berndt, C.C. Thermal processing of hydroxyapatite for coating production. J. Biomed. Mater. Res. 1998, 39, 580–587. [Google Scholar] [CrossRef]
- Steinemann, S.G. Titanium—The materials of choice? Periodontology 2000 1998, 17, 7–21. [Google Scholar] [CrossRef]
- Koutsopoulos, S. Kinetic study on the crystal growth of hydroxyapatite. Langmuir 2002, 17, 8092–8097. [Google Scholar] [CrossRef]
- Blackwood, D.J.; Seah, K.H.W. Galvanostatic pulse deposition of hydroxyapatite for adhesion to titanium for biomedical purposes. Mater. Sci. Eng. C 2010, 30, 561–565. [Google Scholar] [CrossRef]
- Ueda, M.; Ikeda, M.; Ogawa, M. Chemical-hydrothermal combined surface modification of titanium for improvement of osteointegration. Mater. Sci. Eng. C 2009, 29, 994–1000. [Google Scholar]
- Sun, L.; Berndt, C.C.; Gross, K.A.; Kucuk, A. Material fundamentals and clinical performance of plasma-sprayed hydroxyapatite coatings: A review. J. Biomed. Mater. Res. 2001, 58, 570–592. [Google Scholar] [CrossRef]
- Habibovic, P.; Barrere, F.; van Blitterswijk, C.A.; de Groot, K.; Layrolle, P. Biomimetic hydroxyapatite coating on metal implants. J. Am. Ceram. Soc. 2002, 85, 517–522. [Google Scholar]
- Stumm, W. Chemistry of the Solid-Water Interface: Processes at the Mineral-Water and Particle-Water Interface in Natural Systems; Wiley-Interscience: Weinheim, Germany, 1992. [Google Scholar]
- Li, P.; Zhang, F. The electrochemistry of a glass surface and its application to bioactive glass in solution. J. Non Cryst. Solids. 1990, 119, 112–118. [Google Scholar] [CrossRef]
- Calvert, P.; Mann, S. The nagtive side of crystal growth. Nature 1997, 386, 127–128. [Google Scholar] [CrossRef]
- Wang, X.; Hayakawa, S.; Tsurub, K.; Osaka, A. Bioactive titania gel layers formed by chemical treatment of Ti substrate with a H2O2/HCl solution. Biomaterials 2002, 23, 1353–1357. [Google Scholar] [CrossRef]
- Lee, B.H.; Kim, Y.D.; Shin, J.H.; Lee, K.H. Surface modification by alkali and heat treatments in titanium alloys. J. Biomed. Mater. Res. 2002, 61, 466–473. [Google Scholar] [CrossRef]
- Kim, H.M.; Miyaji, F.; Kokubo, T.; Nakamura, T. Preparation of bioactive Ti and its alloys via simple chemical surface treatment. J. Biomed. Mater. Res. 1996, 32, 409–417. [Google Scholar] [CrossRef]
- Wei, M.; Kim, H.M.; Kokubo, T.; Evans, J.H. Optimising the bioactivity of alkaline-treated titanium alloy. Mater. Sci. Eng. C 2002, 20, 125–134. [Google Scholar] [CrossRef]
- Wen, H.B.; de Wijn, J.R.; Liu, Q.; de Groot, K. A simple method to prepare calcium phosphate coatings on Ti6Al4V. J. Mater. Sci. Mater. Med. 1997, 8, 765–770. [Google Scholar] [CrossRef]
- Pham, M.T.; Maitz, M.F.; Matz, W.; Reuther, H.; Richter, E.; Steiner, G. Promoted hydroxyapatite nucleation on titanium ion-implanted with sodium. Thin Solid Films 2000, 379, 50–56. [Google Scholar] [CrossRef]
- Majewski, P.J.; Allidi, G. Synthesis of hydroxyapatite on titanium coated with organic self-assembled monolayers. Mater. Sci. Eng. A 2006, 420, 13–20. [Google Scholar] [CrossRef]
- Huang, S.; Zhou, K.; Liu, Y.; Huang, B. Controlled crystallization of hydroxyapatite under hexadecylamine self-assembled monolayer. Trans. Nonferrous Met. Soc. China 2003, 13, 595–599. [Google Scholar]
- Zhu, P.; Masuda, Y.; Koumoto, K. A novel approach to fabricate Hydroxyapatite coating on titanium substrate in an aqueous solution. J. Ceram. Soc. Jpn. 2001, 109, 676–680. [Google Scholar] [CrossRef]
- Tanahashi, M.; Matsuda, T. Surface functional group dependence on apatite formation on self-assembled monolayers in a simulated body fluid. J. Biomed. Mater. Res. 1997, 34, 305–315. [Google Scholar] [CrossRef]
- Li, P.; Ohtsuki, C.; Kokubo, T.; Nakanishi, K.; Soga, N.; de Groot, K. The role of hydrated silica, titania, and alumina in inducing apatite on implants. J. Biomed. Mater. Res. 1994, 28, 7–15. [Google Scholar]
- Ajami, E.; Aquey-Zinsou, K.F. Formation of OTS self-assembled monolayers at chemically treated titanium surfaces. J. Mater. Sci. Mater. Med. 2011, 22, 1813–1824. [Google Scholar] [CrossRef]
- Zhu, P.; Masuda, Y.; Koumoto, K. The effect of surface charge on hydroxyapatite nucleation. Biomaterials 2004, 25, 3915–3921. [Google Scholar] [CrossRef]
- Hanawa, T.; Kamiura, Y.; Yamamoto, S.; Kohgo, T.; Amemiya, A.; Ukai, H.; Murakami, K.; Asaoka, K. Early bone formation around calcium-ion-implanted titanium inserted into rat tibia. J. Biomed. Mater. Res. 1997, 36, 131–136. [Google Scholar] [CrossRef]
- Xie, Y.; Liu, X.; Chu, P.K.; Ding, C. Nucleation and growth of calcium-phosphate on Ca-implanted titanium surface. Surf. Sci. 2006, 600, 651–656. [Google Scholar] [CrossRef]
- Wan, Y.Z.; Huang, Y.; He, F.; Wang, Y.L.; Zhao, Z.G.; Ding, H.F. Effect of Mg ion implantation on calcium phosphate formation on titanium. Surf. Coating Tech. 2006, 201, 2904–2909. [Google Scholar] [CrossRef]
- Maitz, M.F.; Pham, M.T.; Matz, W.; Reuther, H.; Steiner, G.; Richter, E. Ion beam treatment of titanium surfaces for enhancing deposition of hydroxyapatite from solution. Biomol. Eng. 2002, 19, 269–272. [Google Scholar] [CrossRef]
- Hanawa, T. In vivo metallic biomaterials and surface modification. Mater. Sci. Eng. A 1999, 267, 260–266. [Google Scholar] [CrossRef]
- Feng, B.; Chen, J.Y.; Qi, S.K.; He, L.; Zhao, J.Z.; Zhang, X.D. Carbonate apatite coating on titanium induced rapidly by precalcification. Biomaterials 2002, 23, 173–179. [Google Scholar] [CrossRef]
- Jobin, M.; Taborelli, M.; Descouts, P. Surface properties of electropolished titanium and vanadium. Appl. Surf. Sci. 1993, 72, 363–372. [Google Scholar] [CrossRef]
- Lausmaa, J.; Kasemo, B.; Mattsson, H.; Odelius, H. Multi-technique surface characterization of oxide films on electropolished and anodically oxidized titanium. Appl. Surf. Sci. 1990, 45, 189–200. [Google Scholar] [CrossRef]
- Mathieu, J.B.; Mathieu, H.J.; Landolt, D. Electropolishing of titanium in perchloric acid—Acetic acid solution. J. Electrochem. Soc. 1978, 125, 1039–1043. [Google Scholar] [CrossRef]
- Larsson, C.; Thomsen, P.; Lausmaa, J.; Rodahl, M.; Kasemo, B.; Ericson, L.E. Bone response to surface modified titanium implants: Studies on electropolished implants with different oxide thicknesses and morphology. Biomaterials 1994, 15, 1062–1074. [Google Scholar] [CrossRef]
- Meredith, D.O.; Eschbach, L.; Wood, M.A.; Riehle, M.O.; Curtis, A.S.G.; Richards, R.G. Human fibroblast reactions to standard and electropolished titanium and Ti-6Al-7Nb, and electropolished stainless steel. J. Biomed. Mater. Res. A 2005, 75, 541–555. [Google Scholar]
- Lewandowska, M.; Wlodkowska, M.; Olkowski, R.; Roguska, A.; Polak, B.; Pisarek, M.; Lewandowska-Szumiel, M.; Kurzydłowski, K.J. Chemical surface modifications of titanium implants. Macromol. Symp. 2007, 253, 115–121. [Google Scholar] [CrossRef]
- Fatehi, k.; Moztarzadeh, F.; Solati-Hashjin, M.; Tahriri, M. In vitro biomimetic deposition of apatite on alkaline and heat treated Ti6Al4V alloy surface. Bull. Mater. Sci. 2008, 31, 101–108. [Google Scholar]
- Chen, Y.; Zheng, X.; Ji, H.; Ding, C. Effect of Ti-OH formation on bioactivity of vacuum plasma sprayed titanium coating after chemical treatment. Surf. Coating Tech. 2007, 202, 494–498. [Google Scholar]
- Takadama, H.; Kim, H.M.; Kokubo, T.; Nakamura, T. XPS study of the process of apatite formation on bioactive Ti-6Al-4V alloy in simulated body fluid. Sci. Tech. Adv. Mater. 2001, 2, 389–396. [Google Scholar]
- Ntais, S.; Dracopoulos, V.; Siokou, A. TiCl4(THF)2 impregnation on a flat SiOx/Si(1 0 0) and on polycrystalline Au foil: Determination of surface species using XPS. J. Mol. Catal. A Chem. 2004, 220, 199–205. [Google Scholar] [CrossRef]
- Lausmaa, J. Surface spectroscopic characterization of titanium implant materials. J. Electron Spectros. Relat. Phenom. 1996, 81, 343–361. [Google Scholar] [CrossRef]
- Feng, B.; Chen, J.Y.; QI, S.K.; He, L.; Zhao, J.Z.; Zhang, X.D. Characterization of surface oxide films on titanium and bioactivity. J. Mater. Sci. Mater. Med. 2002, 13, 457–464. [Google Scholar] [CrossRef]
- Takeuchi, M.; Abe, Y.; Yoshida, Y.; Nakayama, Y.; Okazaki, M.; Akagawa, Y. Acid pretreatment of titanium implants. Biomaterials 2003, 24, 1821–1827. [Google Scholar] [CrossRef]
- Pouilleau, J.; Devilliers, D.; Groult, H. Surface study of a titanium-based ceramic electrode material by X-ray photoelectron spectroscopy. J. Mater. Sci. Eng. A 1997, 32, 5645–5651. [Google Scholar]
- Lu, G.; Bernasek, S.L.; Schwartz, J. Oxidation of a polycrystalline titanium surface by oxygen and water. Surf. Sci. 2000, 458, 80–90. [Google Scholar] [CrossRef]
- Shirkhanzadeh, M. XRD and XPS characterization of superplastic TiO2 coatings prepared on Ti6Al4V surgical alloy by an electrochemical method. J. Mater. Sci. Mater. Med. 1995, 6, 206–210. [Google Scholar] [CrossRef]
- Sundgren, J.E.; Bodo, P.; Lundstrom, I. Auger electron spectroscopic studies of the interface between human tissue and implants of titanium and stainless steel. J. Colloid Interface Sci. 1986, 110, 9–20. [Google Scholar] [CrossRef]
- Sham, T.K.; Lazarus, M.S. X-ray photoelectron spectroscopy (XPS) studies of clean and hydrated TiO2 (Rutile) surfaces. Chem. Phys. Lett. 1979, 68, 426–432. [Google Scholar] [CrossRef]
- Boehm, H.P. Acidic and basic properties of hydroxylated metal oxide surfaces. Discuss. Faraday Soc. 1971, 52, 264–275. [Google Scholar] [CrossRef]
- Kokubo, T. Design of bioactive bone substitutes based on mineralization process. Mater. Sci. Eng. C 2005, 25, 97–104. [Google Scholar] [CrossRef]
- Resende, C.X.; Dille, J.; Platt, G.M.; Bastos, N.I.; Soares, G.A. Characterization of coating produced on titanium surface by a designed solution containing calcium and phosphate ions. Mater. Chem. Phys. 2008, 109, 429–435. [Google Scholar] [CrossRef]
- Kokubo, T.; Kim, H.M.; Kawashita, M.; Nakamura, T. Bioactive metals: Preparation and properties. J. Mater. Sci. Mater. Med. 2004, 15, 99–107. [Google Scholar] [CrossRef]
- Wang, X.X.; Hayakawa, S.; Tsuru, K.; Osaka, A. Improvement of bioactivity of H2O2/TaCl2-treated titanium after subsequent heat treatments. J. Biomed. Mater. Res. 2000, 52, 171–176. [Google Scholar] [CrossRef]
- Cui, X.; Kim, H.M.; Kawashita, M.; Wang, L.; Xiong, T.; Kokubo, T.; Nakamura, T. Effect of hot water and heat treatment on the apatite-forming ability of titania films formed on titanium metal via anodic oxidation in acetic acid solutions. J. Mater. Sci. Mater. Med. 2008, 19, 1767–1773. [Google Scholar] [CrossRef]
- Takemoto, M.; Fujibayashi, S.; Neo, M.; Suzuki, J.; Matsushita, T.; Kokubo, T.; Nakamura, T. Osteoinductive porous titanium implants: Effect of sodium removal by dilute HCl treatment. Biomaterials 2006, 27, 2682–2691. [Google Scholar] [CrossRef]
- Barrere, F.; Snell, M.E.; van Blitterswijk, C.A.; De Groot, K.; Layrolle, P. Nano-scale study of the nucleation and growth of calcium phosphate coating on titanium implants. Biomaterials 2004, 25, 2901–2910. [Google Scholar]
- Kokubo, T.; Kim, H.M.; Kawashita, M. Novel bioactive materials with different mechanical properties. Biomaterials 2003, 24, 2161–2175. [Google Scholar] [CrossRef]
- Uchida, M.; Kim, H.M.; Fujibayashi, S.; Nakamura, T. Structural dependence of apatite formation on titania gels in a simulated body fluid. J. Biomed. Mater. Res. 2003, 64A, 164–170. [Google Scholar] [CrossRef]
- Bigi, A.; Boanini, E.B.; Bracci, A.; Facchini, S.; Panzavolta, F.; Segatti, L. Sturba nanocrystalline hydroxyapatite coatings on titanium:a new fast biomimetic method. Biomaterials 2005, 26, 4085–4089. [Google Scholar] [CrossRef]
- Uchida, M.; Kim, H.M.; Kokubo, T.; Fujibayashi, S.; Nakamura, T. Effect of water treatment on the apatite-forming ability of NaOH-treated titanium metal. J. Biomed. Mater. Res. 2002, 63, 522–530. [Google Scholar] [CrossRef]
- Li, F.; Feng, Q.L.; Cui, F.Z.; Li, H.D.; Schubert, H. A simple biomimetic method for calcium phosphate coating. Surf. Coating Tech. 2002, 154, 88–93. [Google Scholar] [CrossRef]
- Davies, J.E. Bone bonding at natural and biomaterial surfaces. Biomaterials 2007, 28, 5058–5067. [Google Scholar] [CrossRef]
- Davies, J.E. Understanding peri-implant endosseous healing. J. Dent. Educ. 2003, 67, 932–949. [Google Scholar]
- You, C.; OH, S.; Kim, S. Influences of heating condition and substrate-surface roughness on the characteristics of sol-gel-derived hydroxyapatite coatings. J. Sol-Gel Sci. Tech. 2001, 21, 49–54. [Google Scholar] [CrossRef]
- Blackwood, D.J.; Seah, K.H. Seah Influence of anodization on the adhesion of calcium and phosphate coatings on titanium substrates. J. Biomed. Mater. Res. 2009, 93A, 1551–1556. [Google Scholar]
- Liu, D.P.; Majewski, P.J.; O'Neill, B.K.; Ngothai, Y.; Colby, C.B. The optimal SAM surface functional group for producing a biomimetic HA coating on Ti. J. Biomed. Mater. Res. 2006, 77A, 763–772. [Google Scholar] [CrossRef]
- Lin, C.M.; Yen, S.K. Characterization and bond strength of electrolytic HA/TiO2 double layers for orthopaedic applications. J. Mater. Sci. Mater. Med. 2005, 16, 889–897. [Google Scholar] [CrossRef]
- Koutsopoulos, S. Synthesis and characterization of hydroxyapatite crystals: A review study on the analytical methods. J. Biomed. Mater. Res. 2002, 62, 600–612. [Google Scholar] [CrossRef]
- Li, H.; Huang, W.; Zhang, Y.; Zhong, M. Biomimetic synthesis of enamel-like hydroxyapatite on self-assembled monolayers. Mater. Sci. Eng. C 2007, 27, 756–761. [Google Scholar] [CrossRef]
- Yousefpour, M.; Afashar, A.; Yang, X.; Li, X.; Yang, B.; Wu, Y.; Chen, J.; Zhang, X. Nano-crystalline growth of electrochemically deposited apatite coating on pure titanium. J. Electroanal. Chem. 2006, 589, 96–105. [Google Scholar] [CrossRef]
- Leeuwenburgh, S.; Layrolle, P.; Barrere, F.; de Bruijn, J.; Schoonman, J.; van Blitterswijk, C.A.; de Groot, K. Osteoclastic resorption of biomimetic calcium phosphate coatings in vitro. J. Biomed. Mater. Res. 2001, 56, 208–215. [Google Scholar] [CrossRef]
- He, L.; Feng, Z. Preparation and characterization of dicalcium phosphate dihydrate coating on enamel. Mater. Lett. 2007, 61, 3923–3926. [Google Scholar] [CrossRef]
- Ermrich, M.; Peters, F. X-ray powder diffraction data of synthetic b-tricalcium phosphate. Z. Kristallogr. Suppl. 2006, 23, 523–528. [Google Scholar] [CrossRef]
- Furuzono, T.; Walsh, D.; Yasuda, S.; Sato, K.; Tanaka, J.; Kishida, A. Preparation of plated β-tricalcium phosphate containing hydroxyapatite for use in bonded inorganic-organic composites. J. Mater. Sci. 2005, 40, 2595–2597. [Google Scholar]
- Rajabi-Zamani, A.H.; Behnamghader, A.; Kazemzadeh, A. Synthesis of nanocrystalline carbonated hydroxyapatite powder via nonalkoxide sol-gel method. Mater. Sci. Eng. C 2008, 28, 1326–1329. [Google Scholar] [CrossRef]
- Kim, H.E.; Himeno, T.; Kawashita, M.; Kokubo, T.; Nakamura, T. The mechanism of biomineralization of bone-like apatite on synthetic hydroxyapatite: An in vitro assessment. J. R. Soc. Interface 2004, 1, 17–22. [Google Scholar] [CrossRef]
- Kim, H.M.; Miyaji, F.; Kokubo, T. Effect of heat treatment on apatite-forming ability of Ti metal induced by alkali treatment. J. Mater. Sci. Mater. Med. 1997, 8, 341–347. [Google Scholar] [CrossRef]
- Schmidt, M. X-ray photoelectron spectroscopy studies on adsorption of amino acids from aqueous solutions onto oxidised titanium surfaces. Arch. Orthop. Trauma Surg. 2001, 121, 403–410. [Google Scholar] [CrossRef]
- Tomazic, B.; Tomson, M.; Nancollas, G.H. Growth of calcium phosphates on hydroxyapatite crystals: The effect of magnesium. Arch. Oral. Biol. 1975, 20, 803–808. [Google Scholar] [CrossRef]
- Barrere, F.; van Blitterswijk, C.A.; de Groot, K.; Layrolle, P. Nucleation of biomimetic Ca-P coatings on Ti6Al4V from a SBF× 5 solution: influence of magnesium. Biomaterials 2002, 23, 2211–2220. [Google Scholar] [CrossRef]
- Salimi, M.H.; Heughebaert, J.C.; Nancollas, G.H. Crystal growth of calcium phosphates in the presence of magnesium ions. Langmuir 1985, 1, 119–122. [Google Scholar] [CrossRef]
- Rey, C.; Bracci, B.; Goehl, T.; Dickson, I.R.; Glimcher, M.J. The carbonate environment in bone mineral: A resolution-enhanced fourier transform infrared spectroscopy study. Calcif. Tissue Int. 1989, 45, 157–164. [Google Scholar] [CrossRef]
- Vignoles, M.; Bonel, G.; Holcomb, D.W.; Young, R.A. Influence of preparation conditions on the composition of type B carbonated hydroxyapatite and on the localization of the carbonate ions. Calcif. Tissue Int. 1988, 43, 33–40. [Google Scholar] [CrossRef]
- Cheng, Z.H.; Yasukawa, A.; Kandori, K.; Ishikawa, T. FTIR study on incorporation of CO2 into calcium hydroxyapatite. J. Chem. Soc. Faraday Trans. 1998, 94, 1501–1505. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, J.; Feng, J.; Cao, Y.; Deng, C.; Zhang, X. Dissolution and mineralization behaviours of HA coatings. Biomaterials 2003, 24, 4741–4748. [Google Scholar] [CrossRef]
- Chang, M.C.; Douglas, W.H.; Tanaka, J. Organic-inorganic interaction and the growth mechanism of hydroxyapatite crystals in gelatin matrices between 37 and 80 °C. J. Mater. Sci. Mater. Med. 2006, 17, 387–396. [Google Scholar] [CrossRef]
- Stoch, A.; Jastrzebski, W.; Brozek, A.; Trybalska, B.; Cichocinska, M.; Szarawara, E. FTIR monitoring of the growth of the carbonate containing apatite layers from simulated and natural body fluids. J. Mol. Struct. 1999, 511–512, 287–294. [Google Scholar]
- Muller, L.; Conforto, E.; Caillard, D.; Muller, F.A. Biomimetic apatite coatings—Carbonate substitution and preferred growth orientation. Biomol. Eng. 2007, 24, 462–466. [Google Scholar] [CrossRef]
- Landi, E.; Tampieri, A.; Celotti, G.; Vichi, L.; Sandri, M. Influence of synthesis and sintering parameters on the characteristics of carbonate apatite. Biomaterials 2004, 25, 1763–1770. [Google Scholar] [CrossRef]
- Piotrowski, O.; Madore, C.; Landolt, D. The mechanism of electropolishing of titanium in methanol-sulfuric acid electrolytes. J. Electrochem. Soc. 1998, 145, 2362–2369. [Google Scholar] [CrossRef]
- Kim, H.M.; Miyazaki, T.; Kokubo, T.; Nakamura, T. Revised simulated body fluid. Key Eng. Mater. 2001, 192–195, 47–50. [Google Scholar]
- Kokubo, T.; Takadama, H. How useful is SBF in predicting in vivo bone bioactivity? Biomaterials 2006, 27, 2907–2915. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ajami, E.; Aguey-Zinsou, K.-F. Calcium Phosphate Growth at Electropolished Titanium Surfaces. J. Funct. Biomater. 2012, 3, 327-348. https://doi.org/10.3390/jfb3020327
Ajami E, Aguey-Zinsou K-F. Calcium Phosphate Growth at Electropolished Titanium Surfaces. Journal of Functional Biomaterials. 2012; 3(2):327-348. https://doi.org/10.3390/jfb3020327
Chicago/Turabian StyleAjami, Elnaz, and Kondo-Francois Aguey-Zinsou. 2012. "Calcium Phosphate Growth at Electropolished Titanium Surfaces" Journal of Functional Biomaterials 3, no. 2: 327-348. https://doi.org/10.3390/jfb3020327
APA StyleAjami, E., & Aguey-Zinsou, K.-F. (2012). Calcium Phosphate Growth at Electropolished Titanium Surfaces. Journal of Functional Biomaterials, 3(2), 327-348. https://doi.org/10.3390/jfb3020327