Electro-Optical Properties of Monolayer and Bilayer Pentagonal BN: First Principles Study

 , ,
, ,
Abstract
:1. Introduction
2. Methods
3. Results
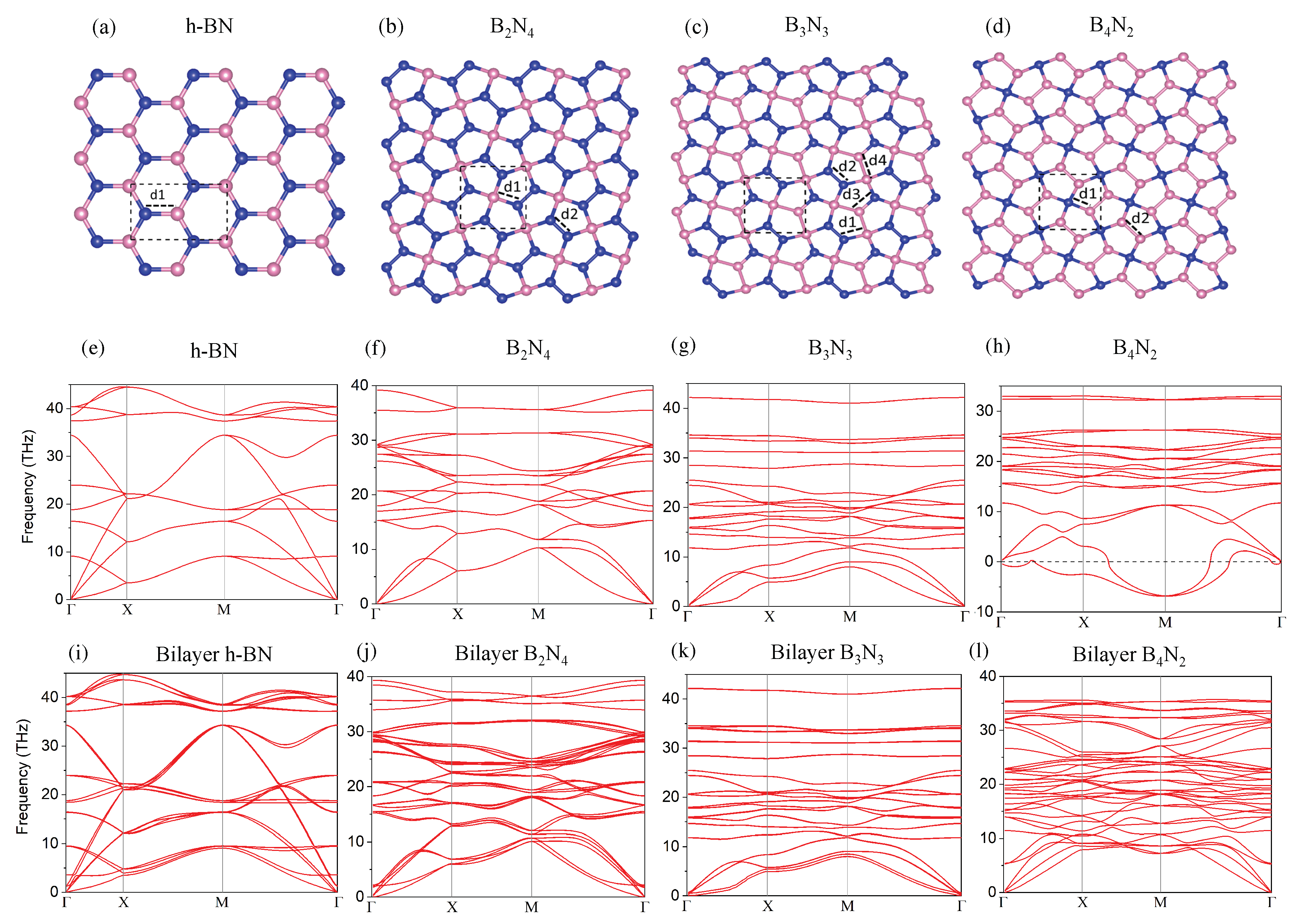
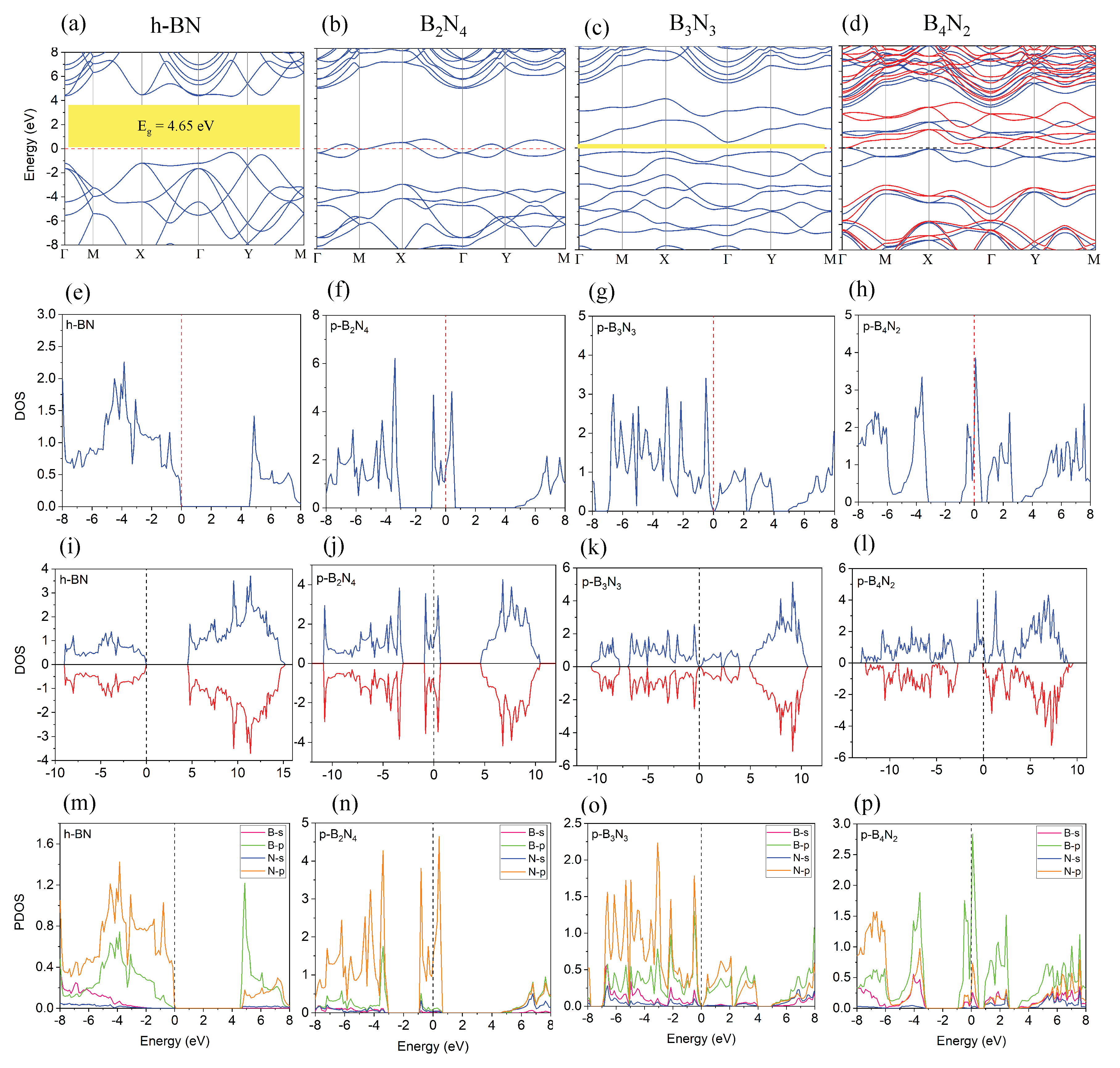
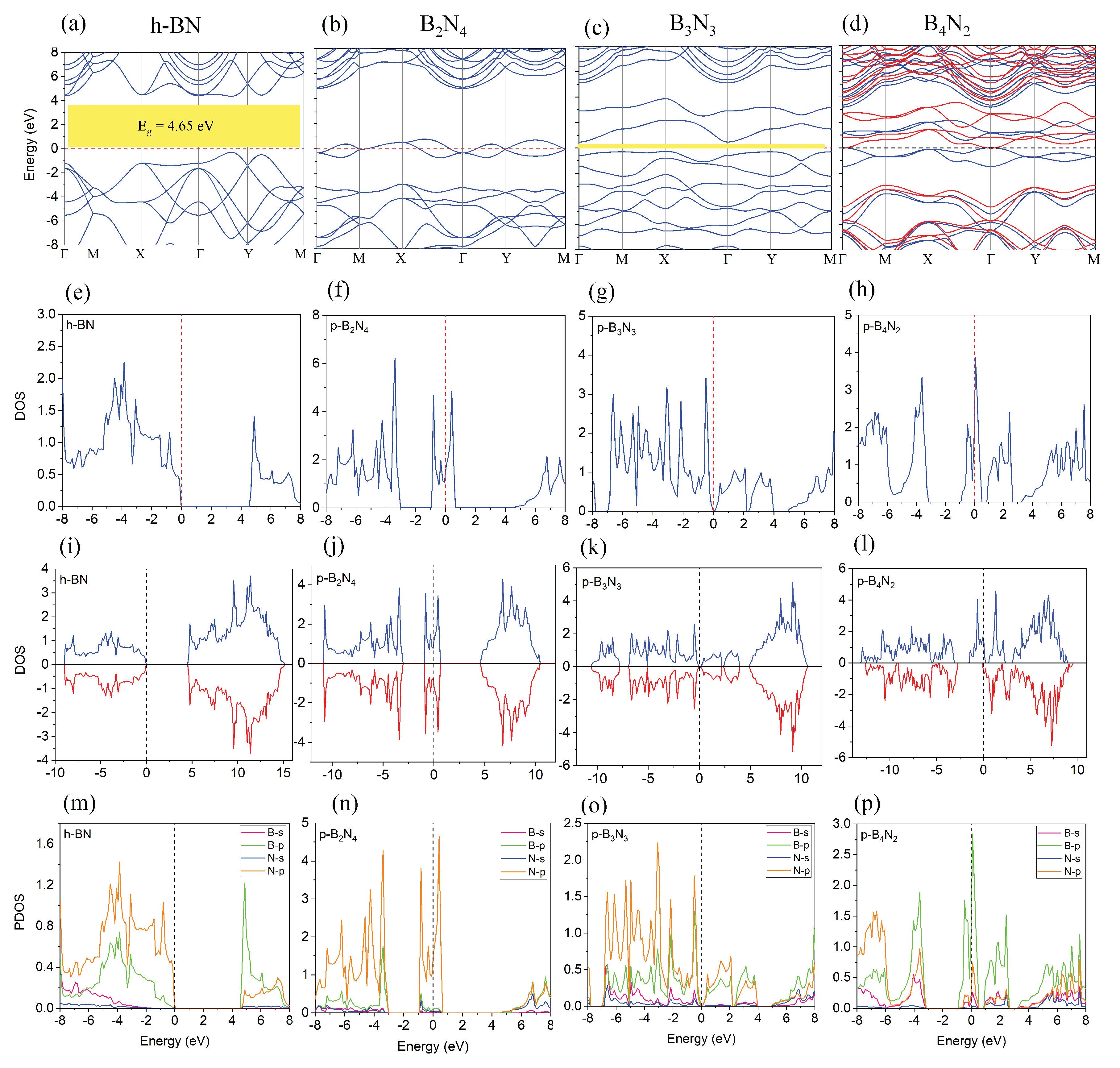
3.1. Monolayer Pentagonal BN
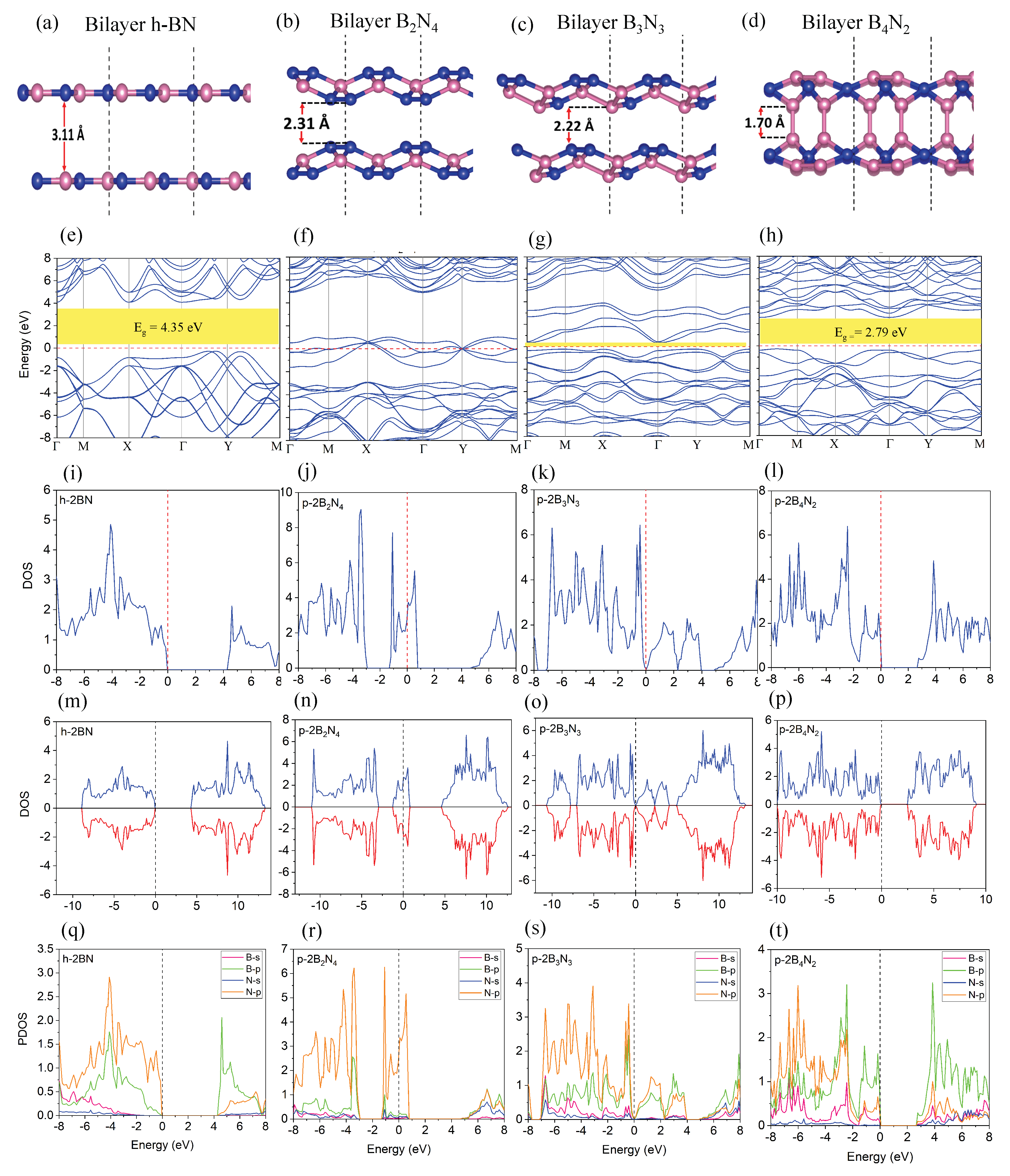
3.2. Bilayer Pentagonal BN
4. Discussion
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Tian, Y.; Xu, B.; Yu, D.; Ma, Y.; Wang, Y.; Jiang, Y.; Hu, W.; Tang, C.; Gao, Y.; Luo, K.; et al. Ultrahard nanotwinned cubic boron nitride. Nature 2013, 493, 385. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Hamsen, C.; Jia, X.; Kim, K.K.; Reina, A.; Hofmann, M.; Hsu, A.L.; Zhang, K.; Li, H.; Juang, Z.-Y.; et al. Synthesis of few-layer hexagonal boron nitride thin film by chemical vapor deposition. J. Nano Lett. 2010, 10, 4134–4139. [Google Scholar] [CrossRef] [PubMed]
- Miyata, N.; Moriki, K.; Mishima, O.; Fujisawa, M.; Hattori, T. Optical constants of cubic boron nitride. Phys. Rev. B 1989, 40, 12028. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.-N.; Ching, W.Y. Calculation of ground-state and optical properties of boron nitrides in the hexagonal, cubic, and wurtzite structures. Phys. Rev. B 1991, 44, 7787. [Google Scholar] [CrossRef]
- Christensen, N.E.; Gorczyca, I. Optical and structural properties of III-V nitrides under pressure. Phys. Rev. B 1994, 50, 4397. [Google Scholar] [CrossRef]
- Li, X.; Zhao, J.; Yang, J. Semihydrogenated BN sheet: A promising visible-light driven photocatalyst for water splitting. J. Sci. Rep. 2013, 3, 1858. [Google Scholar] [CrossRef]
- Kern, G.; Kresse, G.; Hafner, J. Ab initio calculation of the lattice dynamics and phase diagram of boron nitride. Phys. Rev. B 1999, 59, 8551. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, Q.; Kawazoe, Y.; Jena, P. Three-Dimensional Metallic Boron Nitride. J. Am. Chem. Soc. 2013, 135, 18216–18221. [Google Scholar] [CrossRef]
- Shahsavari, R. Intercalated Hexagonal Boron Nitride/Silicates as Bilayer Multifunctional Ceramics. ACS Appl. Mater. Interfaces 2018, 10, 2203–2209. [Google Scholar] [CrossRef]
- Hromadová, L.; Martoňák, R. Pressure-induced structural transitions in BN from ab initio metadynamics. Phys. Rev. B 2011, 84, 224108. [Google Scholar] [CrossRef]
- Wen, B.; Zhao, J.; Melnik, R.; Tian, Y. Body-centered tetragonal B2N2: A novel sp3 bonding boron nitride polymorph. Phys. Chem. Chem. Phys. 2011, 13, 14566. [Google Scholar] [CrossRef]
- He, C.; Sun, L.; Zhang, C.; Peng, X.; Zhang, K.; Zhong, J. Z-BN: A novel superhard boron nitride phase. J. Phys. Chem. Chem. Phys. 2012, 14, 10967. [Google Scholar] [CrossRef] [PubMed]
- Germaneau, É.; Su, G.; Zheng, Q.-R. New boron nitride structures B4N4: A first-principles random searching application. J. Phys. Condens. Matter 2013, 25, 125504. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Yu, D.; Zhao, Z.; Fu, S.; Xiong, M.; Wang, Q.; Gao, Y.; Luo, K.; He, J.; Tian, Y. First-principles study of O-BN: A sp3-bonding boron nitride allotrope. J. Appl. Phys. 2012, 112, 053518. [Google Scholar] [CrossRef]
- Jiang, X.; Zhao, J.; Ahuja, R. A novel superhard BN allotrope under cold compression of h-BN. J. Phys. Condens. Matter 2013, 25, 122204. [Google Scholar] [CrossRef]
- Blase, X.; Rubio, A.; Louie, S.G.; Cohen, M.L. Stability and Band Gap Constancy of Boron Nitride Nanotubes. Europhys. Lett. 1994, 28, 335. [Google Scholar] [CrossRef] [Green Version]
- Rubio, A.; Corkill, J.L.; Cohen, M.L. Theory of graphitic boron nitride nanotubes. Phys. Rev. B 1994, 49, 5081. [Google Scholar] [CrossRef] [Green Version]
- Xiang, H.J.; Yang, J.; Hou, J.G.; Zhu, Q. First-principles study of small-radius single-walled BN nanotubes. Phys. Rev. B 2003, 68, 035427. [Google Scholar] [CrossRef]
- Shayeganfar, F.; Javad Beheshtian, J.; Shahsavari, R. First-Principles Study of Water Nanotubes Captured Inside Carbon/Boron Nitride Nanotubes. Langmuir 2018, 34, 11176–11187. [Google Scholar] [CrossRef]
- Enyashin, A.N.; Ivanovskii, A.L. Graphene-like BN allotropes: Structural and electronic properties from DFTB calculations. Chem. Phys. Lett. 2011, 509, 143. [Google Scholar] [CrossRef] [Green Version]
- Butt, H.; Butler, T.; Montelongo, Y.; Rajesekharan, R.; Wilkinson, T.D.; Amaratunga, G.A.J. Continuous diffraction patterns from circular arrays of carbon nanotubes. Appl. Phys. Lett. 2012, 101, 251102. [Google Scholar] [CrossRef] [Green Version]
- Won, K.; Palani, A.; Butt, H.; Hands, P.J.W.; Rajeskharan, R.; Dai, Q.; Ahmed Khan, A.; Amaratunga, G.A.J.; Coles, H.J.; Wilkinson, T.D. Electrically Switchable Diffraction Grating Using a Hybrid Liquid Crystal and Carbon Nanotube-Based Nanophotonic Device. Adv. Opt. Mater. 2013, 1, 368–373. [Google Scholar] [CrossRef] [Green Version]
- Rajasekharan, R.; Bay, C.; Dai, Q.; Jon Freeman, J.; Wilkinsonet, T.D. Electrically reconfigurable nanophotonic hybrid grating lens array. Appl. Phys. Lett. 2010, 96, 233108. [Google Scholar] [CrossRef]
- Zhang, Z.; Guo, W. Intrinsic Metallic and Semiconducting Cubic Boron Nitride Nanofilms. Nano Lett. 2012, 12, 3650. [Google Scholar] [CrossRef]
- Wang, Y.; Ding, Y.; Ni, J. Fluorination-induced half-metallicity in zigzag boron nitride nanoribbons: First-principles calculations. Phys. Rev. B 2010, 81, 193407. [Google Scholar] [CrossRef]
- Lopez-Bezanilla, A.; Huang, J.; Terrones, H.; Sumpter, B.G. Boron Nitride Nanoribbons Become Metallic. Nano Lett. 2011, 11, 3267. [Google Scholar] [CrossRef]
- Barone, V.; Peralta, J.E. Magnetic Boron Nitride Nanoribbons with Tunable Electronic Properties. Nano Lett. 2008, 8, 2210–2214. [Google Scholar] [CrossRef] [Green Version]
- Zheng, F.; Zhou, G.; Liu, Z.; Wu, J.; Duan, W.; Gu, B.-L.; Zhang, S.B. Half metallicity along the edge of zigzag boron nitride nanoribbons. Phys. Rev. B 2008, 78, 205415. [Google Scholar] [CrossRef]
- Shayeganfar, F.; Rahimi tabar, M.R.; Simchi, A.; Beheshtian, J. Effects of functionalization and side defects on single-photon emission in boron nitride quantum dots. Phys. Rev. B 2017, 96, 165307. [Google Scholar] [CrossRef]
- Zhang, Z.; Guo, W. Energy-gap modulation of BN ribbons by transverse electric fields: First-principles calculations. Phys. Rev. B 2008, 77, 075403. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; Zhi, C.; Zhang, Z.; Wei, X.; Wang, X.; Guo, W.; Bando, Y.; Golberg, D. “White Graphenes”: Boron Nitride Nanoribbons via Boron Nitride Nanotube Unwrapping. Nano Lett. 2010, 10, 5049–5055. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. J. Comput. Mat. Sci. 1996, 6, 15. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Li, J.; Fan, X.; Wei, Y.; Chen, G. Penta-BxNy sheet: A density functional theory study of two-dimensional material. Sci. Rep. 2016, 6, 31840. [Google Scholar] [CrossRef] [Green Version]
- Ehrenreich, H.; Cohen, M.H. Self-Consistent Field Approach to the Many-Electron Problem. Phys. Rev. 1959, 115, 786–790. [Google Scholar] [CrossRef]
- Abt, R.; Draxl, C.A.; Knoll, P. Optical response of high temperature superconductors by full potential LAPW band structure calculations. Physica B 1994, 194–196, 1451–1452. [Google Scholar] [CrossRef]
- Sun, Z.; Yuan, K.; Zhang, X.; Qin, G.; Gong, X.; Tang, D. Disparate strain response of the thermal transport properties of bilayer penta-graphene as compared to that of monolayer penta-graphene. Phys. Chem. Chem. Phys. 2019, 21, 15647–15655. [Google Scholar] [CrossRef]
- Ramazani, A.; Shayeganfar, F.; Jalilian, J.; Fang, N. Exciton-plasmon polariton coupling and hot carrier generation in two-dimensional SiB semiconductors: A first-principles study. Nanophotonics 2020, 9, 337–349. [Google Scholar] [CrossRef] [Green Version]
- Shayeganfar, F.; Rochefort, A. Electronic Properties of Self-Assembled Trimesic Acid Monolayer on Graphene. Langmuir 2014, 30, 9707–9716. [Google Scholar] [CrossRef]
- Grosso, G.; Parravicini, G. Solid State Physics, 2nd ed.; Academic Press: London, UK, 2003. [Google Scholar]
- Zhang, K.; Feng, Y.; Wang, F.; Yanga, Z.; Wang, J. Two dimensional hexagonal boron nitride (2D-hBN): Synthesis, properties and applications. Mater. Chem. C 2017, 5, 11992. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397. [Google Scholar] [CrossRef]
- Savin, A.; Jepsen, O.; Flad, J.; Andersen, O.K.; Preuss, H.; von Schnering, H.G. Electron Localization in Solid-State Structures of the Elements: The Diamond Structure. Angew. Chem. Int. Ed. 1992, 31, 187. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683. [Google Scholar] [CrossRef]
- Kortus, J.; Mazin, I.I.; Belashchenko, K.D.; Antropov, V.P.; Boyer, L.L. Superconductivity of Metallic Boron in MgB2. Phys. Rev. Lett. 2001, 86, 4656. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.P. Role of Boron p-Electrons and Holes in Superconducting MgB2, and Other Diborides: A Fully Relaxed, Full-Potential Electronic Structure Study. Phys. Rev. Lett. 2001, 87, 087004. [Google Scholar] [CrossRef] [Green Version]
- Kiran, B.; Phukan, A.K.; Jemmis, E.D. Is borazine aromatic? Unusual parallel behavior between hydrocarbons and corresponding B-N analogues. Inorg. Chem. 2001, 40, 3615. [Google Scholar] [CrossRef]
- Islas, R.; Chamorro, E.; Robles, J.; Heine, T.; Santos, J.; Merino, G. Borazine: To be or not to be aromatic. Struct. Chem. 2007, 18, 833. [Google Scholar] [CrossRef]
- Eremets, M.I.; Troyan, I.A. Conductive dense hydrogen. Nat. Mater. 2011, 10, 927. [Google Scholar] [CrossRef]
- Yao, Y.; Tse, J.S.; Klug, D.D. Structures of Insulating Phases of Dense Lithium. Phys. Rev. Lett. 2009, 102, 115503. [Google Scholar] [CrossRef]
- Guillaume, C.L.; Gregoryanz, E.; Degtyareva, O.; McMahon, M.I.; Hanfland, M.; Evans, S.; Guthrie, M.; Sinogeikin, S.V.; Mao, H.K. Cold melting and solid structures of dense lithium. Nat. Phys. 2011, 7, 211. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.; Wang, Y.; Zhu, L.; Ma, Y. Predicted Novel High-Pressure Phases of Lithium. Phys. Rev. Lett. 2011, 106, 015503. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lattice (Å) | |||||
|---|---|---|---|---|---|
| a = 4.35, b = 2.51 | 1.45 | — | — | — | |
| a = b = 3.62 | 1.55 | 1.34 | — | — | |
| a = b = 3.75 | 1.60 | 1.34 | 1.34 | 1.78 | |
| a = b = 3.79 | 1.57 | 1.59 | — | — |
| Energy | Interaction Energy | Fermi Energy | Band Gap | |
|---|---|---|---|---|
| (eV) | −35.45 | — | −4.48 | 4.65 |
| (eV) | −71.19 | −0.27 | −3.16 | 4.35 |
| (eV) | −47.96 | — | −3.33 | 0 |
| (eV) | −96.35 | −0.43 | −1.51 | 0 |
| (eV) | −45.41 | — | −3.80 | 0.11 |
| (eV) | −91.20 | −0.37 | −2.11 | 0.092 |
| (eV) | −41.43 | — | −2.05 | 0 |
| (eV) | −91.31 | −8.44 | −1.77 | 2.79 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amiri, M.; Beheshtian, J.; Shayeganfar, F.; Faghihnasiri, M.; Shahsavari, R.; Ramazani, A. Electro-Optical Properties of Monolayer and Bilayer Pentagonal BN: First Principles Study. Nanomaterials 2020, 10, 440. https://doi.org/10.3390/nano10030440
Amiri M, Beheshtian J, Shayeganfar F, Faghihnasiri M, Shahsavari R, Ramazani A. Electro-Optical Properties of Monolayer and Bilayer Pentagonal BN: First Principles Study. Nanomaterials. 2020; 10(3):440. https://doi.org/10.3390/nano10030440
Chicago/Turabian StyleAmiri, Mehran, Javad Beheshtian, Farzaneh Shayeganfar, Mahdi Faghihnasiri, Rouzbeh Shahsavari, and Ali Ramazani. 2020. "Electro-Optical Properties of Monolayer and Bilayer Pentagonal BN: First Principles Study" Nanomaterials 10, no. 3: 440. https://doi.org/10.3390/nano10030440