Effective Platinum-Copper Catalysts for Methanol Oxidation and Oxygen Reduction in Proton-Exchange Membrane Fuel Cell


 ,
,  , , ,
, , ,  ,
,
Abstract

1. Introduction
2. Materials and Methods
3. Results and Discussion
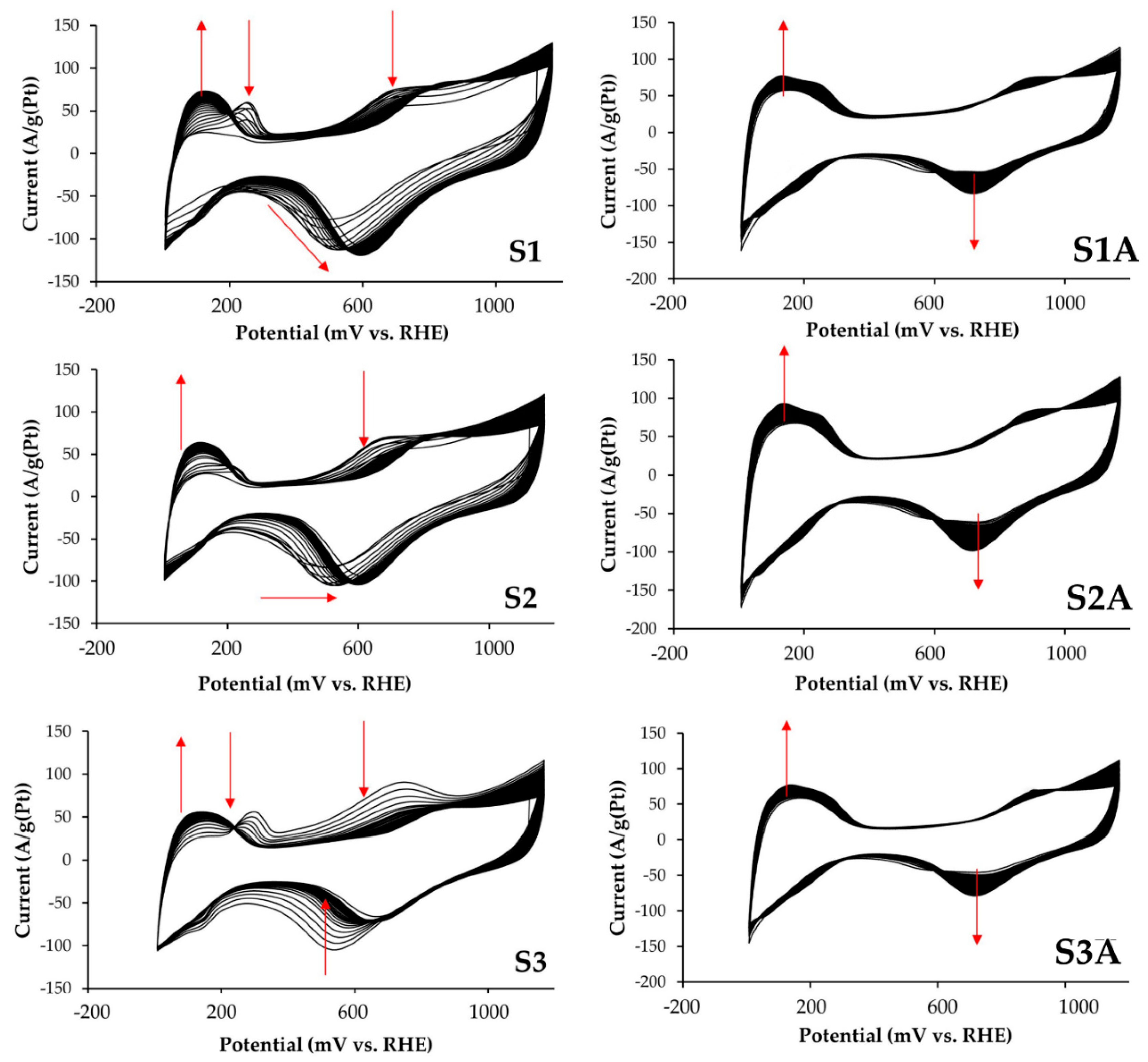
3.1. Methanol Electro-Oxidation at the Alloyed and De-Alloyed Platinum-Copper Catalysts
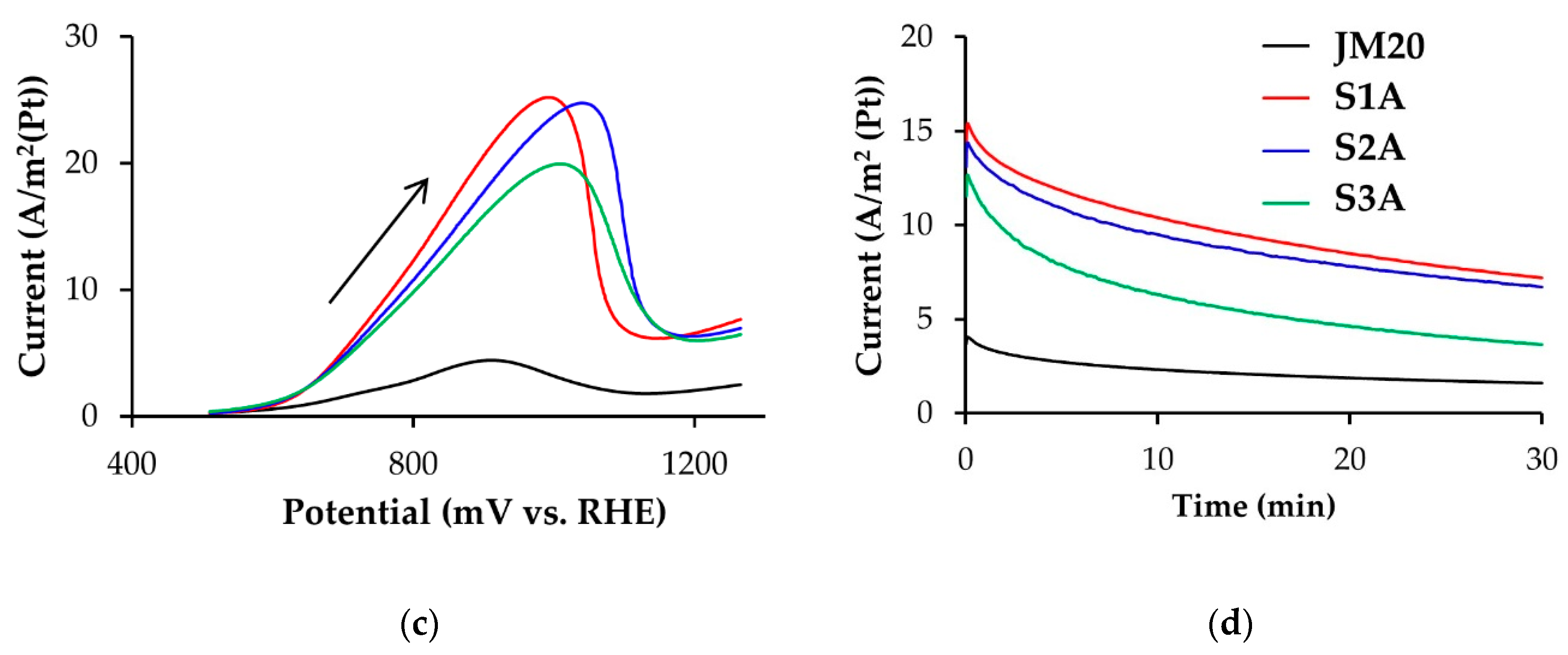
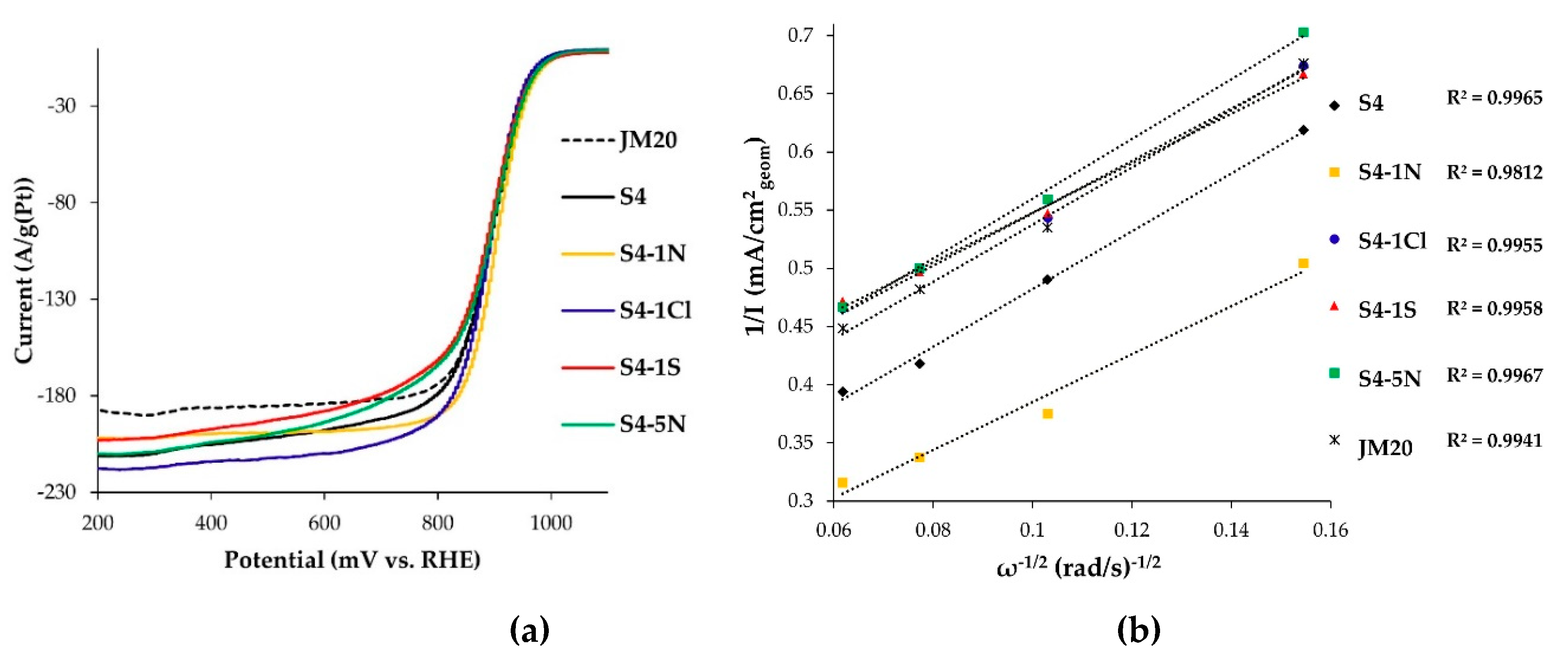
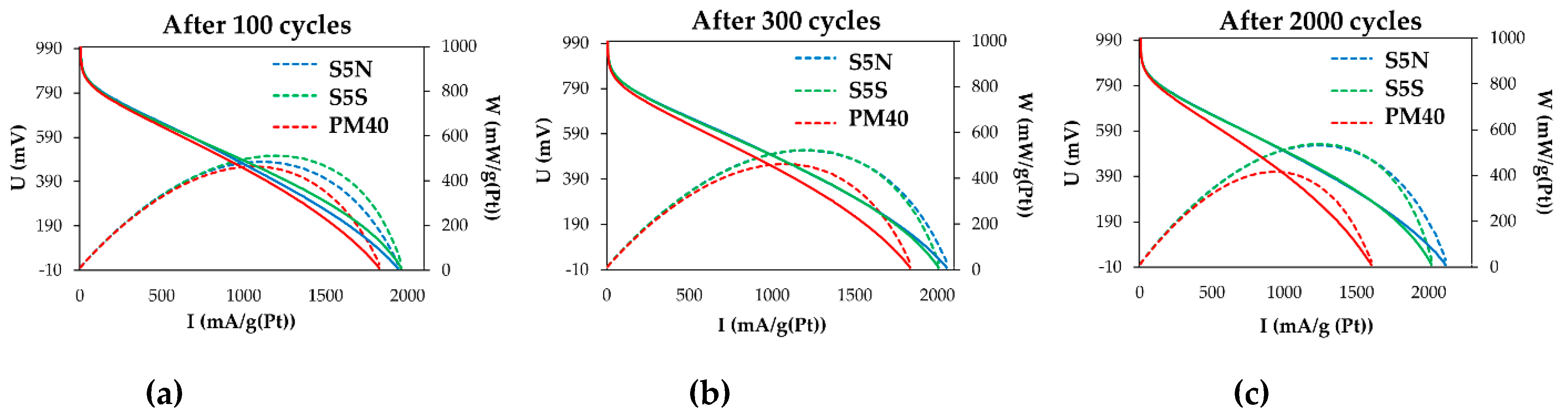
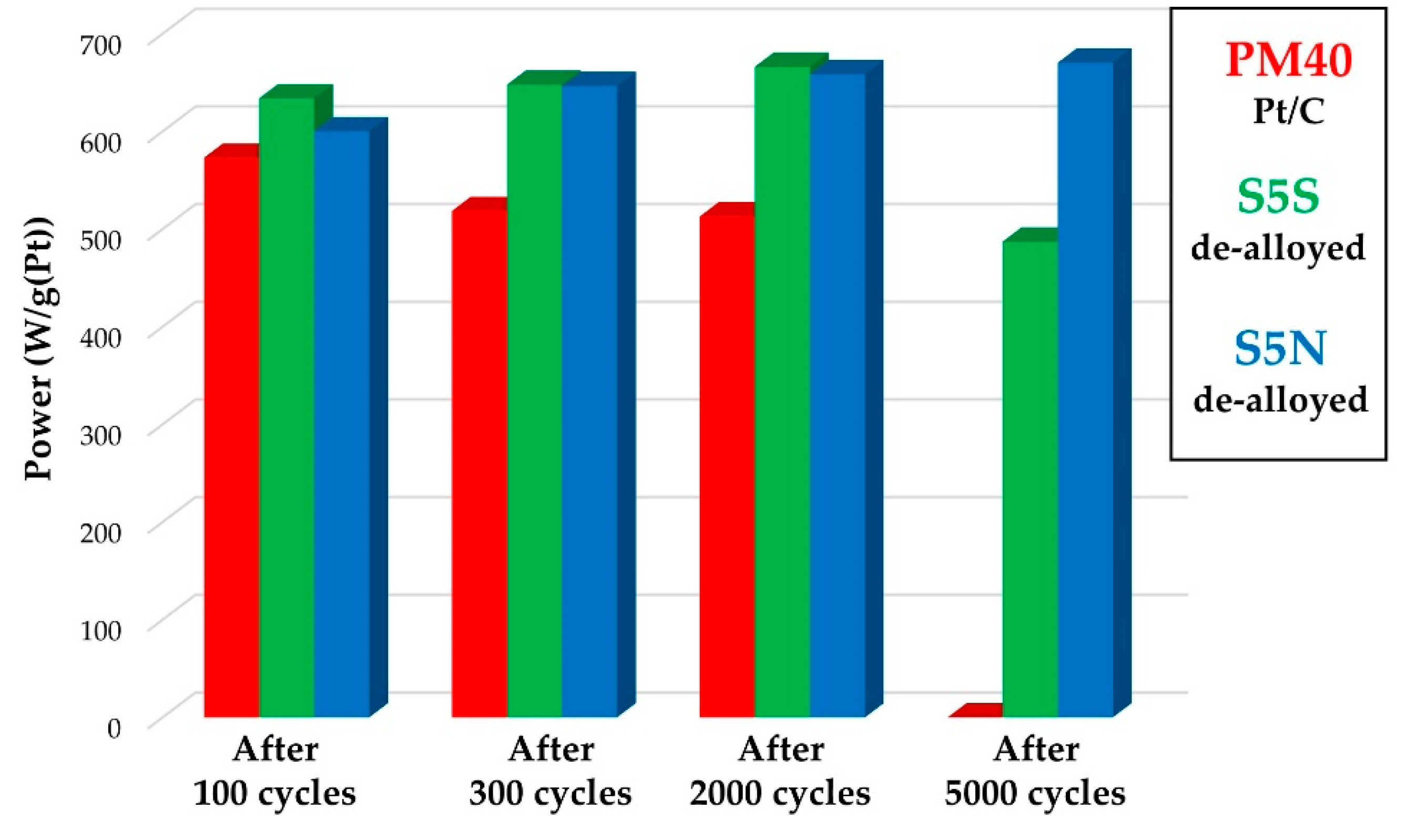
3.2. Electroreduction of Oxygen on Alloyed and De-Alloyed Platinum-Copper Catalysts
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- O’Hayre, R.P. Fuel Cell Fundamentals, 2nd ed.; Wiley & Sons Ltd.: New York, NY, USA, 2009; pp. 57–67. [Google Scholar]
- Qin, C.; Wang, J.; Yang, D.; Li, B.; Zhang, C. Proton Exchange Membrane Fuel Cell Reversal: A Review. Catalysts 2016, 6, 197. [Google Scholar] [CrossRef]
- Kongkanand, A.; Mathias, M.F. The Priority and Challenge of High-Power Performance of Low-Platinum Proton-Exchange Membrane Fuel Cells. J. Phys. Chem. Lett. 2016, 7, 1127–1137. [Google Scholar] [CrossRef] [PubMed]
- Crawley, G. Proton exchange membrane (PEM) fuel cells: Opening doors to fuel cell commercialization. Fuel Cell Today 2006, 1–12. [Google Scholar]
- Gong, L.; Yang, Z.; Li, K.; Xing, W.; Liu, C.; Ge, J. Recent development of methanol electrooxidation catalysts for direct methanol fuel cell. J. Energy Chem. 2018, 27, 1618–1628. [Google Scholar] [CrossRef]
- Basri, S.; Kamarudin, S.K.; Daud, W.R.W.; Yaakub, Z. Nanocatalyst for direct methanol fuel cell (DMFC). Int. J. Hydrogen Energy 2010, 35, 7957–7970. [Google Scholar] [CrossRef]
- Xia, Z.; Zhang, X.; Sun, H.; Wang, S.; Sun, G. Recent advances in multi-scale design and construction of materials for direct methanol fuel cells. Nano Energy 2019, 65, 104048. [Google Scholar] [CrossRef]
- Holton, O.T.; Stevenson, J.W. The Role of Platinum in Proton Exchange Membrane Fuel Cells. Platin. Met. Rev. 2013, 57, 259–271. [Google Scholar] [CrossRef]
- Hyun, K.; Lee, J.H.; Yoon, C.; Kwon, Y. The Effect of Platinum Based Bimetallic Electrocatalysts on Oxygen Reduction Reaction of Proton Exchange Membrane Fuel Cells. Int. J. Electrochem. Sci. 2013, 8, 11752–11767. [Google Scholar]
- Long, N.V.; Yang, Y.; Thi, C.M.; Minh, N.V.; Cao, Y.; Nogami, M. The development of mixture, alloy, and core-shell nanocatalysts with nanomaterial supports for energy conversion in low-temperature fuel cells. Nano Energy 2013, 2, 636–676. [Google Scholar] [CrossRef]
- Bele, M.; Gatalo, M.; Jovanovič, P.; Ruiz-Zepeda, F.; Šala, M.; Šest, E.; Gaberšček, M. Insight on Single Cell Proton Exchange Membrane Fuel Cell Performance of Pt-Cu/C Cathode. Catalysts 2019, 9, 544. [Google Scholar] [CrossRef]
- Lv, H.; Li, D.; Strmcnik, D.; Paulikas, A.P.; Markovic, N.M.; Stamenkovic, V.R. Recent advances in the design of tailored nanomaterials for efficient oxygen reduction reaction. Nano Energy 2016, 29, 149–165. [Google Scholar] [CrossRef]
- Caballero-Manrique, G.; Brillas, E.; Centellas, F.; Garrido, J.; Rodríguez, R.; Cabot, P.-L. Electrochemical Oxidation of the Carbon Support to Synthesize Pt(Cu) and Pt-Ru(Cu) Core-Shell Electrocatalysts for Low-Temperature Fuel Cells. Catalysts 2015, 5, 815–837. [Google Scholar] [CrossRef]
- Kaewsai, D.; Hunsom, M. Comparative Study of the ORR Activity and Stability of Pt and PtM (M = Ni, Co, Cr, Pd) Supported on Polyaniline/Carbon Nanotubes in a PEM Fuel Cell. Nanomaterials 2018, 8, 299. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.N.; Awasthi, R.; Sharma, C.S. Review: An overview of recent development of platinum based cathode materials for direct methanol fuel cells. Int. J. Electrochem. Sci. 2014, 9, 5607–5639. [Google Scholar]
- Ramli, Z.A.C.; Kamarudin, S.K. Platinum-Based Catalysts on Various Carbon Supports and Conducting Polymers for Direct Methanol Fuel Cell Applications: a Review. Nanoscale Res. Lett. 2018, 13, 410–435. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, H.; Sun, P.; Chen, T. Exceptional methanol electro-oxidation activity by bimetallic concave and dendritic Pt-Cu nanocrystals catalysts. J. Power Sources 2014, 245, 663–670. [Google Scholar] [CrossRef]
- Gómez–Marín, A.M.; Ticianelli, E.A. A reviewed vision of the oxygen reduction reaction mechanism on Pt-based catalysts. Curr. Opin. Electrochem. 2018, 9, 129–136. [Google Scholar] [CrossRef]
- Neergat, M.; Rahul, R. Unsupported Cu-Pt Core-Shell Nanoparticles: Oxygen Reduction Reaction (ORR) Catalyst with Better Activity and Reduced Precious Metal Content. J. Electrochem. Soc. 2012, 159, F234–F241. [Google Scholar] [CrossRef]
- Gasteiger, H.A.; Kocha, S.S.; Sompalli, B.; Wagner, F.T. Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl. Catal. B Environ. 2005, 56, 9–35. [Google Scholar] [CrossRef]
- Oezaslan, M.; Hasche, F.; Strasser, P. PtCu3, PtCu and Pt3Cu alloy nanoparticle electrocatalysts for oxygen reduction reaction in alkaline and acidic media. J. Electrochem. Soc. 2012, 159, B444–B454. [Google Scholar] [CrossRef]
- Alekseenko, A.A.; Guterman, V.E.; Belenov, S.V.; Menshikov, V.S.; Tabachkova, N.Y.; Safronenko, O.I.; Moguchikh, E.A. Pt/C electrocatalysts based on the nanoparticles with the gradient structure. Int. J. Hydrogen Energy 2018, 43, 3676–3687. [Google Scholar] [CrossRef]
- Wu, D.; Yang, Y.; Dai, C.; Cheng, D. Enhanced oxygen reduction activity of PtCu nanoparticles by morphology tuning and transition-metal doping. Int. J. Hydrogen Energy 2020, 45, 4427–4434. [Google Scholar] [CrossRef]
- Shabani, B.; Hafttananian, M.; Khamani, S.; Ramiar, A.; Ranjbar, A.A. Poisoning of proton exchange membrane fuel cells by contaminants and impurities: Review of mechanisms, effects, and mitigation strategies. J. Power Sources 2019, 427, 21–48. [Google Scholar] [CrossRef]
- Zamel, N.; Li, X. Effect of contaminants on polymer electrolyte membrane fuel cells. Prog. Energy Combust. Sci. 2011, 37, 292–329. [Google Scholar] [CrossRef]
- Cheng, X.; Shi, Z.; Glass, N.; Zhang, L. A review of PEM hydrogen fuel cell contamination: Impacts, mechanisms, and mitigation. J. Power Sources 2007, 165, 739–756. [Google Scholar] [CrossRef]
- Kühl, S.; Strasser, P. Oxygen Electrocatalysis on Dealloyed Pt Nanocatalysts. Top. Catal. 2016, 59, 1628–1637. [Google Scholar] [CrossRef]
- Gatalo, M.; Bele, M.; Ruiz-Zepeda, F.; Šest, E.; Šala, M.; Kamšek, A.R.; Maselj, N.; Galun, T.; Hodnik, N.; Gaberšček, M. Double Passivation Water Based Galvanic Displacement Method for Reproducible Gram Scale Production of High Performance Pt-alloy Electrocatalysts. Angew. Chem. Int. Edit. 2019, 58, 18096–18101. [Google Scholar] [CrossRef]
- Guterman, V.E.; Belenov, S.V.; Alekseenko, A.A.; Tabachkova, N.Y.; Volochaev, V.A. The relationship between activity and stability of deposited platinum-carbon electrocatalysts. Russ. J. Electrochem. 2017, 53, 531–539. [Google Scholar] [CrossRef]
- Ghavidel, Z.M.R.; Monteverde Videla, A.H.A.; Specchia, S.; Easton, E.B. The relationship between the structure and ethanol oxidation activity of Pt-Cu/C alloy catalysts. Electrochim. Acta 2017, 230, 58–72. [Google Scholar] [CrossRef]
- Marcu, A.; Toth, G.; Srivastava, R.; Strasser, P. Preparation, characterization and degradation mechanisms of PtCu alloy nanoparticles for automotive fuel cells. J. Power Sources 2012, 208, 288–295. [Google Scholar] [CrossRef]
- Barim, S.B.; Bozbag, S.E.; Deljoo, B.; Aindow, M.; Erkey, C. Highly Active Carbon Supported PtCu Electrocatalysts for PEMFCs by in situ Supercritical Deposition Coupled with Electrochemical Dealloying. Fuel Cells 2019. [Google Scholar] [CrossRef]
- Strasser, P.; Koh, S.; Anniyev, T.; Greeley, J.; More, K.; Yu, C.; Nilsson, A. Lattice-strain control of the activity in dealloyed core–shell fuel cell catalysts. Nat. Chem. 2010, 26, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Mani, P.; Srivastava, R.; Strasser, P. Dealloyed binary PtM3 (M = Cu, Co, Ni) and ternary PtNi3M (M = Cu, Co, Fe, Cr) electrocatalysts for the oxygen reduction reaction: Performance in polymer electrolyte membrane fuel cells. J. Power Sources 2011, 196, 666–673. [Google Scholar] [CrossRef]
- Gatalo, M.; Moriau, L.; Petek, U.; Ruiz-Zepeda, F.; Šala, M.; Grom, M.; Galun, T.; Jovanovič, P.; Pavlišič, A.; Bele, M.; et al. CO-assisted ex-situ chemical activation of Pt-Cu/C oxygen reduction reaction electrocatalyst. Electrochim. Acta 2019, 306, 377–386. [Google Scholar] [CrossRef]
- Volochaev, V.A.; Belenov, S.V.; Alekseenko, A.A.; Guterman, V.E. On the possibilities of recognizing the architecture of binary Pt–M nanoparticles. Nanotechnol. Russ. 2017, 5-6, 227–235. [Google Scholar] [CrossRef]
- Shinozaki, K.; Zack, J.W.; Pylypenko, S.; Pivovar, B.S.; Kocha, S.S. Oxygen reduction reaction measurements on platinum electrocatalysts utilizing rotating disk electrode technique: II. Influence of ink formulation, catalyst layer uniformity and thickness. J. Electrochem. Soc. 2015, 162, F1384–F1396. [Google Scholar] [CrossRef]
- Van der Vliet, D.; Strmcnik, D.S.; Wang, C.; Stamenkovic, V.R. On the importance of correcting for the uncompensated Ohmic resistance in model experiments of the oxygen reduction reaction. J. Electroanal. Chem. 2010, 647, 29–34. [Google Scholar] [CrossRef]
- Khudhayer, W.J.; Kariuki, N.N.; Wang, X.; Myers, D.J. Oxygen Reduction Reaction Electrocatalytic Activity of Glancing Angle Deposited Platinum Nanorod. J. Electrochem. Soc 2011, 158, 1029–1041. [Google Scholar] [CrossRef]
- Shinozaki, K.; Zack, J.W.; Pylypenko, S.; Richards, R.M.; Pivovar, B.S.; Kocha, S.S. Benchmarking the oxygen reduction reaction activity of Pt-based catalysts using standardized rotating disk electrode methods. Int. J. Hydrogen Energy 2015, 40, 16820–16830. [Google Scholar] [CrossRef]
- Trindell, J.A.; Duan, Z.; Henkelman, G.; Crooks, R.M. Well-Defined Nanoparticle Electrocatalysts for the Refinement of Theory. Chem. Rev. 2020, 120, 814–850. [Google Scholar] [CrossRef]
- Ishikawa, H.; Sugawara, Y.; Inoue, G.; Kawase, M. Effects of Pt and ionomer ratios on the structure of catalyst layer: A theoretical model for polymer electrolyte fuel cells. J. Power Sources 2018, 374, 196–204. [Google Scholar] [CrossRef]
- Nechitailov, A.A.; Glebova, N.V.; Tomasov, A.A.; Krasnova, A.O.; Zelenina, N.K. Study of the Heterogeneity of a Mixed-Conducting Electrochemical Electrode. Tech. Phys. 2019, 64, 899–907. [Google Scholar] [CrossRef]
- Fuel Cell Technical Team Roadmap. p. 20. Available online: https://www.energy.gov/sites/prod/files/2014/02/f8/fctt_roadmap_june2013.pdf (accessed on 10 December 2019).
- Pryadchenko, V.V.; Srabionyan, V.V.; Belenov, S.V.; Volochaev, V.A.; Kurzin, A.A.; Avakyan, L.A.; Zizak, I.; Guterman, V.E.; Bugaev, L.A. Bimetallic PtCu Nanoparticles in PtCu/C Electrocatalysts: Structural and Electrochemical Characterization. Appl. Catal. A Gen. 2016, 525, 226–236. [Google Scholar] [CrossRef]
- Guterman, V.E.; Belenov, S.V.; Krikov, V.V.; Vysochina, L.L.; Yohannes, W.; Tabachkova, N.Y.; Balakshina, E.N. Reasons for the differences in the kinetics of thermal oxidation of Pt/C nanostructured materials. J. Phys. Chem. C 2014, 118, 23835. [Google Scholar] [CrossRef]
- Leontyev, I.N.; Leontyeva, D.V.; Kuriganova, A.B.; Popov, Y.V.; Maslova, O.A.; Glebova, N.V.; Smirnova, N.V. Characterization of the electrocatalytic activity of carbon-supported platinum-based catalysts by thermal gravimetric analysis. Mendeleev Commun. 2015, 25, 468–469. [Google Scholar] [CrossRef]
- Guterman, V.E.; Belenov, S.V.; Alekseenko, A.A.; Moguchikh, E.A.; Kirakosyan, S.A.; Danilenko, M.V. New platinum-containing electrocatalysts for PEM Fuel Cells. In Proceedings of the XXI Mendeleev Congress on General and Applied Chemisrtry, Saint Petersburg, Russia, 9–13 September 2019. [Google Scholar]
- Alekseenko, A.A.; Moguchikh, E.A.; Safronenko, O.I.; Guterman, V.E. Durability of de-alloyed PtCu/C electrocatalysts. Int. J. Hydrogen Energy 2018, 43, 22885–22895. [Google Scholar] [CrossRef]
- Hodnik, N.; Cherevko, S. Spot the Difference at the Nanoscale: Identical Location Electron Microscopy in Electrocatalysis. Cur. Opin. Electrochem. 2019, 15, 73–82. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Metal Component Composition of the Catalysts (According to XRFA) | Platinum Loading, ω (Pt), % wt. | 2θ (111), grad | Average Crystallite Size, DAv *, nm | |
|---|---|---|---|---|---|
| In “as-Prepared” State | After Electrochemical Standardization | ||||
| S1 | Pt37Cu63 | Pt67Cu33 | 19.9 ± 0.5 | 41.2 | 3.0 ± 0.2 |
| S2 | Pt31Cu69 | Pt63Cu38 | 23.4 ± 0.5 | 41.3 | 3.0 ± 0.2 |
| S3 | Pt26Cu74 | Pt67Cu33 | 26.6 ± 0.5 | 41.2 | 2.8 ± 0.2 |
| S1A | Pt53Cu47 | Pt63Cu38 | 18.5 ± 0.5 | 40.9 | 2.7 ± 0.2 |
| S2A | Pt53Cu47 | Pt67Cu33 | 22.4 ± 0.5 | 41.0 | 3.0 ± 0.2 |
| S3A | Pt56Cu44 | Pt67Cu33 | 26.4 ± 0.5 | 40.8 | 2.7 ± 0.2 |
| JM20 (Pt/C) | Pt | Pt | 20.0 ± 0.5 | 39.9 | 2.0 ± 0.2 |
| Sample | ECSA (Hads/des), m2/g(Pt) | ECSA (CO), m2/g(Pt) | QCH3OH * 102, Cl/g(Pt) | Imax oxidation CH3OH, A/g(Pt) | Chronoamperometry Results at E = 0.70 V | |
|---|---|---|---|---|---|---|
| Iinitial, A/g(Pt) | Ifinal, A/g(Pt) | |||||
| S1 | 39 ± 4 | 31 ± 3 | 127.8 | 970 | 492.3 | 230.6 |
| S2 | 31 ± 3 | 33 ± 3 | 116.6 | 816 | 464.4 | 239.0 |
| S3 | 37 ± 4 | 29 ± 3 | 70.1 | 600 | 432.3 | 82.8 |
| S1A | 38 ± 4 | 31 ± 3 | 96.0 | 868 | 529.5 | 248.1 |
| S2A | 43 ± 4 | 37 ± 4 | 107.8 | 997 | 578.7 | 269.9 |
| S3A | 33 ± 3 | 32 ± 3 | 72.3 | 647 | 411.7 | 117.7 |
| JM20 | 81 ± 8 | 78 ± 8 | 32.9 | 350 | 291.6 | 126.5 |
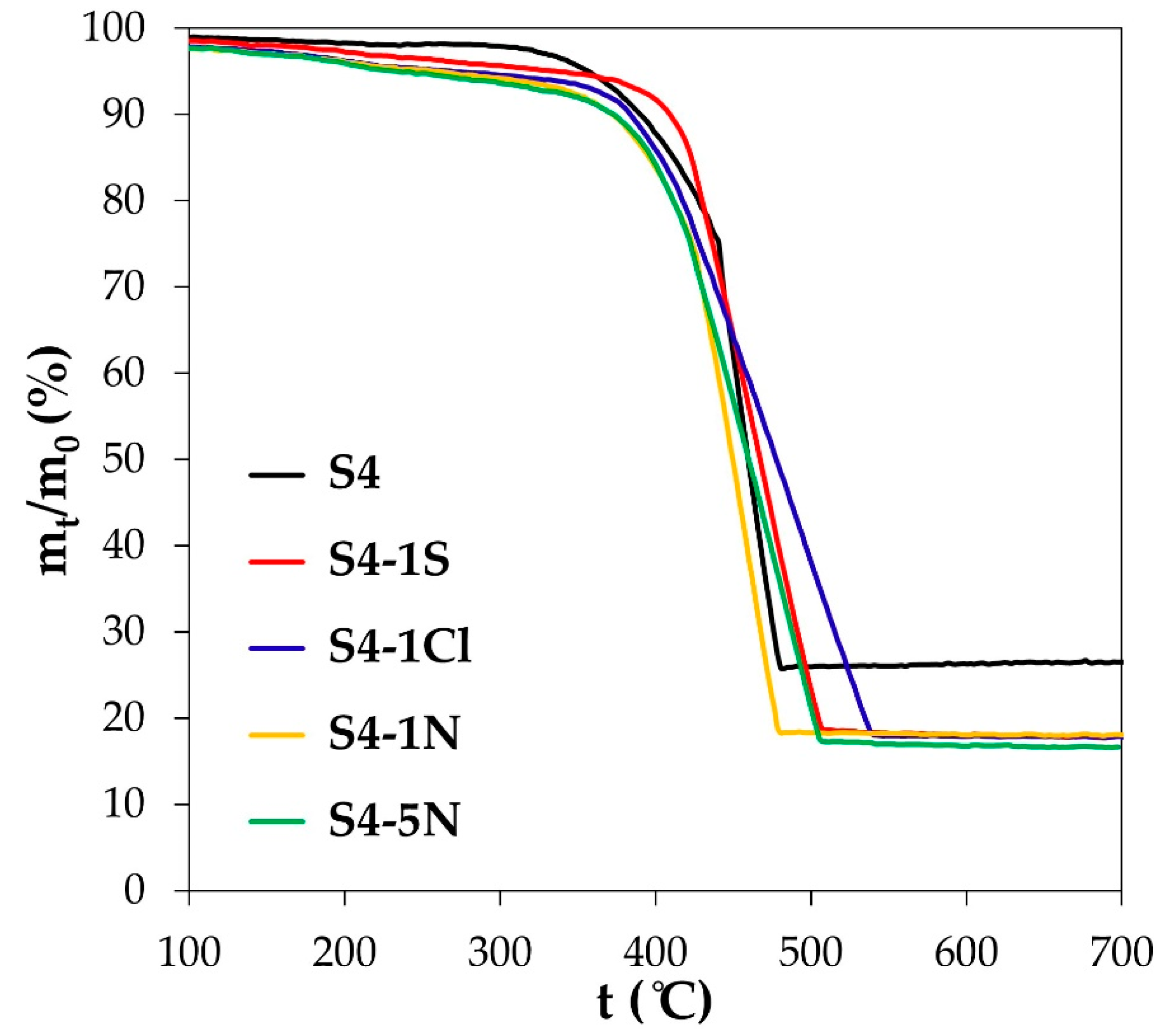
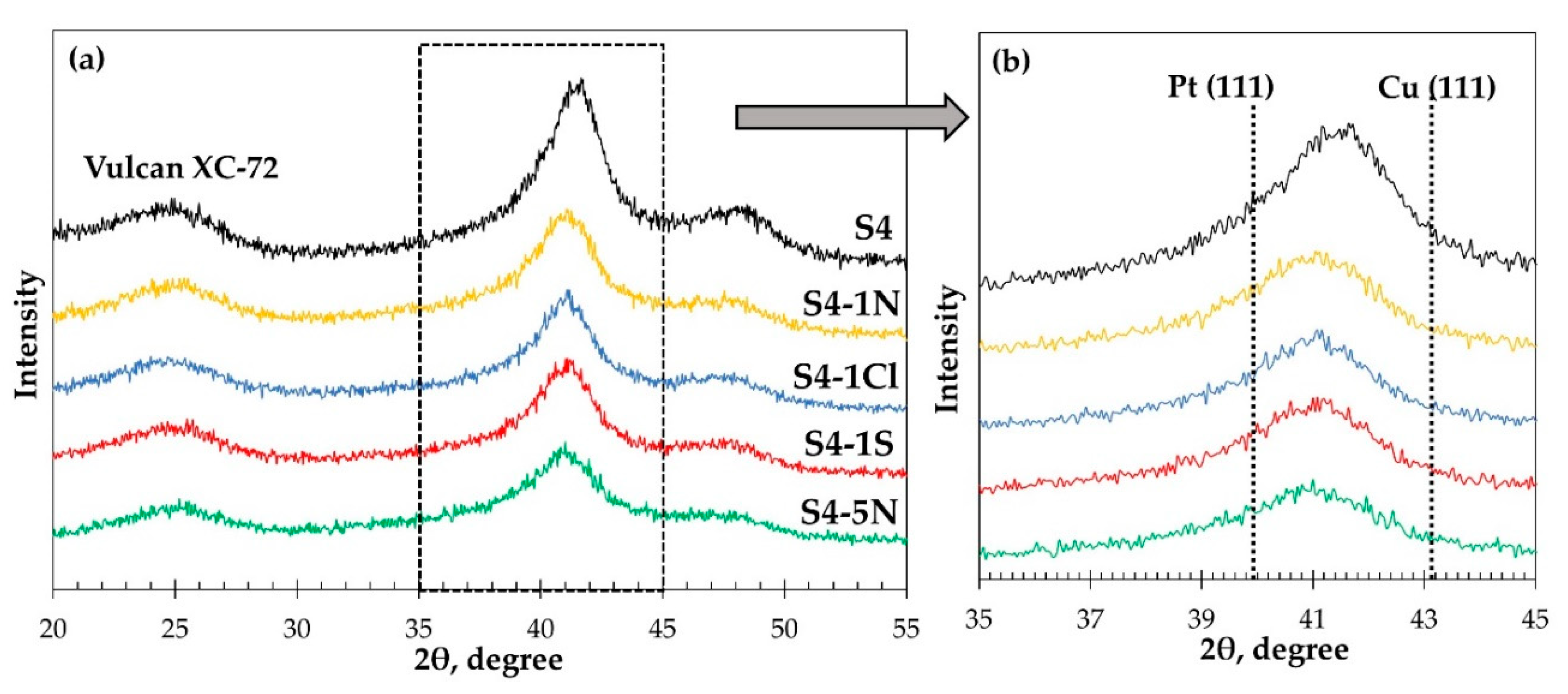
| Sample | Composition of the Solution for Treatment | Composition of Metallic Component | Metals Loading, % wt. | Average size of Crystallites (XRD), nm | |
|---|---|---|---|---|---|
| Metals | Platinum | ||||
| S4 | - | Pt31Cu69 | 23.4 ± 0.2 | 13.6 ± 0.2 | 2.8 ± 0.2 |
| S4-1N | 1M HNO3 | Pt57Cu43 | 17.4 ± 0.2 | 13.9 ± 0.2 | 2.4 ± 0.2 |
| S4-1Cl | 1M HClO4 | Pt57Cu43 | 17.2 ± 0.2 | 13.8 ± 0.2 | 2.6 ± 0.2 |
| S4-1S | 1M H2SO4 | Pt58Cu42 | 17.8 ± 0.2 | 14.4 ± 0.2 | 2.5 ± 0.2 |
| S4-5N | 5M HNO3 | Pt63Cu37 | 16.6 ± 0.2 | 13.9 ± 0.2 | 1.8 ± 0.2 |
| Sample | Pt and Cu Ratio in the Sample after Completion of Electrochemical Measurements | ECSA, m2/g(Pt) | E1/2, V (1600 min−1) | Kinetic Current, jк (at E = 0.90 V) | n | |
|---|---|---|---|---|---|---|
| A/g (Pt) | A/m2 (Pt) | |||||
| S4 | Pt67Cu33 | 39.4 | 0.90 | 161 | 4.1 | 3.8 |
| S4-1N | Pt65Cu35 | 41.2 | 0.90 | 200 | 4.9 | 4.4 |
| S4-1Cl | Pt69Cu31 | 35.7 | 0.89 | 130 | 3.6 | 4.0 |
| S4-1S | Pt68Cu32 | 35.2 | 0.90 | 117 | 3.3 | 4.2 |
| S4-5N | Pt72Cu28 | 39.2 | 0.90 | 140 | 3.6 | 3.5 |
| JM20 | Pt/C | 81.8 | 0.89 | 156 | 1.9 | 4.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menshchikov, V.; Alekseenko, A.; Guterman, V.; Nechitailov, A.; Glebova, N.; Tomasov, A.; Spiridonova, O.; Belenov, S.; Zelenina, N.; Safronenko, O. Effective Platinum-Copper Catalysts for Methanol Oxidation and Oxygen Reduction in Proton-Exchange Membrane Fuel Cell. Nanomaterials 2020, 10, 742. https://doi.org/10.3390/nano10040742
Menshchikov V, Alekseenko A, Guterman V, Nechitailov A, Glebova N, Tomasov A, Spiridonova O, Belenov S, Zelenina N, Safronenko O. Effective Platinum-Copper Catalysts for Methanol Oxidation and Oxygen Reduction in Proton-Exchange Membrane Fuel Cell. Nanomaterials. 2020; 10(4):742. https://doi.org/10.3390/nano10040742
Chicago/Turabian StyleMenshchikov, Vladislav, Anastasya Alekseenko, Vladimir Guterman, Andrey Nechitailov, Nadezhda Glebova, Aleksandr Tomasov, Olga Spiridonova, Sergey Belenov, Natalia Zelenina, and Olga Safronenko. 2020. "Effective Platinum-Copper Catalysts for Methanol Oxidation and Oxygen Reduction in Proton-Exchange Membrane Fuel Cell" Nanomaterials 10, no. 4: 742. https://doi.org/10.3390/nano10040742
APA StyleMenshchikov, V., Alekseenko, A., Guterman, V., Nechitailov, A., Glebova, N., Tomasov, A., Spiridonova, O., Belenov, S., Zelenina, N., & Safronenko, O. (2020). Effective Platinum-Copper Catalysts for Methanol Oxidation and Oxygen Reduction in Proton-Exchange Membrane Fuel Cell. Nanomaterials, 10(4), 742. https://doi.org/10.3390/nano10040742