
Galactomannan-Decorated Lipidic Nanocarrier for Gene Supplementation Therapy in Fabry Disease

 , , , , and
, , , , and
Abstract
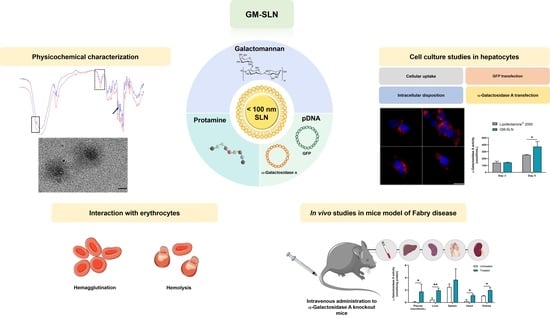
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of SLNs
2.3. Physical Stability of SLNs
2.4. Formulation of the SLNs-Based Vector with Galactomannan (GM-SLN)
2.5. Characterization of SLNs and the GM-SLN Vector: Particle Size, Polydispersity Index and ζ-Potential Measurements
2.6. Fourier Transform Infrared Spectroscopy (FT-IR) of SLNs and the GM-SLN Vector
2.7. Cryo-Transmission Electron Microscopy (Cryo-TEM) Images
2.8. Binding, Protection and Release of the pDNA
2.9. In Vitro Studies in Hep G2 Cells
2.9.1. Cell Culture and Transfection Protocol
2.9.2. Cellular Uptake
2.9.3. Intracellular Disposition
2.9.4. GFP Transfection Efficacy and Cell Viability
2.9.5. α-Galactosidase A Transfection
2.10. Interaction with Erythrocytes: Hemolysis and Hemagglutination
2.11. Animal Experimentation
2.11.1. In Vivo Intravenous Administration to α-Gal A KO Mice
2.12. α-Galactosidase A Activity Assay
2.13. Data Analysis and Statistics
3. Results
3.1. Preparation of SLNs
3.2. Physical Stability of SLNs
3.3. Characterization of the GM-SLN Vector: Particle Size, Polydispersity Index and ζ-Potential Measurements
3.4. Fourier Transform Infrared Spectroscopy (FT-IR) of SLNs and the GM-SLN Vector
3.5. Cryo-Transmission Electron Microscopy (Cryo-TEM) Images
3.6. Binding, Protection and Release of the pDNA
3.7. In Vitro Studies in Hep G2 Cells
3.7.1. Cellular Uptake
3.7.2. Intracellular Disposition
3.7.3. GFP Transfection Efficacy and Cell Viability
3.7.4. α-Galactosidase A Transfection
3.8. Interaction with Erythrocytes: Hemolysis and Hemagglutination
3.9. In Vivo Intravenous Administration of the GM-SLN Vector to α-Gal A KO Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Bernardes, T.P.; Foresto, R.D.; Kirsztajn, G.M. Fabry disease: Genetics, pathology, and treatment. Rev. Assoc. Med. Bras. 2020, 66, 10–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azevedo, O.; Gago, M.F.; Miltenberger-Miltenyi, G.; Sousa, N.; Cunha, D. Fabry Disease Therapy: State-of-the-Art and Current Challenges. Int. J. Mol. Sci. 2020, 22, 206. [Google Scholar] [CrossRef] [PubMed]
- Felis, A.; Whitlow, M.; Kraus, A.; Warnock, D.G.; Wallace, E. Current and Investigational Therapeutics for Fabry Disease. Kidney Int. Rep. 2020, 5, 407–413. [Google Scholar] [CrossRef]
- Miller, J.J.; Kanack, A.J.; Dahms, N.M. Progress in the understanding and treatment of Fabry disease. Biochim. Biophys. Acta-Gen. Subj. 2020, 1864, 129437. [Google Scholar] [CrossRef] [PubMed]
- McCafferty, E.H.; Scott, L.J. Migalastat: A Review in Fabry Disease. Drugs 2019, 79, 543–554. [Google Scholar] [CrossRef] [Green Version]
- Kant, S.; Atta, M.G. Therapeutic advances in Fabry disease: The future awaits. Biomed. Pharmacother. 2020, 131, 110779. [Google Scholar] [CrossRef]
- Domm, J.M.; Wootton, S.K.; Medin, J.A.; West, M.L. Gene therapy for Fabry disease: Progress, challenges, and outlooks on gene-editing. Mol. Genet. Metab. 2021, 134, 117–131. [Google Scholar] [CrossRef]
- Xu, S.; Lun, Y.; Brignol, N.; Hamler, R.; Schilling, A.; Frascella, M.; Sullivan, S.; Boyd, R.E.; Chang, K.; Soska, R.; et al. Coformulation of a novel human α-galactosidase a with the pharmacological chaperone AT1001 leads to improved substrate reduction in fabry mice. Mol. Ther. 2015, 23, 1169–1181. [Google Scholar] [CrossRef] [Green Version]
- Kizhner, T.; Azulay, Y.; Hainrichson, M.; Tekoah, Y.; Arvatz, G.; Shulman, A.; Ruderfer, I.; Aviezer, D.; Shaaltiel, Y. Characterization of a chemically modified plant cell culture expressed human α-Galactosidase-A enzyme for treatment of Fabry disease. Mol. Genet. Metab. 2015, 114, 259–267. [Google Scholar] [CrossRef]
- Bénichou, B.; Goyal, S.; Sung, C.; Norfleet, A.M.; O’Brien, F. A retrospective analysis of the potential impact of IgG antibodies to agalsidase β on efficacy during enzyme replacement therapy for Fabry disease. Mol. Genet. Metab. 2009, 96, 4–12. [Google Scholar] [CrossRef]
- Tesmoingt, C.; Lidove, O.; Reberga, A.; Thetis, M.; Ackaert, C.; Nicaise, P.; Arnaud, P.; Papo, T. Enzyme therapy in Fabry disease: Severe adverse events associated with anti-agalsidase cross-reactive IgG antibodies. Br. J. Clin. Pharmacol. 2009, 68, 765–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linthorst, G.E.; Hollak, C.E.M.; Donker-Koopman, W.E.; Strijland, A.; Aerts, J.M.F.G. Enzyme therapy for Fabry disease: Neutralizing antibodies toward agalsidase alpha and beta. Kidney Int. 2004, 66, 1589–1595. [Google Scholar] [CrossRef] [Green Version]
- DeRosa, F.; Smith, L.; Shen, Y.; Huang, Y.; Pan, J.; Xie, H.; Yahalom, B.; Heartlein, M.W. Improved Efficacy in a Fabry Disease Model Using a Systemic mRNA Liver Depot System as Compared to Enzyme Replacement Therapy. Mol. Ther. 2019, 27, 878–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Yin, L.; Theisen, M.; Zhuo, J.; Siddiqui, S.; Levy, B.; Presnyak, V.; Frassetto, A.; Milton, J.; Salerno, T.; et al. Systemic mRNA Therapy for the Treatment of Fabry Disease: Preclinical Studies in Wild-Type Mice, Fabry Mouse Model, and Wild-Type Non-human Primates. Am. J. Hum. Genet. 2019, 104, 625–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, G.; Maruyama, H.; Ishii, S.; Shimotori, M.; Kameda, S.; Kono, T.; Miyazaki, J.I.; Kulkarni, A.B.; Gejyo, F. Naked plasmid DNA-based α-galactosidase a gene transfer partially reduces systemic accumulation of globotriaosylceramide in Fabry mice. Mol. Biotechnol. 2008, 38, 109–119. [Google Scholar] [CrossRef]
- Sands, M.S.; Davidson, B.L. Gene therapy for lysosomal storage diseases. Mol. Ther. 2006, 13, 839–849. [Google Scholar] [CrossRef]
- Huston, M.W.; Yasuda, M.; Pagant, S.; Martin, S.S.; Cao, L.; Falese, L.; Meyer, K.; Desnick, R.J.; Wechsler, T. Liver-targeted AAV gene therapy vectors produced by a clinical scale manufacturing process result in high, continuous therapeutic levels of enzyme activity and effective substrate reduction in mouse model of Fabry disease. Mol. Genet. Metab. 2019, 126, S77. [Google Scholar] [CrossRef]
- Jeyakumar, J.; Kia, A.; McIntosh, J.; Verhoef, D.; Kalcheva, P.; Hosseini, P.; Sheridan, R.; Corbau, R.; Nathwani, A. Liver-directed gene therapy corrects Fabry disease in mice. Mol. Genet. Metab. 2019, 126, S80. [Google Scholar] [CrossRef]
- Yasuda, M.; Huston, M.W.; Pagant, S.; Gan, L.; St. Martin, S.; Sproul, S.; Richards, D.; Ballaron, S.; Hettini, K.; Ledeboer, A.; et al. AAV2/6 Gene Therapy in a Murine Model of Fabry Disease Results in Supraphysiological Enzyme Activity and Effective Substrate Reduction. Mol. Ther.-Methods Clin. Dev. 2020, 18, 607–619. [Google Scholar] [CrossRef]
- Jung, S.C.; Han, I.P.; Limaye, A.; Xu, R.; Gelderman, M.P.; Zerfas, P.; Tirumalai, K.; Murray, G.J.; During, M.J.; Brady, R.O.; et al. Adeno-associated viral vector-mediated gene transfer results in long-term enzymatic and functional correction in multiple organs of Fabry mice. Proc. Natl. Acad. Sci. USA 2001, 98, 2676–2681. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, K.; Hirai, Y.; Ishizaki, M.; Takahashi, H.; Hanawa, H.; Fukunaga, Y.; Shimada, T. Long-term inhibition of glycosphingolipid accumulation in Fabry model mice by a single systemic injection of AAV1 vector in the neonatal period. Mol. Genet. Metab. 2009, 96, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Amalfitano, A.; Rastall, D. Recent advances in gene therapy for lysosomal storage disorders. Appl. Clin. Genet. 2015, 8, 157. [Google Scholar] [CrossRef] [Green Version]
- Malaviya, M.; Shiroya, M. Systemic gene delivery using lipid envelope systems and its potential in overcoming challenges. Int. J. Pharm. Drug Anal. 2021, 9, 46–55. [Google Scholar] [CrossRef]
- Thi, T.T.H.; Suys, E.J.A.; Lee, J.S.; Nguyen, D.H.; Park, K.D.; Truong, N.P. Lipid-Based Nanoparticles in the Clinic and Clinical Trials: From Cancer Nanomedicine to COVID-19 Vaccines. Vaccines 2021, 9, 359. [Google Scholar] [CrossRef] [PubMed]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic: An update post COVID-19 vaccines. Bioeng. Transl. Med. 2021, 6, e10246. [Google Scholar] [CrossRef]
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 2021, 6, 1078–1094. [Google Scholar] [CrossRef]
- Musielak, E.; Feliczak-Guzik, A.; Nowak, I. Synthesis and Potential Applications of Lipid Nanoparticles in Medicine. Materials 2022, 15, 682. [Google Scholar] [CrossRef]
- Gaspar, D.P.; Almeida, A.J. Surface-Functionalized Lipid Nanoparticles for Site-Specific Drug Delivery. In Surface Modification of Nanoparticles for Targeted Drug Delivery; Pathak, Y.V., Ed.; Springer International Publishing: Cham, Switerland, 2019; pp. 73–98. ISBN 978-3-030-06115-9. [Google Scholar]
- Delgado, D.; Gascón, A.R.; Del Pozo-Rodríguez, A.; Echevarría, E.; Ruiz De Garibay, A.P.; Rodríguez, J.M.; Solinís, M.Á. Dextran-protamine-solid lipid nanoparticles as a non-viral vector for gene therapy: In vitro characterization and in vivo transfection after intravenous administration to mice. Int. J. Pharm. 2012, 425, 35–43. [Google Scholar] [CrossRef]
- Perez Ruiz de Garibay, A.; Delgado, D.; del Pozo, A.; Solinís, M.A.; Rodriguez Gascón, A. Multicomponent nanoparticles as nonviral vectors for the treatment of Fabry disease by gene therapy. Drug Des. Devel. Ther. 2012, 6, 303. [Google Scholar] [CrossRef] [Green Version]
- Ruiz De Garibay, A.P.; Solinís, M.A.; Del Pozo-Rodríguez, A.; Apaolaza, P.S.; Shen, J.S.; Rodríguez-Gascón, A. Solid lipid nanoparticles as non-viral vectors for gene transfection in a cell model of fabry disease. J. Biomed. Nanotechnol. 2015, 11, 500–511. [Google Scholar] [CrossRef]
- Rodríguez-Castejón, J.; Alarcia-Lacalle, A.; Gómez-Aguado, I.; Vicente-Pascual, M.; Solinís Aspiazu, M.Á.; del Pozo-Rodríguez, A.; Rodríguez-Gascón, A. α-Galactosidase A Augmentation by Non-Viral Gene Therapy: Evaluation in Fabry Disease Mice. Pharmaceutics 2021, 13, 771. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, F.; Gordts, S.; Muthuramu, I.; De Geest, B. The Liver as a Target Organ for Gene Therapy: State of the Art, Challenges, and Future Perspectives. Pharmaceuticals 2012, 5, 1372–1392. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Huang, G.; Huang, H. Sugar ligand-mediated drug delivery. Future Med. Chem. 2020, 12, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, T.; Murray, G.J.; Swaim, W.D.; Longenecker, G.; Quirk, J.M.; Cardarelli, C.O.; Sugimoto, Y.; Pastan, I.; Gottesman, M.M.; Brady, R.O.; et al. α-Galactosidase A deficient mice: A model of fabry disease. Proc. Natl. Acad. Sci. USA 1997, 94, 2540–2544. [Google Scholar] [CrossRef] [Green Version]
- Vicente-Pascual, M.; Albano, A.; Solinís, M.; Serpe, L.; Rodríguez-Gascón, A.; Foglietta, F.; Muntoni, E.; Torrecilla, J.; Del Pozo-Rodríguez, A.; Battaglia, L. Gene delivery in the cornea: In vitro & ex vivo evaluation of solid lipid nanoparticle-based vectors. Nanomedicine 2018, 13, 1847–1864. [Google Scholar] [CrossRef]
- Sloot, Y.J.E.; Rabold, K.; Ulas, T.; De Graaf, D.M.; Heinhuis, B.; Händler, K.; Schultze, J.L.; Netea, M.G.; Smit, J.W.A.; Joosten, L.A.B.; et al. Interplay between thyroid cancer cells and macrophages: Effects on IL-32 mediated cell death and thyroid cancer cell migration. Cell. Oncol. 2019, 42, 691–703. [Google Scholar] [CrossRef] [Green Version]
- Tsai, Y.C.; Tsai, T.H.; Chang, C.P.; Chen, S.F.; Lee, Y.M.; Shyue, S.K. Linear correlation between average fluorescence intensity of green fluorescent protein and the multiplicity of infection of recombinant adenovirus. J. Biomed. Sci. 2015, 22, 31. [Google Scholar] [CrossRef] [Green Version]
- Sandhu, K.S.; Al-Rubeai, M. Monitoring of the adenovirus production process by flow cytometry. Biotechnol. Prog. 2008, 24, 250–261. [Google Scholar] [CrossRef]
- Kurosaki, T.; Kitahara, T.; Fumoto, S.; Nishida, K.; Yamamoto, K.; Nakagawa, H.; Kodama, Y.; Higuchi, N.; Nakamura, T.; Sasaki, H. Chondroitin sulfate capsule system for efficient and secure gene delivery. J. Pharm. Pharm. Sci. 2010, 13, 351–361. [Google Scholar] [CrossRef]
- Misra, S.; Chopra, K.; Sinha, V.R.; Medhi, B. Galantamine-loaded solid–lipid nanoparticles for enhanced brain delivery: Preparation, characterization, in vitro and in vivo evaluations. Drug Deliv. 2016, 23, 1434–1443. [Google Scholar] [CrossRef] [Green Version]
- Ruseska, I.; Fresacher, K.; Petschacher, C.; Zimmer, A. Use of Protamine in Nanopharmaceuticals—A Review. Nanomaterials 2021, 11, 1508. [Google Scholar] [CrossRef] [PubMed]
- Delgado, D.; Del Pozo-Rodríguez, A.; Solinís, M.Á.; Rodríguez-Gascón, A. Understanding the mechanism of protamine in solid lipid nanoparticle-based lipofection: The importance of the entry pathway. Eur. J. Pharm. Biopharm. 2011, 79, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, V.D.; Jani, G.K.; Moradiya, N.G.; Randeria, N.P.; Nagar, B.J.; Naikwadi, N.N.; Variya, B.C. Galactomannan: A versatile biodegradable seed polysaccharide. Int. J. Biol. Macromol. 2013, 60, 83–92. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, A.A.; Devarajan, P.V. Asialoglycoprotein receptor mediated hepatocyte targeting—Strategies and applications. J. Control. Release 2015, 203, 126–139. [Google Scholar] [CrossRef]
- del Pozo-Rodríguez, A.; Solinís, M.Á.; Rodríguez-Gascón, A. Applications of lipid nanoparticles in gene therapy. Eur. J. Pharm. Biopharm. 2016, 109, 184–193. [Google Scholar] [CrossRef]
- Duan, Y.; Dhar, A.; Patel, C.; Khimani, M.; Neogi, S.; Sharma, P.; Siva Kumar, N.; Vekariya, R.L. A brief review on solid lipid nanoparticles: Part and parcel of contemporary drug delivery systems. RSC Adv. 2020, 10, 26777–26791. [Google Scholar] [CrossRef]
- Somsak, P.; Sriwattana, S.; Prinyawiwatkul, W. Ultrasonic-assisted chitin nanoparticle and its application as saltiness enhancer. Int. J. Food Sci. Technol. 2021, 56, 608–617. [Google Scholar] [CrossRef]
- Rajivgandhi, G.N.; Ramachandran, G.; Alharbi, N.S.; Kadaikunnan, S.; Khaleed, J.M.; Manokaran, N.; Li, W.J. Substantial effect of Cr doping on the antimicrobial activity of ZnO nanoparticles prepared by ultrasonication process. Mater. Sci. Eng. B Solid-State Mater. Adv. Technol. 2021, 263, 114817. [Google Scholar] [CrossRef]
- Ramalingam, M.; Kokulnathan, T.; Tsai, P.C.; Valan Arasu, M.; Al-Dhabi, N.A.; Prakasham, K.; Ponnusamy, V.K. Ultrasonication-assisted synthesis of gold nanoparticles decorated ultrathin graphitic carbon nitride nanosheets as a highly efficient electrocatalyst for sensitive analysis of caffeic acid in food samples. Appl. Nanosci. 2021, 1, 3. [Google Scholar] [CrossRef]
- Fröhlich, E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int. J. Nanomed. 2012, 7, 5577–5591. [Google Scholar] [CrossRef] [Green Version]
- Miller, C.R.; Bondurant, B.; McLean, S.D.; McGovern, K.A.; O’Brien, D.F. Liposome-cell interactions in vitro: Effect of liposome surface charge on the binding and endocytosis of conventional and sterically stabilized liposomes. Biochemistry 1998, 37, 12875–12883. [Google Scholar] [CrossRef] [PubMed]
- Beloqui, A.; Solinís, M.A.; Delgado, A.; Évora, C.; Del Pozo-Rodríguez, A.; Rodríguez-Gascón, A. Biodistribution of Nanostructured Lipid Carriers (NLCs) after intravenous administration to rats: Influence of technological factors. Eur. J. Pharm. Biopharm. 2013, 84, 309–314. [Google Scholar] [CrossRef] [PubMed]
- del Pozo-Rodríguez, A.; Delgado, D.; Solinís, M.A.; Gascón, A.R.; Pedraz, J.L. Solid lipid nanoparticles for retinal gene therapy: Transfection and intracellular trafficking in RPE cells. Int. J. Pharm. 2008, 360, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Saffari, M.; Moghimi, H.R.; Dass, C.R. Barriers to liposomal gene delivery: From application site to the target. Iran. J. Pharm. Res. 2016, 15, 3–17. [Google Scholar] [PubMed]
- Wang, T.; Larcher, L.; Ma, L.; Veedu, R. Systematic Screening of Commonly Used Commercial Transfection Reagents towards Efficient Transfection of Single-Stranded Oligonucleotides. Molecules 2018, 23, 2564. [Google Scholar] [CrossRef] [Green Version]
- Zheng, M.; Pan, M.; Zhang, W.; Lin, H.; Wu, S.; Lu, C.; Tang, S.; Liu, D.; Cai, J. Poly(α-l-lysine)-based nanomaterials for versatile biomedical applications: Current advances and perspectives. Bioact. Mater. 2021, 6, 1878–1909. [Google Scholar] [CrossRef]
- van den Berg, A.I.S.; Yun, C.O.; Schiffelers, R.M.; Hennink, W.E. Polymeric delivery systems for nucleic acid therapeutics: Approaching the clinic. J. Control. Release 2021, 331, 121–141. [Google Scholar] [CrossRef]
- Takakura, Y.; Nishikawa, M.; Yamashita, F.; Hashida, M. Influence of Physicochemical Properties on Pharmacokinetics of Non-viral Vectors for Gene Delivery. J. Drug Target. 2002, 10, 99–104. [Google Scholar] [CrossRef]
- Eliyahu, N.; Servel, N.; Domb, A.J.; Barenholz, Y. Lipoplex-induced hemagglutination: Potential involvement in intravenous gene delivery. Gene Ther. 2002, 9, 850–858. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Huang, L. Recent advances in nonviral vectors for gene delivery. Acc. Chem. Res. 2012, 45, 971–979. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef] [PubMed]
- Alroy, J.; Sabnis, S.; Kopp, J.B. Renal Pathology in Fabry Disease. J. Am. Soc. Nephrol. 2002, 13, S134–S138. [Google Scholar] [CrossRef] [PubMed]
- Branton, M.H.; Schiffmann, R.; Sabnis, S.G.; Murray, G.J.; Quirk, J.M.; Altarescu, G.; Goldfarb, L.; Brady, R.O.; Balow, J.E.; Austin, H.A.; et al. Natural history of fabry renal disease: Influence of α-galactosidase a activity and genetic mutations on clinical course. Medicine 2002, 81, 122–138. [Google Scholar] [CrossRef]
- Veen, S.J.; Hollak, C.E.M.; Kuilenburg, A.B.P.; Langeveld, M. Developments in the treatment of Fabry disease. J. Inherit. Metab. Dis. 2020, 43, 908–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massaro, G.; Geard, A.F.; Liu, W.; Coombe-Tennant, O.; Waddington, S.N.; Baruteau, J.; Gissen, P.; Rahim, A.A. Gene Therapy for Lysosomal Storage Disorders: Ongoing Studies and Clinical Development. Biomolecules 2021, 11, 611. [Google Scholar] [CrossRef]
- Takahashi, H.; Hirai, Y.; Migita, M.; Seino, Y.; Fukuda, Y.; Sakuraba, H.; Kase, R.; Kobayashi, T.; Hashimoto, Y.; Shimada, T. Long-term systemic therapy of Fabry disease in a knockout mouse by adeno-associated virus-mediated muscle-directed gene transfer. Proc. Natl. Acad. Sci. USA 2002, 99, 13777–13782. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Z-Average (d.nm) | PDI | ζ-Potential (mV) | |
|---|---|---|---|
| pcDNA3-EGFP | |||
| GM-SLN | 98.3 ± 0.9 | 0.20 ± 0.02 | +35.2 ± 1.4 |
| pR-M10-αGal A | |||
| GM-SLN | 97.3 ± 2.8 | 0.17 ± 0.02 | +33.6 ± 1.6 |
| % of Wild-Type | Plasma | Liver | Spleen | Heart | Kidney |
| 18 | 6 | 7 | 28 | 14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Castejón, J.; Gómez-Aguado, I.; Beraza-Millor, M.; Solinís, M.Á.; del Pozo-Rodríguez, A.; Rodríguez-Gascón, A. Galactomannan-Decorated Lipidic Nanocarrier for Gene Supplementation Therapy in Fabry Disease. Nanomaterials 2022, 12, 2339. https://doi.org/10.3390/nano12142339
Rodríguez-Castejón J, Gómez-Aguado I, Beraza-Millor M, Solinís MÁ, del Pozo-Rodríguez A, Rodríguez-Gascón A. Galactomannan-Decorated Lipidic Nanocarrier for Gene Supplementation Therapy in Fabry Disease. Nanomaterials. 2022; 12(14):2339. https://doi.org/10.3390/nano12142339
Chicago/Turabian StyleRodríguez-Castejón, Julen, Itziar Gómez-Aguado, Marina Beraza-Millor, María Ángeles Solinís, Ana del Pozo-Rodríguez, and Alicia Rodríguez-Gascón. 2022. "Galactomannan-Decorated Lipidic Nanocarrier for Gene Supplementation Therapy in Fabry Disease" Nanomaterials 12, no. 14: 2339. https://doi.org/10.3390/nano12142339