
Comparative Analysis of LiMPO4 (M = Fe, Co, Cr, Mn, V) as Cathode Materials for Lithium-Ion Battery Applications—A First-Principle-Based Theoretical Approach

Abstract
:1. Introduction
2. Materials and Computational Methodology
2.1. Battery Mechanism
2.2. Material Specifications
2.3. Simulation Framework
2.4. Parameter Definitions
3. Results and Discussion
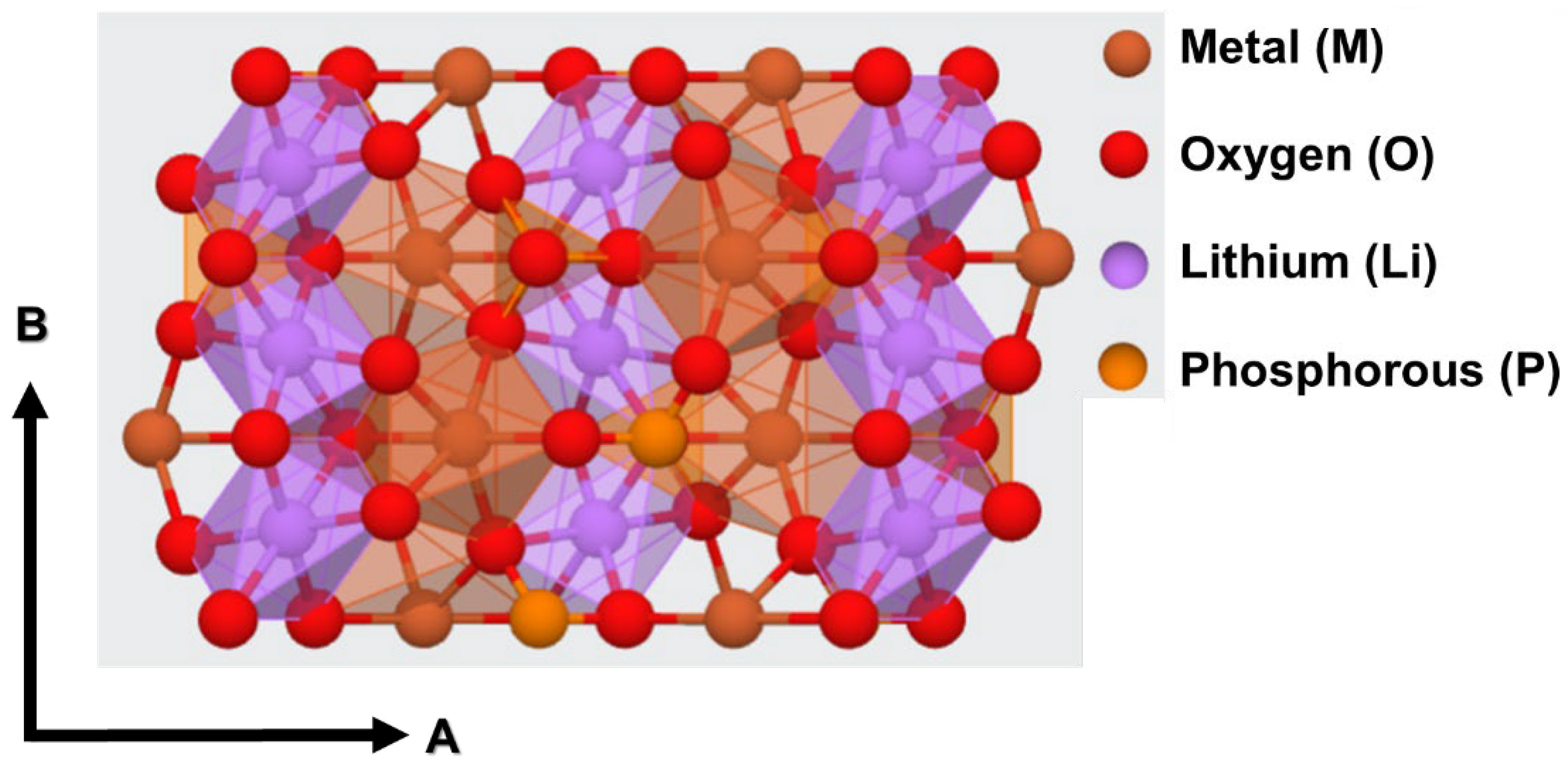
3.1. Structural Properties of LiMPO4 (M = Fe, Co, Cr, Mn, and V)
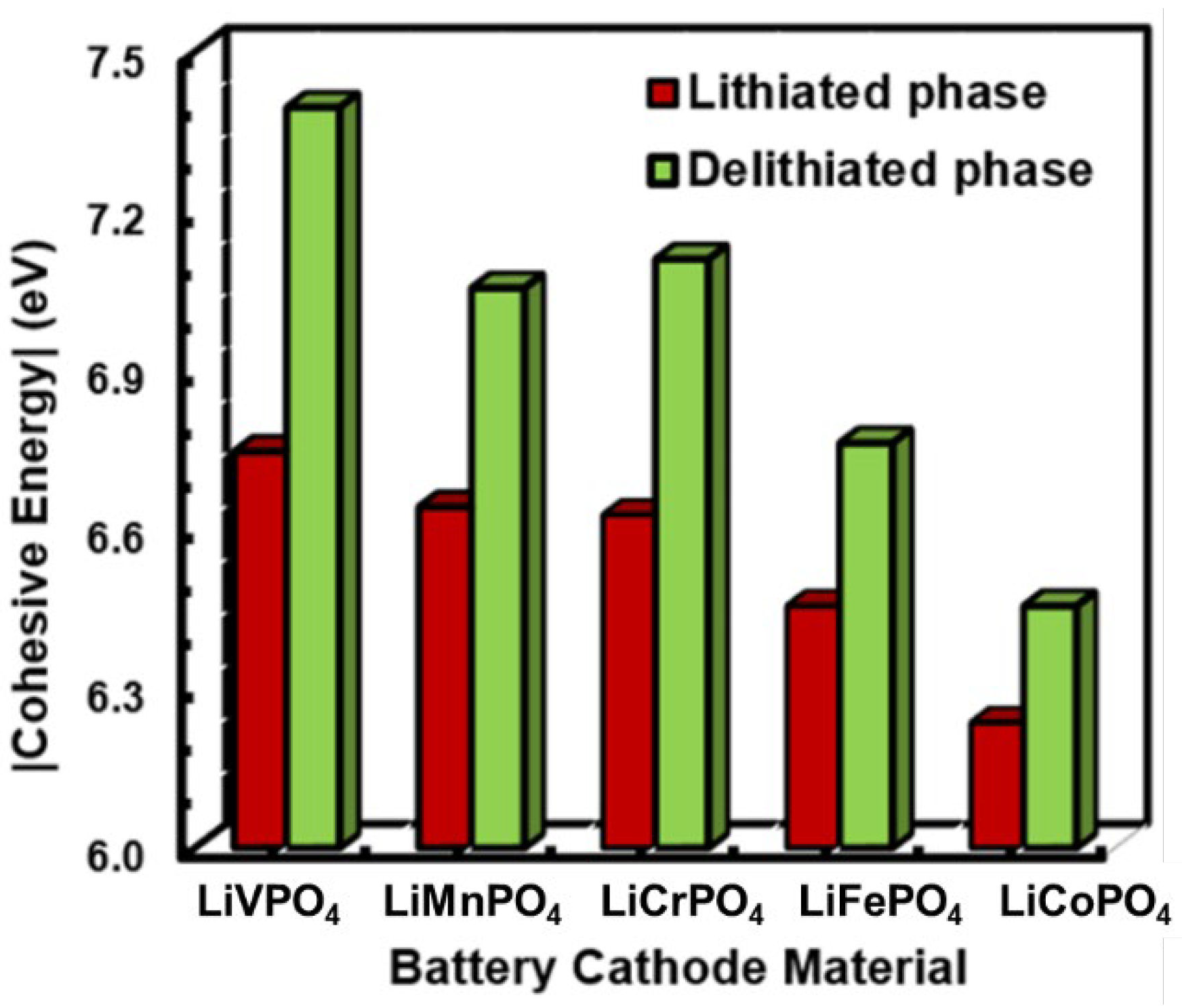
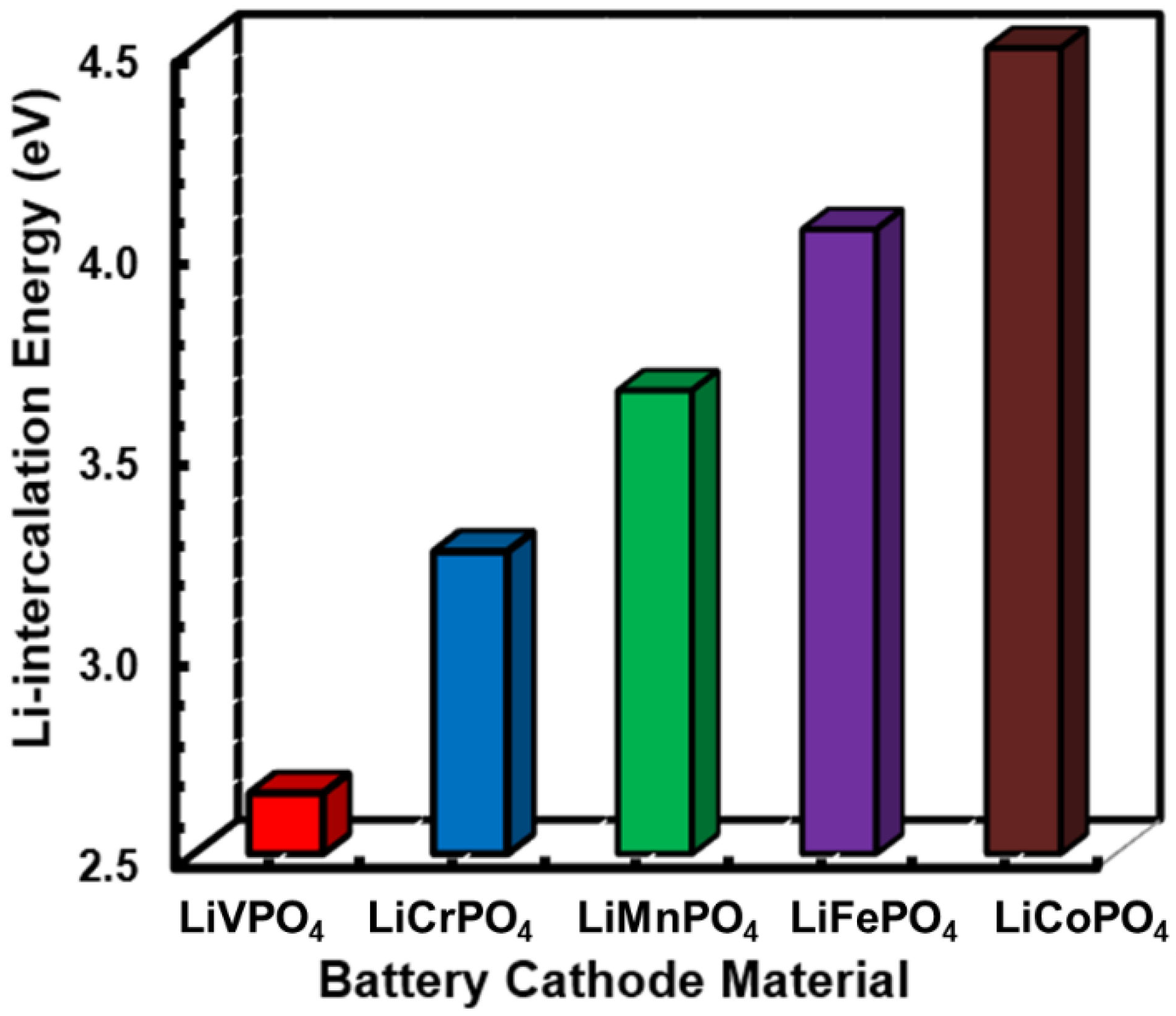
3.2. Electrochemical Properties of LiMPO4 (M = Fe, Co, Cr, Mn, and V)
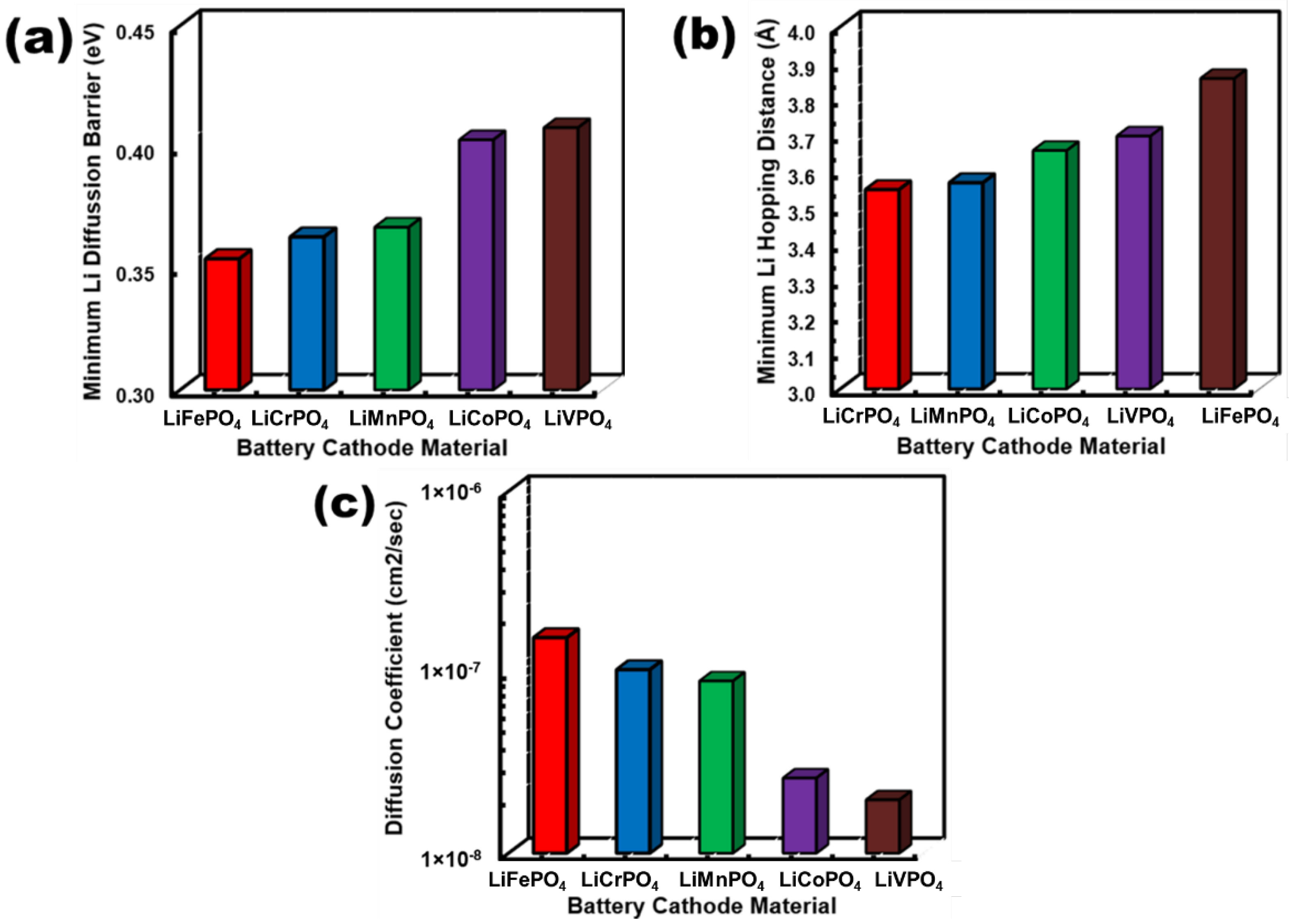
3.3. Electrical Characteristics of LiMPO4 (M = Fe, Co, Cr, Mn, and V)
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Dunn, B.; Kamath, H.; Tarascon, J.M. Electrical Energy Storage for the Grid: A Battery of Choices. Science 2011, 334, 928–935. [Google Scholar] [CrossRef] [PubMed]
- Nitta, N.; Wu, F.; Lee, J.T.; Yushin, G. Li-ion battery materials: Present and future. Mater. Today 2015, 18, 252–264. [Google Scholar] [CrossRef]
- Kim, T.; Song, W.; Son, D.Y.; Ono, L.K.; Qi, Y. Lithium-ion batteries: Outlook on present, future, and hybridized technologies. J. Mater. Chem. A 2019, 7, 2942–2964. [Google Scholar] [CrossRef]
- Islam, M.S.; Fisher, C.A. Lithium and sodium battery cathode materials: Computational insights into voltage, diffusion and nanostructural properties. Chem. Soc. Rev. 2014, 43, 185–204. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Yamada, S.; Jalem, R.; Kasuga, T. Density functional studies of olivine-type LiFePO4 and NaFePO4 as positive electrode materials for rechargeable lithium and sodium ion batteries. Solid State Ion. 2016, 286, 40–44. [Google Scholar] [CrossRef]
- Chen, Y.; Sang, M.; Jiang, W.; Wang, Y.; Zou, Y.; Lu, C.; Ma, Z. Fracture predictions based on a coupled chemo-mechanical model with strain gradient plasticity theory for film electrodes of Li-ion batteries. Eng. Fract. Mech. 2021, 253, 107866. [Google Scholar] [CrossRef]
- Ma, Z.S.; Xie, Z.C.; Wang, Y.; Zhang, P.P.; Pan, Y.; Zhou, Y.C.; Lu, C. Failure modes of hollow core-shell structural active materials during the lithiation-delithiation process. J. Power Sources 2015, 290, 114–122. [Google Scholar] [CrossRef]
- Chen, C.F.; Barai, P.; Mukherjee, P.P. An overview of degradation phenomena modelling in lithium-ion battery electrodes. Curr. Opin. Chem. Eng. 2016, 13, 82–90. [Google Scholar] [CrossRef]
- Ma, Z.; Wu, H.; Wang, Y.; Pan, Y.; Lu, C. An electrochemical-irradiated plasticity model for metallic electrodes in lithium-ion batteries. Int. J. Plast. 2017, 88, 188–203. [Google Scholar] [CrossRef]
- Tian, Y.; Zeng, G.; Rutt, A.; Shi, T.; Kim, H.; Wang, J.; Koettgen, J.; Sun, Y.; Ouyang, B.; Chen, T.; et al. Promises and challenges of next-generation “beyond Li-ion” batteries for electric vehicles and grid decarbonization. Chem. Rev. 2020, 121, 1623–1669. [Google Scholar] [CrossRef]
- Urban, A.; Seo, D.H.; Ceder, G. Computational understanding of Li-ion batteries. NPJ Comput. Mater. 2016, 2, 16002. [Google Scholar] [CrossRef]
- Padhi, A.; Nanjundaswamy, K.S.; Goodenough, J.B. Phospho-Olivines as Positive-Electrode Materials for Rechargeable Lithium Batteries. J. Electrochem. Soc. 1997, 144, 1188–1194. [Google Scholar] [CrossRef]
- Morgan, D.; Ven, A.; Ceder, G. Li conductivity in LixMPO4 (M = Mn, Fe, Co, Ni) olivine materials. Electrochem. Solid-State Lett. 2004, 7, 4032–4037. [Google Scholar] [CrossRef]
- Islam, M.S.; Driscoll, D.J.; Fisher, C.A.; Slater, P.R. Atomic-scale investigation of defects, dopants, and lithium transport in the LiFePO4 olivine-type battery material. Chem. Mater. 2005, 17, 5085–5092. [Google Scholar] [CrossRef]
- Ficher, C.A.J.; Prieto, V.M.H.; Islam, M.S. Lithium battery materials LiMPO4 (M= Mn, Fe, Co and Ni): Insights into defect association, transport mechanisms and doping behaviour. Chem. Mater. 2008, 20, 5907–5915. [Google Scholar] [CrossRef]
- Yang, J.; Tse, J.S. Li ion diffusion mechanisms in LiFePO4: An ab initio molecular dynamics study. J. Phys. Chem. A 2011, 115, 13045–13049. [Google Scholar] [CrossRef]
- Dathar, G.K.P.; Sheppard, D.; Stevenson, K.J.; Henkelman, G. Calculations of Li-ion diffusion in olivine phosphates. Chem. Mater. 2011, 23, 4032–4037. [Google Scholar] [CrossRef]
- Wu, K.C.; Hsieh, C.H.; Chang, B.K. First principles calculations of lithium diffusion near the surface and in the bulk of Fe-doped LiCoPO4. Phys. Chem. Chem. Phys. 2022, 24, 1147–1155. [Google Scholar] [CrossRef]
- Oukakhou, S.; Maymoun, M.; Elomrani, A.; Sbiaai, K.; Hasnaoui, A. Enhancing the Electrochemical Performance of Olivine LiMnPO4 as Cathode Materials for Li-ion Batteries by Ni-Fe codoping. ACS Appl. Energy Mater. 2022. [Google Scholar] [CrossRef]
- Ping, L.Z.; Ming, Z.Y.; Jun, Z.Y. First-principles studies of Mn-doped LiCoPO4. Chin. Phys. B 2011, 20, 018201. [Google Scholar] [CrossRef]
- Zhang, H.; Gong, Y.; Li, J.; Du, K.; Cao, Y.; Li, J. Selecting substituent element for LiMnPO4 cathode materials combined with density functional theory (DFT) calculations and experiments. J. Alloys Compd. 2019, 793, 360–368. [Google Scholar] [CrossRef]
- Yang, L.; Deng, W.; Xu, W.; Tian, Y.; Wang, A.; Wang, B.; Zou, G.; Hou, H.; Deng, W.; Ji, X. Olivine LiMnxFe1−xPo4 cathode materials for lithium ion batteries: Restricted factors of rate performances. J. Mater. Chem. A 2021, 9, 14214–14232. [Google Scholar] [CrossRef]
- Kuganathan, N.; Chroneos, A. Formation, doping, and lithium incorporation in LiFePO4. AIP Adv. 2022, 12, 045225. [Google Scholar] [CrossRef]
- Alfaruqi, M.H.; Kim, S.; Park, S.; Lee, S.; Lee, J.; Hwang, J.Y.; Sun, Y.K.; Kim, J. Density functional theory investigation of mixed transition metals in olivine and tavorite cathode materials for Li-ion batteries. ACS Appl. Mater. Interfaces 2020, 12, 16376–16386. [Google Scholar] [CrossRef]
- Molenda, J.; Kulka, A.; Milewska, A.; Zając, W.; Świerczek, K. Structural, transport and electrochemical properties of LiFePO4 substituted in lithium and iron sublattices (Al, Zr, W, Mn, Co and Ni). Materials 2013, 6, 1656–1687. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, C.; Shi, S.; Wang, Z.; Huang, X.; Chen, L. First-principles study of Li ion diffusion in LiFePO4. Phys. Rev. B 2004, 69, 104303. [Google Scholar] [CrossRef]
- “Quantum ATK”. Available online: https://www.synopsys.com/silicon/quantumatk.html (accessed on 1 January 2022).
- Berdiyorov, G.R.; Madjet, M.E.; Mohmoud, K.A. First-Principles Density Functional Theory Calculations of Bilayer Membranes Heterostructures of Ti3C2T2 (MXene)/Graphene and AgNPs. Membranes 2021, 11, 543. [Google Scholar] [CrossRef] [PubMed]
- Stanley, J.C.; Mayr, F.; Gagliardi, A. Machine Learning Stability and Bandgaps of Lead-Free Perovskites for Photovoltaics. Adv. Theory Simul. 2020, 3, 1900178. [Google Scholar] [CrossRef]
- Palepu, J.; Anand, P.P.; Parshi, P.; Jain, V.; Tiwari, A.; Bhattacharya, S.; Chakraborthy, S.; Kanungo, S. Compartitive analysis of strain engineering on the electronic properties of homogenous and heterostructure bilayers od MoX2 (X = S, Se, Te). Micro Nanostruct. 2022, 168, 207334. [Google Scholar] [CrossRef]
- Tiwari, A.; Palepu, J.; Choudhury, A.; Bhattacharya, S.; Kanungo, S. Theoretical analysis of the NH3, NO, and NO2 adsorption on boron-nitrogen and boron-phosphorous co-doped monolayer graphene-A comparative study. FlatChem 2022, 34, 100392. [Google Scholar] [CrossRef]
- Yang, C.C.; Li, S. Cohesive energy: The intrinsic dominant of thermal stability and structural evolution in Sn from size scales of bulk to dimer. J. Phys. Chem. C 2009, 113, 14207–14212. [Google Scholar] [CrossRef]
- Klerk, N.J.J.de.; Maas, E.V.D.; Wagemaker, M. Analysis of Diffusion in Solid-State Electrolytes through MD Simulations, improvement of the Li-Ion Conductivity in β-Li3PS4 as an Example. ACS Appl. Energy Mater. 2018, 1, 3230–3242. [Google Scholar] [CrossRef] [PubMed]
- Koettgen, J.; Zacherle, T.; Grieshammer, S.; Martin, M. Ab initio calculation of the attempt frequency of oxygen diffusion in pure and samarium doped ceria. Phys. Chem. Chem. Phys. 2017, 19, 9957–9973. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, G.R.; Islam, M.S. Anti-site defects and ion migration in the LiFe0.5Mn0.5PO4 mixed-metal cathode material. Chem. Mater. 2010, 22, 1242–1248. [Google Scholar] [CrossRef]
- Clementi, E.; Raimondi, D.L.; Reinhardt, W.P. Atomic Screening Constants from SCF Functions. II. Atoms with 37 To 86 Electrons. J. Chem. Phys. 2004, 47, 1300. [Google Scholar] [CrossRef]
- Fan, C.L.; Lin, C.R.; Han, S.C.; Chen, J.; Li, L.F.; Bai, Y.M.; Zhang, K.H.; Zhange, X. Structure, conductive mechanisn and electrochemical of LiFePO4/C doped with Mg2+, Cr3+ and Ti4+ by a carbothermal reduction method. New J. Chem. 2014, 38, 795–801. [Google Scholar] [CrossRef]
- Strobridge, F.C.; Clement, R.J.; Leskes, M.; Middlemiss, D.S.; Borekwicz, O.J.; Wiaderek, K.M.; Chapman, K.W. Identifying the structure of the Intermediate, Li2/3CoPO4, formed during Electrochemical cycling of LiCoPO4. Chem. Mater. 2014, 26, 6193–6205. [Google Scholar] [CrossRef]
- Sgroi, M.F.; Lazzaroni, R.; Beljonne, D.; Pullini, D. Doping LiMnPO4 with Cobalt and Nickel: A First Principle study. Batteries 2017, 3, 11. [Google Scholar] [CrossRef]
- Satyavani, T.V.S.L.; Kiran, B.R.; Kumar, V.R.; Kumar, A.S.; Naidu, S.V. Effect on particle size on dc conductivity, activation energy and diffusion coefficient of lithium iron phosphate in Li-ion cells. Eng. Sci. Technol. Int. J. 2016, 19, 40–44. [Google Scholar] [CrossRef]
- Chen, S.; Zhao, Z.; Gu, X. The Research on Characteristics of Li-NiMnCo Lithium-Ion Batteries in Electric Vehicles. J. Energy 2020, 2020, 3721047. [Google Scholar] [CrossRef]
- Zhang, R.; Xia, B.; Li, B.; Cao, L.; Lai, Y.; Zheng, W.; Wang, H.; Wang, W.; Wang, M. A Study on the Open Circuit Voltage and State of the Charge Characterization of High Capacity Lithium-Ion Battery Under Different Temperatures. Energies 2018, 11, 2408. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Materials | Lattice Constant, a (Å) | Lattice Constant, b (Å) | Lattice Constant, c (Å) |
|---|---|---|---|
| LiFePO4 | 9.851 | 5.773 | 4.672 |
| LiCoPO4 | 9.885 | 5.882 | 4.702 |
| LiMnPO4 | 9.953 | 5.830 | 4.689 |
| LiCrPO4 | 10.041 | 5.882 | 4.707 |
| LiVPO4 | 10.276 | 5.981 | 4.709 |
| Materials | Li-O Bond Length Range (Å) | Mulliken Charge on Li (e−) |
|---|---|---|
| LiFePO4 | 2.07–2.13 | 0.009 |
| LiCoPO4 | 2.09–2.13 | 0.009 |
| LiMnPO4 | 2.07–2.17 | 0.052 |
| LiCrPO4 | 2.07–2.20 | 0.054 |
| LiVPO4 | 2.09–2.21 | 0.083 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanungo, S.; Bhattacharjee, A.; Bahadursha, N.; Ghosh, A. Comparative Analysis of LiMPO4 (M = Fe, Co, Cr, Mn, V) as Cathode Materials for Lithium-Ion Battery Applications—A First-Principle-Based Theoretical Approach. Nanomaterials 2022, 12, 3266. https://doi.org/10.3390/nano12193266
Kanungo S, Bhattacharjee A, Bahadursha N, Ghosh A. Comparative Analysis of LiMPO4 (M = Fe, Co, Cr, Mn, V) as Cathode Materials for Lithium-Ion Battery Applications—A First-Principle-Based Theoretical Approach. Nanomaterials. 2022; 12(19):3266. https://doi.org/10.3390/nano12193266
Chicago/Turabian StyleKanungo, Sayan, Ankur Bhattacharjee, Naresh Bahadursha, and Aritra Ghosh. 2022. "Comparative Analysis of LiMPO4 (M = Fe, Co, Cr, Mn, V) as Cathode Materials for Lithium-Ion Battery Applications—A First-Principle-Based Theoretical Approach" Nanomaterials 12, no. 19: 3266. https://doi.org/10.3390/nano12193266