
First-Principles Insights on the Formation Mechanism of Innermost Layers of Solid Electrolyte Interphases on Carbon Anodes for Lithium-Ion Batteries


1
Physics Department, King Fahd University of Petroleum & Minerals, Dhahran 31261, Saudi Arabia
2
K.A. CARE Energy Research & Innovation Center at Dhahran, Dhahran 31261, Saudi Arabia
3
Interdisciplinary Research Center for Hydrogen and Energy Storage, King Fahd University of Petroleum and Minerals, Dhahran 31261, Saudi Arabia
Nanomaterials 2022, 12(20), 3654; https://doi.org/10.3390/nano12203654
Submission received: 20 August 2022
/
Revised: 16 October 2022
/
Accepted: 17 October 2022
/
Published: 18 October 2022
(This article belongs to the Special Issue Advanced Functional Nanomaterials for Efficient Energy Conversion and Storage)
Abstract
:A solid electrolyte interphase (SEI) plays an essential role in the functionality and service life of ion batteries, where the structure and formation mechanism are still in the midst. Here, we investigate the initial decomposition and reactions of ethylene carbonate (EC) on the surface of a graphite anode using first-principles calculations. EC initially decomposes via the homolytic ring opening with the product of radical anion CH2CH2OCO2−. Bonding with Li, it forms a co-plane structure of CH2CH2OCO2Li, with a binding energy of 1.35 eV. The adsorption energy is −0.91 eV and −0.24 eV on the graphite zigzag edge surface and basal surface, respectively. Two CH2CH2OCO2Li molecules react to form a two-head structure of lithium ethylene dicarbonate (CH2OCO2Li)2, namely LEDC, which further forms a network preferring zigzag edge surfaces. Our results suggest that the first and innermost layers of the solid electrolyte interphase are CH2CH2OCO2Li sticking and networking on the zigzag edges of the surfaces of graphite anodes.
1. Introduction
Lithium-ion batteries have broad applications in portable electronic devices today due to their merits, such as a small volume, high operating voltage, high energy density, low self-discharge rates, long service life, and no memory effect [1,2,3,4,5,6]. The importance of lithium-ion batteries is also reflected by the 2019 Nobel Prize. A typical lithium-ion battery system consists of an anode, cathode, and non-aqueous liquid electrolyte. A solid electrolyte interphase (SEI) refers to the solid layer covering the anode and is widely believed to form due to the decomposition of electrolytes [7,8,9,10,11,12]. Significant to the function of Li-ion batteries, the SEI is electrically insulating yet sufficiently conductive to lithium ions [13]. Moreover, it prevents further decomposition of the electrolyte during the normal use of Li-ion batteries. Therefore, the SEI plays a critical role in influencing the performance, including cycle life, self-discharge, safety, faradic efficiency, and irreversible capacity, of Li-ion batteries [9,14,15,16,17,18,19,20,21].
The formation process of the SEI strongly depends on the type of anode and electrolyte materials, as well as the process of fabrication. Nowadays, many Li-ion rechargeable batteries employ carbon graphite as their anode materials and mixtures of ethylene carbonate (EC) with linear carbonates, such as dimethyl carbonate (DMC) and diethyl carbonate (DEC), as electrolytes. Consequently, the mechanisms of SEI formation between the EC-based electrolytes and carbon graphite has been extensively studied both experimentally [14,22,23,24,25,26,27] and theoretically [28,29,30,31,32,33,34,35,36,37,38,39], summarized in the reviews in Refs. [7,8,9,10,11,12,40,41]. It has been believed that only EC decomposes and contributes to the SEI formation, while the other components of linear carbonates merely improve the viscosity and conductivity of the electrolyte [28,29,30,31,32,33,34,35,36,37,38,39]. As a result, studies have mainly focused on the decomposition process of EC molecules. For example, Aurbach et al. investigated the surface chemistry of lithium electrodes in alkyl carbonate solutions using surface-sensitive Fourier-transform infrared (FTIR) spectroscopy and found that Li2CO3 and ROCO2Li were the major decomposition products of EC [42]. Furthermore, Aurbach et al. [24] experimentally explored reactions between EC and nucleophiles (such as lithium tert-butoxide) on lithiated graphite, and identified Li2CO3 and lithium ethylene dicarbonate (CH2OCO2Li)2 (namely LEDC) as the major decomposition products of EC under this condition. Based on their experimental evidence, Aurbach et al. proposed a three-step reduction mechanism (two-electron path) for EC decomposition on lithiated graphite electrodes. Later, Li and Balbuena showed this three-step reduction mechanism of EC thermodynamically feasible using ab initio density functional theory (DFT) and conventional transition state theory (CTST) computation methods [30]. A recent experiment suggested that the majority of LEDC in the SEI has been called into question [43].
Previous DFT studies [7,32,38] indicated that the cleavage of the O2-C2 or O3-C3 bond in Li+-coordinated EC is thermodynamically and kinetically more favorable than the cleavage of the O2-C1 or O3-C1 bond (Figure 1a). Therefore, the cleavage of the O2-C2 or O3-C3 bond should be the primary reductive reaction of EC in Li-ion batteries. Following this bond cleavage, radical anion CH2CH2OCO2− can further react with Li+ to form lithium ethylene dicarbonate ((CH2OCO2Li)2) [32,39,44,45], which could constitute the main composition of the SEI [41]. It appears that this theoretical prediction is consistent with some experimental findings. For instance, Aurbach et al. [26] found that no matter what salt was used in an EC:DMC 1:1 electrolyte solution, (CH2OCHO2Li)2 could be found as one of the EC reduction products in the SEI, covering the graphite electrodes in EC:DMC 1:1 electrolyte solutions. In a more elaborated study, Zhuang et al. [46] compared the FTIR spectrum from synthesized (CH2OCHO2Li)2 and the SEI formed in 1.2 M LiPF6/EC:EMC electrolyte solution on a Ni electrode, concluding that (CH2OCHO2Li)2 was the dominant surface species on the electrodes. However, an experiment by Hardwick et al. showed that (CH2OCHO2Li)2 was absent in the SEI formed on seriously damaged graphite anodes and the resultant SEI was more resistive than those on normal graphite anodes [47]. Besides monomers of (CH2OCO2Li)2, DFT calculations [7,33] also found that it was possible for even the dimers, trimers, tetramers, and higher order n-polymers of lithium ethylene dicarbonate to form on the graphite anode of the lithium-ion batteries’ surfaces through intermolecular association interactions (in the form of O···Li···O). Using ab initio molecular dynamics simulations, the voltage dependence of the interfacial electrochemical process has been investigated at lithium-intercalated graphite edge planes, which concluded that electrochemical reduction exhibits potential-dependent kinetics [48]. Using the DFT and the implicit solvation theory, the lithium insertion and desorption reaction at the SEI has been studied with insights on the charge transfers [49]. A classic molecular dynamics study suggested that, during the initial dissolution step, the Li cation prefers to shed DMC molecules as opposed to losing the EC [50].
To gain knowledge about the molecular structure of the SEI, in this work we focused on studying the adsorption configuration and strength of major EC decomposition products on the surface of carbon graphite. The paper is organized as following: Section 2 describes our computational method; Section 3 presents our computation results and discussions; and Section 4 gives the conclusions.
2. Computation Methods
We used the density functional theory (DFT) method to determine the optimal structures of EC, CH2CH2OCO2Li, and (CH2OCO2Li)2, and the adsorption of these molecules onto the basal and edge surfaces of graphite. The DFT calculations were performed using the Vienna Ab initio Simulation Package (VASP) [51,52] code which is based on the Kohn–Sham density functional theory (KS-DFT) [53] with the generalized gradient approximations, as parameterized by Perdew and Wang, for the exchange–correlation functions [54]. The electrons explicitly included in the calculations were the (2s22p2) electrons of carbon, (2s1) electron of lithium, (1s1) electron of hydrogen, and (2s22p4) of oxygen. The core electrons (1s2) of carbon, lithium, and oxygen were replaced by the projector augmented wave (PAW) and pseudopotentials [55]. The cutoff energy of the plane-waves was 400 eV. The convergence criteria of the electronic self-consistent iteration were 0.0001 eV. The atomic forces were relaxed to be smaller than 0.01 eV/Å. We used a 3 × 3 × 1 Monkhorst-Pack k-mesh for Brillouin-zone integration for molecular adsorption on graphite surfaces. The size effect and k-mesh convergence were examined as summarized in Supplemental Table S1 in the Supplementary Information.
3. Results
3.1. Isolated Molecules and Pristine Surfaces
3.1.1. EC Molecules
In the optimized configuration of the isolated EC molecule (shown in Figure 1a), three carbonate atoms and three oxygen atoms lay in the same plane. For this optimized EC molecule, our DFT calculations predicted the bond lengths of RC1-O1 = 1.21 Å, RC1-O2 = 1.38 Å, RC2-O2 = 1.44 Å, RC2-C3 = 1.54 Å, and the bond angles of ∠O1C1C2 = 125°, ∠C1O2C2 = 110°. (C1, C2, O1, and O2 refer to the atoms as marked in Figure 1). Our computational results agree well with the previous DFT prediction of RC2-C3 = 1.53 Å [56], RC2-O2 = 1.43 Å [30], and the experimental data of RC2-C3 = 1.522 Å and RC2-O2 = 1.457 Å [57].
3.1.2. CH2CH2OCO2Li Molecule
The first step in the decomposition process of EC is to break the ring structure. This decomposition is energetically favorable [7] and leads to the formation of a radical anion. CH2CH2OCO2−. In lithium-ion batteries, a single radical anion.CH2CH2OCO2− can further react with a Li+ to form the CH2CH2OCO2Li molecule. The configuration of the CH2CH2OCO2Li molecule is determined by the minimization of the total energy of the molecule. The seven examined atomistic structures of CH2CH2OCO2Li are displayed in Figure S1 in the Supplementary Information. Our DFT calculations found that the lowest-energy configuration of the CH2CH2OCO2Li molecule would assume a structure as shown in Figure 1b, in which all the C, O, and Li atoms lie in the same plane. In this optimal structure of the CH2CH2OCO2Li molecule, Li+ strongly binds to the two O atoms and one C atom with bond lengths of RLi-O1 = 1.85 Å, RLi-O2 = 1.86 Å, and RLi-C1 = 2.08 Å. Our results are consistent with the previous computational values of RLi-O1 = 1.848 Å and RLi-O2 = 1.864 Å [38].
For the EC decomposition reaction , we estimated its reaction energy to be −1.35 eV. The reaction energy was calculated as the difference in potential energies between the product (CH2CH2OCO2Li) and the reactants (isolated EC molecule and single Li atom) and as . A negative reaction energy here indicates that this reaction was exothermic.
3.1.3. (CH2OCO2Li)2 Molecule
It is also believed that, in Li-ion batteries with two radical anions, CH2CH2OCO2− could react with two Li+ to form a (CH2OCO2Li)2 molecule by the chemical reaction [58].
In order to find the lowest-energy molecular structure, we have performed structure optimization calculations for the five high-symmetry configurations of the (CH2OCO2Li)2 molecule using the DFT method. In this work, we found that the gauche configuration (shown in Figure 1c) of the (CH2OCO2Li)2 molecule had the lowest potential energy.
In the optimized gauche configuration, the two CH2OCO2 side chains face each other when viewing along the central C2-C3 bond. Moreover, each Li atom binds with two oxygen atoms that belong to the two different CH2OCO2 side chains. The other four low-energy configurations of the (CH2OCO2Li)2 molecule are listed in Figure 1d–g. Figure 1d shows a cis configuration, in which the two CH2OCO2Li side chains form a dihedral angle of 60° around the central C2-C3 bond. The potential energy of this cis configuration was found to be 1.11 eV higher than that of the gauche configuration. Figure 1e shows a trans configuration, in which the two CH2OCO2Li clusters form a dihedral angle of 180° around the central C-C bond. For this configuration of the (CH2OCO2Li)2 molecule, we calculated the bond lengths to be RLi-O1 = 1.85 Å, RLi-O2 = 1.85 Å, RLi-C1 = 2.08 Å, RC1-O3 = 1.36 Å, and RC2-O3 = 1.52 Å. Our computational results agree well with the previous DFT predictions of RLi1-O1 = 1.856 Å, RLi1-O2 = 1.868 Å, RLi1-C1 = 2.095 Å, and RC1-O3 = 1.346 Å [33]. The potential energy of this trans configuration is 1.12 eV higher than that of the gauche configuration.
The (CH2OCO2Li)2 molecule can form another cis configuration, which is distinct from the one in Figure 1d in that the two CH2OCO2Li clusters are twisted around the Csp2-Osp3 bond by 90° as shown in Figure 1f. The potential energy of this configuration was found to be 1.12 eV higher than that of the gauche configuration. Figure 1g shows a twisted trans configuration, in which the two CH2OCO2Li clusters are twisted around the Csp2-Osp3 bond by 90°, and whose potential energy was 1.14 eV higher than that of the gauche configuration. Thus, our result of the transition energy from trans (Figure 1e) to gauche (Figure 1c) was 1.12 eV, which agrees well with the theoretical value of 1.048 eV [45].
For the EC decomposition reaction, , we estimated its reaction energy to be −7.69 eV. The reaction energy was calculated as the difference in potential energies between the products (the gauche configuration of (CH2OCO2Li)2 and the isolated C2H4 molecule) and the reactants (the isolated EC molecule and a single Li atom). The detailed calculations are elaborated in the Supplementary Information. Hence, our calculation results imply that, after decomposing into CH2CH2OCO2Li, electrolyte EC can further decompose to form (CH2OCO2Li)2 and release 4.99 eV energy in Li-ion batteries.
3.1.4. Graphite Surfaces
Graphite has a layered structure where the carbon atoms in each layer are bonded in hexagonal arrays, and the isolated suspended single layer is named graphene [59,60,61,62,63,64,65,66,67,68]. In this work, we modeled graphite using a multi-layer graphene with a stacking order of AB (hexagonal graphite or α-phase). Shown in Figure 1h, the atomic structure of such bulk graphite was determined by two parameters: a (the edge length of the primary unit cell (rhombohedra cell) on basal plane) and H (the height perpendicular to basal plane, Figure 1i). From our DFT calculations, we predicted a to be 2.464 Å and H to be 7.197 Å in our modeled bulk graphite. Compared with the experimental measurement data (a = 2.461 Å and H = 6.710 Å) [69], our theoretical calculations overestimated the value of a by 0.1 % and the value of H by 7.3%.
Figure 1h–i show that the graphite with a layer structure has two types of surfaces: basal plane surfaces and edge surfaces. The edge surfaces of graphite could assume armchair or zigzag forms [70,71,72]. It is known that the basal plane and edge surfaces of graphite could exhibit different electrochemical behaviors [73]. Hence, we modeled both types of graphite surfaces in this work. The basal surface of graphite was modeled using a super cell containing a single graphene layer (54 carbon atoms) and a vacuum region in the direction normal to the surface. In our DFT calculations of isolated and molecule-adsorbed basal plane surfaces, we used a 4 × 4 × 1 Monkhorst-Pack k-mesh for Brillouin-zone integration. The armchair edge surface was modeled using a super cell containing four graphene half-layers (24 carbon atoms at each layer) and a vacuum region in the direction normal to the surface. The armchair edge of graphite is marked with a black dotted line in Figure 1j. The zigzag edge surface was modeled using a super cell containing four graphene half-layers (16 carbon atoms at each layer) and a vacuum region in the direction normal to the surface. The zigzag edge of graphite is marked with a red dashed line in Figure 1j. In our DFT calculations of isolated and molecule-adsorbed armchair and zigzag edge surfaces, we used a 3 × 3 × 1 Monkhorst-Pack k-mesh for Brillouin-zone integration.
3.2. Molecule Adsorption on Graphite Surfaces
The adsorption energy is the energy difference between the adsorption system (the complex consisting of multiple components) and the isolated configurations of the individual components. Taking (CH2OCO2Li)2 adsorbing on the graphite surface as an example, the adsorption energy is calculated as . The adsorption energy indicates the adhesion strength of the adsorbate molecule on the surface. A negative adsorption energy implies that the adsorbate molecule would be energetically favorable to be adducted to the surface, while a positive adsorption energy means that the adsorbate molecule would not energetically prefer contact with the surface. Consequently, a necessary condition to form a stable SEI protective layer in Li-ion batteries is the negative adsorption energy of EC decomposition molecules on the graphite surface. The more negative these adsorption energies are, the stronger the binding strength between the SEI and the graphite anodes will be.
In order to determine the adsorption energies of the EC decomposition molecules on the graphite surfaces, we used DFT computations to predict the optimized structures and energies of the CH2CH2OCO2Li and (CH2OCO2Li)2 molecules adsorbed on the basal and edge surfaces of graphite. We further compared the attained energies of the adsorption configuration with the energies of the isolated molecules and surfaces. The adsorption structures of EC on graphite surfaces are displayed in Supplementary Figure S2 in the Supplementary Information. The adsorption of CH2CH2OCO2Li and (CH2OCO2Li)2 molecules on graphite surfaces is elaborated as follows.
3.2.1. Adsorption of CH2CH2OCO2Li Molecule on Graphite Surfaces
To evaluate the adhesion strength between the CH2CH2OCO2Li molecule and the graphite surfaces, we placed the lowest-energy configuration of the CH2CH2OCO2Li molecule on various locations of the graphite surface and optimized the structures using DFT calculations. In our modeled graphite surfaces, we included a 20 Å thick vacuum region in their normal direction.
Basal Plane Surface
We examined three distinct configurations of the CH2CH2OCO2Li molecule adsorbed on the basal plane surface of graphite. In the lowest-energy configuration (Figure 2a), the Li atom in the CH2CH2OCO2Li molecule lies 2.11 Å right above the hollow site; thus, it directly interacts with the carbon-ring atoms forming the hollow site. The distance between the Li atom and the carbon-ring atoms is 2.58 Å. The corresponding adsorption energy was determined to be −0.24 eV. Figure 2b shows the configuration that the CH2CH2OCO2Li molecule lies parallel to the basal plane and the Li atom lies above the hollow site. The adsorption energy associated with this configuration is −0.04 eV. Figure 2c shows the configuration that the CH2CH2OCO2Li molecule is flipped upside-down from Figure 2a, with an adsorption energy of 0.01 eV. Because the configuration of Figure 2a gives the lowest adsorption energy, it would be the most possible configuration for the CH2CH2OCO2Li molecule on the basal plane surface.
Edge Surfaces
We studied the adsorption of the CH2CH2OCO2Li molecule on the two kinds of edge surfaces of graphite, (a) zigzag edge surface and (b) armchair edge surface, as shown in Figure 2d–g. Their corresponding side-views are also shown on the side for a clear view. We found very strong adsorptions of CH2CH2OCO2Li on the zigzag edge surface of graphite, of −0.91 eV. The adsorption on the armchair edge surface was −0.54 eV. In this orientation, the C1-O2-O3 planes were parallel to the graphite layers, which left the channel for the migration of Li atoms and intercalation, while blocking other solvent molecules.
3.2.2. Adsorption of (CH2OCO2Li)2 Molecule on Graphite Surfaces
Minimizing the effect of periodic images, we set the thickness of the vacuum region to 30 Å in the super cells of our modeled graphite surfaces (both basal and edge surfaces) for the adsorption of (CH2OCO2Li)2. Although five configurations of (CH2OCO2Li)2 were studied in the previous section, only the two typical configurations, gauche and trans, were subjected to the study of the adsorption on the basal plane surface, and only the trans configuration was selected to study the adsorption on edge surfaces for simplicity and representativeness.
Basal Plane Surface
We studied the adsorption of two configurations (gauche and trans) of (CH2OCO2Li)2 on the graphite basal surface. When one lithium atom of gauche configuration sits above the hollow site of a carbon hexagon ring (center), there are three possible configurations where the other lithium atom is most likely to stand. These configurations are: (1) hollow site (H), which is on the center of another carbon hexagon ring; (2) top site (T), which is on top of a carbon atom; and (3) bridge site (B) which is the middle of the two nearest carbon neighbors. Figure 3a shows the initial gauche configuration on the basal plane surface where two lithium atoms are in the Hollow-Hollow configuration of the carbon hexagon ring. Figure 3b,c shows the Hollow-Top and Hollow-Bridge configurations, respectively. Figure 3d shows the trans configuration on the basal surface. We found adsorptions of −0.03, −0.04, −0.04, and −0.17 eV for configurations Figure 3a–d, respectively. The minimum distance between lithium and carbon atoms on the basal plane was 4.08, 4.05, 4.07, and 2.68 Å, respectively. This result indicates that the adsorption of (CH2OCO2Li)2 on the graphite basal surface is weak. Compared to gauche, the trans configuration has the stronger adsorption on the basal plane surface.
Edge Surface
The adsorption of (CH2OCO2Li)2 on the edge surface of graphite was studied on two kinds of edges, zigzag and armchair, with the trans configuration. Previous test examinations suggest that the parallel orientation, where the C1-O1-O2 plane of (CH2OCO2Li)2 is parallel to graphite basal plane, is energetically favorable. As a result, the adsorption calculations of (CH2OCO2Li)2 on zigzag and armchair edge surfaces were only carried on the parallel orientation of the trans configuration. Figure 4a,b are the setup of the adsorption calculations for the zigzag and armchair edge surfaces, respectively. We found that the adsorption energy was −0.49 eV on the zigzag edge surface, which was larger than the armchair edge surface, which was −0.32 eV. This result suggests that the adsorption of (CH2OCO2Li)2 on the zigzag edge surface is the most favorable adsorption between the basal surface, zigzag edge surface, and armchair edge surface.
3.3. Networking Adsorption on Graphite Surfaces
We studied several possible networks of (CH2OCO2Li)2 on the graphite surface. Such a network of (CH2OCO2Li)2 may form the backbone of the SEI layer. Therefore, the study of the formation of such a network and their adsorption on graphite surfaces is of great importance.
3.3.1. Network on Basal Surface
We built up the model with one (CH2OCO2Li)2 linear molecule sitting 3.0 Å above the hollow site of the hexagon. Two graphene layers and a 30 Å vacuum region were used to present the basal surface on graphite. The nearest neighbor (CH2OCO2Li)2 molecule was 4.27 Å away.
After full relaxation, we found that the Li atoms moved to the middle point between the (CH2OCO2Li)2 molecules and a network was formed. The Li atoms bonded with oxygen atoms from other (CH2OCO2Li)2 molecules and formed the network as shown in Figure 5. The Li-O bond was found to be 2.16 Å. The minimum distance between the lithium and carbon atoms in graphite was 3.88 Å. The adsorption energy of such networked (CH2OCO2Li)2 was −0.01 eV with a reaction energy of −1.39 eV. It is worth noting that the reaction energy is the energy difference between the product and the reactants = E(products) − E(reactants). For the adsorption systems, the reaction energy includes the adsorption energy (see the calculation examples in Supplementary Information).
3.3.2. Network on Zigzag Edge Surface
The networking of (CH2OCO2Li)2 was studied on the zigzag edge surface. The surface in this model was selected to have an area of 7.4 Å × 7.4 Å. There were two layers of carbon atoms in one unit cell and 25 carbon atoms on each layer. There were 10 Å thick vacuum regions to present the surface. We used a 4 × 4 × 1 Monkhorst-Pack k-mesh for Brillouin-zone integration.
To reduce numerical errors during geometry optimization caused by the unphysical placement of the (CH2OCO2Li)2 molecules, we initially displaced the (CH2OCO2Li)2 molecules far away from the zigzag edge surface, about 4.0 Å, which is larger than any possible bond length among these elements between Li, O, C, and H. It turns out this is a trick to obtain a physical and reasonable optimized geometry. It is critical to set an initial large distance between (CH2OCO2Li)2 molecules and the zigzag surface, about 4.0 Å.
After full relaxation, the (CH2OCO2Li)2 molecules formed a network on the zigzag edge surface of graphite, as shown in Figure 4. The Li and a carbon atom on graphite formed a bond with a bond length of 2.14 Å, which was also the distance between (CH2OCO2Li)2 and the graphite zigzag edge surface. The Li-O bond was 1.95 Å. The network formed in one row along layers (perpendicular to the layer). The adsorption energy of such networked (CH2OCO2Li)2 on the graphite zigzag edge surface was −1.08 eV with a reaction energy of −1.66 eV.
3.3.3. Network on Armchair Edge Surface
Similarly, the networking of (CH2OCO2Li)2 was studied on the armchair edge surface. After full relaxation, the (CH2OCO2Li)2 molecules formed a network on the armchair edge surface of graphite, as shown in Figure 6c,d.
The minimum distance between the Li atom and a carbon atom on graphite is 2.45 Å, which was also the distance between (CH2OCO2Li)2 to the graphite armchair edge surface. The Li-O bond close to the surface was 1.9 Å, but 2.0 Å on another end. The network formed in 2D, with both rows and columns on the surfaces. The adsorption energy of such networked (CH2OCO2Li)2 on the graphite armchair edge surface was −0.32 eV, with a reaction energy of −1.89 eV.
4. Discussions
Compared with the basal plane surfaces, the adsorptions of CH2CH2OCO2Li and (CH2OCO2Li)2 on the edge surfaces were much stronger. The reason for such a strong adsorption may lay in the presence of the dangling bonds of carbon atoms on the edge surfaces. For the same reason, the zigzag edge surfaces have a stronger adsorption to (CH2OCO2Li)2 than the armchair edge surfaces because the dangling bond density is higher. The ratio of dangling bonds per unit length in one layer of the zigzag edge surface to that of the armchair surface is . The precise quantity relationship between the dangling bond density and the adsorption energy needs further study and analysis. Despite this, we still can qualitatively conclude that (CH2OCO2Li)2, a main product of the initial electric chemical reaction, is energetically favorable to be adsorbed on the edge surface of graphite. Such an adsorption may form the first layer of the SEI on the edge surface and provide the base of other layers’ growth in further SEI forming. Compared with the isolated EC and Li molecules, the total energy gained during the binding and adsorption was 1.58 eV in this reaction path.
To state clearly, the results of the adsorption energies of CH2CH2OCO2Li and (CH2OCO2Li)2 on the graphite basal, zigzag edge, armchair edge surfaces are listed in Table 1. The strong adsorptions on edge surfaces are because of the dangling bonds of carbon atoms on the surface. The large adsorption energy of −0.91 eV on the zigzag edge surface indicates the strong bonding between CH2CH2OCO2Li and the graphite surface, making this layer formed by CH2CH2OCO2Li electronically insulating. This layer of CH2CH2OCO2Li makes the innermost layer of the SEI and becomes the base for the growth of further SEI layers. This strong adsorption suggests that the SEI film is favorably formed on the edge surface. Li acts as the root and medium for the initial decomposition of EC and adsorption onto the graphite surface, forming the first and innermost layer of the SEI.
Based on our DFT calculations of the adsorption strength of EC, (CH2OCO2Li)2, CH2CH2OCO2Li on the graphite basal surface and the zigzag and armchair edge surfaces, we can propose a mechanism for the formation of the SEI on carbon anodes in E-based electrolytes. An EC molecule initially decomposes via a hemolytic ring opening through an O2C2 bond cleavage, the product of which being a radical anion, CH2CH2OCO2−, which further reacts with Li to form a stable structure of CH2CH2OCO2Li. In turn, it can be absorbed onto the graphite surface, with −0.24 eV on the basal surface, −0.54 eV on the armchair edge surface, and −0.91 eV on the zigzag edge surface. The absorbed CH2CH2OCO2Li molecule forms the first and innermost layer of the SEI. The orientation with the molecule plane (all atoms O3-C2-O3-C1-O1-O2 are coplanar), parallel to the graphite layer (see Figure 4d), ensures the function of SEI film, conducting to Li/Li+, but preventing any co-insertion of solution species.
CH2CH2OCO2Li is composed of the same elements and bonds with lithium ethylene dicarbonate (CH2OCO2Li)2, a well-known SEI component [13,46]. As a result, CH2CH2OCO2Li has an identical FTIR spectrum, which means that the data from experimental FTIR spectroscopy, for example in Ref. [41], also suggests the existence of CH2CH2OCO2Li in SEI film. As such, we believe that CH2CH2OCO2Li and (CH2OCO2Li)2 may co-exist in SEI film, in which CH2CH2OCO2Li is the inner layer. There are some experimental observations that the SEI films will cleave from the graphite surface once the battery is overcharged. We think this is because the overcharge impairs the adsorption binding between the CH2CH2OCO2Li and the graphite surface. As such, these observations also support our model. Recent in situ neutron reflectometry measurements have illustrated that the SEI layer has a LiOCO2C2H5 layer with a thickness of about 0.5 nm after 20 cyclic voltammetry cycles, later growing to about 1.0 nm on the copper surface. Considering the lengths of the molecules of CH2CH2OCO2Li and (CH2OCO2Li)2, which are 0.6 nm and 1.0 nm, respectively, this neutron reflectometry experiment evidences our theoretical findings.
One interesting thing that needs to be mentioned here is that the parallel orientation is more energetically favorable when the plane of C1-O1-O2 is parallel to the graphite layers. Our proposed network is formed through the “zipping” of carbonate branches through lithium ions, which is similar to the 3D network structure of the SEI proposed by Shkrob et al. from a spectroscopic study [74]. Such geometry makes the adsorption of (CH2OCO2Li)2 an extension or “growth” of the graphite layers, which allows Li intercalation into the graphite but blocks other molecular migration.
5. Conclusions
We have investigated the initial decomposition of EC and the formation of the innermost SEI layers on the surfaces of carbon anodes of Li-ion batteries using first-principles calculations. We have studied the adsorptions of EC, (CH2OCO2Li)2, and CH2CH2OCO2Li on the basal, zigzag, and armchair surfaces of graphite. We found that the homolytic ring opening of EC is the first step toward its decomposition. We examined the interaction between the initial product, CH2CH2OCO2− and Li cations, and the graphite surface. We found a stable structure of CH2CH2OCO2Li, where all carbon atoms, oxygen atoms, and the lithium atom are in one plane, with a binding energy of 1.35 eV. This structure can be well adsorbed onto the graphite basal surface with an adsorption energy of −0.24 eV, with the Li atom 2.11 Å above the center of hexagon carbon atoms on the graphene.
The adsorption of CH2CH2OCO2Li will decrease with the concentration. CH2CH2OCO2Li has a strong adsorption on the graphite zigzag edge surface with an adsorption energy of −0.91 eV, due to the dangling carbon bonds, and suggests that the SEI film is primarily formed on the edge surface with CH2CH2OCO2Li as the innermost layer. During the study, we noticed that CH2CH2OCO2Li has the same character bonds, C-H, C-O, and C=C, as those of (CH2OCO2Li)2. These two molecules should have an identical FTIR spectra. As a result, it is hard to distinguish them by FTIR spectra, which is a main experimental method in studying the components of the SEI. We studied the adsorption of (CH2OCO2Li)2 and CH2CH2OCO2Li on the graphite surface using first-principles calculations. We propose an SEI-forming mechanism with the adsorption and bonding molecule CH2CH2OCO2Li with carbon atoms. Our proposed network is formed through a “zipping” of carbonate branches through lithium ions, agreeing with the 3D network structure of the SEI proposed by a spectroscopic study [74]. Such an SEI-forming mechanism on the graphite surface builds up the innermost layer of the SEI in EC-based electrolytes in a lithium-ion battery.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/nano12203654/s1, Table S1: Size effect and k-mesh convergence for adsorption energy of EC on graphite surface; Figure S1: Various atomistic configurations of CH2CH2OCO2Li; Figure S2: Adsorption of EC on basal surface of graphite; Calculation of the adsorption energy and reaction energy.
Funding
This research was funded by the Deanship of Scientific Research (DSR) at King Fahd University of Petroleum & Minerals (KFUPM) through project No. DF201020.
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Acknowledgments
We would like to thank Guofeng Wang for helpful discussions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Armand, M.; Tarascon, J.M. Building better batteries. Nature 2008, 451, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Balbuena, P.B.; Wang, Y. Lithium Battery Solid Electrolyte Interphase; Balbuena, P.B., Wang, Y.X., Eds.; Imperial College Press: London, UK, 2004. [Google Scholar]
- Wang, Y.; Liu, B.; Li, Q.; Cartmell, S.; Ferrara, S.; Deng, Z.D.; Xiao, J. Lithium and lithium ion batteries for applications in microelectronic devices: A review. J. Power Sources 2015, 286, 330–345. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Zhu, Z.; Yan, Q.; Yu, S.; He, X.; Chen, Y.; Zhang, R.; Ma, L.; Liu, T.; Li, M.; et al. A disordered rock salt anode for fast-charging lithium-ion batteries. Nature 2020, 585, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Kotobuki, M.; Song, S.; Lai, M.O.; Lu, L. Review on solid electrolytes for all-solid-state lithium-ion batteries. J. Power Sources 2018, 389, 198–213. [Google Scholar] [CrossRef]
- Lu, J.; Wu, T.; Amine, K. State-of-the-art characterization techniques for advanced lithium-ion batteries. Nat. Energy 2017, 2, 17011. [Google Scholar] [CrossRef]
- Balbuena, P. Lithium-Ion Batteries: Solid-Electrolyte Interphase; Imperial College Press: London, UK, 2004. [Google Scholar]
- Winter, M. The solid electrolyte interphase—The most important and the least understood solid electrolyte in rechargeable Li batteries. Z. Fur Phys. Chem. 2009, 223, 1395–1406. [Google Scholar] [CrossRef]
- Wang, A.; Kadam, S.; Li, H.; Shi, S.; Qi, Y. Review on modeling of the anode solid electrolyte interphase (SEI) for lithium-ion batteries. NPJ Comput. Mater. 2018, 4, 15. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Mao, B.; Stoliarov, S.I.; Sun, J. A review of lithium ion battery failure mechanisms and fire prevention strategies. Prog. Energy Combust. Sci. 2019, 73, 95–131. [Google Scholar] [CrossRef]
- Horstmann, B.; Single, F.; Latz, A. Review on multi-scale models of solid-electrolyte interphase formation. Curr. Opin. Electrochem. 2019, 13, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Verma, P.; Maire, P.; Novák, P. A review of the features and analyses of the solid electrolyte interphase in Li-ion batteries. Electrochim. Acta 2010, 55, 6332–6341. [Google Scholar] [CrossRef]
- Heiskanen, S.K.; Kim, J.; Lucht, B.L. Generation and Evolution of the Solid Electrolyte Interphase of Lithium-Ion Batteries. Joule 2019, 3, 2322–2333. [Google Scholar] [CrossRef]
- Arora, P. Capacity Fade Mechanisms and Side Reactions in Lithium-Ion Batteries. J. Electrochem. Soc. 1998, 145, 3647–3667. [Google Scholar] [CrossRef] [Green Version]
- Smart, M.C.; Ratnakumar, B.V.; Surampudi, S.; Wang, Y.; Zhang, X.; Greenbaum, S.G.; Hightower, A.; Ahn, C.C.; Fultz, B. Irreversible Capacities of Graphite in Low-Temperature Electrolytes for Lithium-Ion Batteries. J. Electrochem. Soc. 2019, 146, 3963–3969. [Google Scholar] [CrossRef]
- Zhang, S.; Ding, M.S.; Xu, K.; Allen, J.; Jow, T.R. Understanding solid electrolyte interface film formation on graphite electrodes. Electrochem. Solid-State Lett. 2001, 4, A206. [Google Scholar] [CrossRef]
- Yan, C.; Yao, Y.X.; Cai, W.L.; Xu, L.; Kaskel, S.; Park, H.S.; Huang, J.Q. The influence of formation temperature on the solid electrolyte interphase of graphite in lithium ion batteries. J. Energy Chem. 2020, 49, 335–338. [Google Scholar] [CrossRef]
- Cheng, X.B.; Zhang, R.; Zhao, C.Z.; Wei, F.; Zhang, J.G.; Zhang, Q. A review of solid electrolyte interphases on lithium metal anode. Adv. Sci. 2015, 3, 1500213. [Google Scholar] [CrossRef]
- Shen, X.; Zhang, R.; Chen, X.; Cheng, X.B.; Li, X.; Zhang, Q. The Failure of Solid Electrolyte Interphase on Li Metal Anode: Structural Uniformity or Mechanical Strength? Adv. Energy Mater. 2020, 10, 1903645. [Google Scholar] [CrossRef]
- Gao, Y.; Yan, Z.; Gray, J.L.; He, X.; Wang, D.; Chen, T.; Huang, Q.; Li, Y.C.; Wang, H.; Kim, S.H.; et al. Polymer-inorganic solid-electrolyte interphase for stable lithium metal batteries under lean electrolyte conditions. Nat. Mater. 2019, 18, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Abate, I.I.; Sivonxay, E.; Shyam, B.; Jia, C.; Moritz, B.; Devereaux, T.P.; Persson, K.A.; Steinrück, H.G.; Toney, M.F. Solid Electrolyte Interphase on Native Oxide-Terminated Silicon Anodes for Li-Ion Batteries. Joule 2019, 3, 762–781. [Google Scholar] [CrossRef] [Green Version]
- Shu, Z.X.; McMillan, R.S.; Murray, J.J. Electrochemical Intercalation of Lithium into Graphite. J. Electrochem. Soc. 2019, 140, 922–927. [Google Scholar] [CrossRef]
- Aurbach, D.; Ein-Eli, Y.; Chusid (Youngman), O.; Carmeli, Y.; Babai, M.; Yamin, H. The Correlation Between the Surface Chemistry and the Performance of Li-Carbon Intercalation Anodes for Rechargeable ‘Rocking-Chair’ Type Batteries. J. Electrochem. Soc. 2019, 141, 603–611. [Google Scholar] [CrossRef]
- Aurbach, D.; Levi, M.D.; Levi, E.; Schechter, A. Failure and stabilization mechanisms of graphite electrodes. J. Phys. Chem. B 1997, 101, 2195–2206. [Google Scholar] [CrossRef]
- Peled, E. An Advanced Tool for the Selection of Electrolyte Components for Rechargeable Lithium Batteries. J. Electrochem. Soc. 1998, 145, 3482–3486. [Google Scholar] [CrossRef]
- Aurbach, D.; Markovsky, B.; Weissman, I.; Levi, E.; Ein-Eli, Y. On the correlation between surface chemistry and performance of graphite negative electrodes for Li ion batteries. Electrochim. Acta 1999, 45, 67–86. [Google Scholar] [CrossRef]
- Aurbach, D.; Moshkovich, M.; Cohen, Y.; Schechter, A. Study of surface film formation on noble-metal electrodes in alkyl carbonates/Li salt solutions, using simultaneous in situ AFM, EQCM, FTIR, and EIS. Langmuir 1999, 15, 2947–2960. [Google Scholar] [CrossRef]
- Endo, E.; Ata, M.; Tanaka, K.; Sekai, K. Electron Spin Resonance Study of the Electrochemical Reduction of Electrolyte Solutions for Lithium Secondary Batteries. J. Electrochem. Soc. 2019, 145, 3757–3764. [Google Scholar] [CrossRef]
- Endo, E.; Tanaka, K.; Sekai, K. Initial Reaction in the Reduction Decomposition of Electrolyte Solutions for Lithium Batteries. J. Electrochem. Soc. 2000, 147, 4029. [Google Scholar] [CrossRef]
- Li, T.; Balbuena, P.B. Theoretical studies of the reduction of ethylene carbonate. Chem. Phys. Lett. 2000, 317, 421–429. [Google Scholar] [CrossRef]
- Márquez, A.; Balbuena, P.B. Molecular Dynamics Study of Graphite/Electrolyte Interfaces. J. Electrochem. Soc. 2001, 148, A624. [Google Scholar] [CrossRef]
- Wang, Y.; Nakamura, S.; Ue, M.; Balbuena, P.B. Theoretical studies to understand surface chemistry on carbon anodes for lithium-ion batteries: Reduction mechanisms of ethylene carbonate. J. Am. Chem. Soc. 2001, 123, 11708–11718. [Google Scholar] [CrossRef]
- Wang, Y.X.; Balbuena, P.B. Associations of lithium alkyl dicarbonates through O···Li···O interactions. J. Phys. Chem. A 2002, 106, 9582–9594. [Google Scholar] [CrossRef]
- Wang, Y.; Balbuena, P.B. Theoretical insights into the reductive decompositions of propylene carbonate and vinylene carbonate: Density functional theory studies. J. Phys. Chem. B 2002, 106, 4486–4495. [Google Scholar] [CrossRef]
- Wang, Y.; Nakamura, S.; Tasaki, K.; Balbuena, P.B. Theoretical studies to understand surface chemistry on carbon anodes for lithium-ion batteries: How does vinylene carbonate play its role as an electrolyte additive? J. Am. Chem. Soc. 2002, 124, 4408–4421. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Kong, W.; Li, H.; Huang, X.; Chen, L. Experimental and theoretical studies on reduction mechanism of vinyl ethylene carbonate on graphite anode for lithium ion batteries. Electrochem. Commun. 2004, 6, 126–131. [Google Scholar] [CrossRef]
- Han, Y.K.; Lee, S.U.; Ok, J.H.; Cho, J.J.; Kim, H.J. Theoretical studies of the solvent decomposition by lithium atoms in lithium-ion battery electrolyte. Chem. Phys. Lett. 2002, 360, 359–366. [Google Scholar] [CrossRef]
- Han, Y.K.; Lee, S.U. Performance of density functional for calculation of reductive ring-opening reaction energies of Li+-EC and Li+-VC. Theor. Chem. Acc. 2004, 112, 106–112. [Google Scholar] [CrossRef]
- Wang, Y.; Balbuena, P.B. Theoretical studies on cosolvation of Li ion and solvent reductive decomposition in binary mixtures of aliphatic carbonates. Int. J. Quantum Chem. 2005, 102, 724–733. [Google Scholar] [CrossRef]
- Aurbach, D. Review of selected electrode-solution interactions which determine the performance of Li and Li ion batteries. J. Power Sources 2000, 89, 206–218. [Google Scholar] [CrossRef]
- Xu, K. Nonaqueous liquid electrolytes for lithium-based rechargeable batteries. Chem. Rev. 2004, 104, 4303–4417. [Google Scholar] [CrossRef]
- Aurbach, D.; Ein-Ely, Y.; Zaban, A. The Surface Chemistry of Lithium Electrodes in Alkyl Carbonate Solutions. J. Electrochem. Soc. 2019, 141, L1–L3. [Google Scholar] [CrossRef]
- Wang, L.; Menakath, A.; Han, F.; Wang, Y.; Zavalij, P.Y.; Gaskell, K.J.; Borodin, O.; Iuga, D.; Brown, S.P.; Wang, C.; et al. Identifying the components of the solid—Electrolyte interphase in Li-ion batteries. Nat. Chem. 2019, 11, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Tasaki, K.; Kanda, K.; Kobayashi, T.; Nakamura, S.; Ue, M. Theoretical Studies on the Reductive Decompositions of Solvents and Additives for Lithium-Ion Batteries near Lithium Anodes. J. Electrochem. Soc. 2006, 153, A2192. [Google Scholar] [CrossRef]
- Tasaki, K. Solvent decompositions and physical properties of decomposition compounds in Li-ion battery electrolytes studied by DFT calculations and molecular dynamics simulations. J. Phys. Chem. B 2005, 109, 2920–2933. [Google Scholar] [CrossRef]
- Zhuang, G.V.; Xu, K.; Yang, H.; Jow, T.R.; Ross, P.N. Lithium ethylene dicarbonate identified as the primary product of chemical and electrochemical reduction of EC in 1.2 M LiPF 6/EC:EMC electrolyte. J. Phys. Chem. B 2005, 109, 17567–17573. [Google Scholar] [CrossRef]
- Hardwick, L.J.; Marcinek, M.; Beer, L.; Kerr, J.B.; Kostecki, R. An Investigation of the Effect of Graphite Degradation on Irreversible Capacity in Lithium-ion Cells. J. Electrochem. Soc. 2008, 155, A442. [Google Scholar] [CrossRef] [Green Version]
- Leung, K. Predicting the voltage dependence of interfacial electrochemical processes at lithium-intercalated graphite edge planes. Phys. Chem. Chem. Phys. 2015, 17, 1637–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haruyama, J.; Ikeshoji, T.; Otani, M. Analysis of Lithium Insertion/Desorption Reaction at Interfaces between Graphite Electrodes and Electrolyte Solution Using Density Functional + Implicit Solvation Theory. J. Phys. Chem. C 2018, 122, 9804–9810. [Google Scholar] [CrossRef]
- Borodin, O.; Bedrov, D. Interfacial structure and dynamics of the lithium alkyl dicarbonate SEI components in contact with the lithium battery electrolyte. J. Phys. Chem. C 2014, 118, 18362–18371. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metalamorphous-semiconductor transition in germanium. Phys. Rev. B Condens. Matter 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter Mater. Phys. 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Ouatani, L.; Dedryvère, R.; Siret, C.; Biensan, P.; Reynaud, S.; Iratçabal, P.; Gonbeau, D. The Effect of Vinylene Carbonate Additive on Surface Film Formation on Both Electrodes in Li-Ion Batteries. J. Electrochem. Soc. 2009, 156, A103. [Google Scholar] [CrossRef]
- Matias, P.M.; Jeffrey, G.A.; Wingert, L.M.; Ruble, J.R. Single crystal neutron diffraction analysis (15 K) and ab initio molecular orbital calculations for ethylene carbonate. J. Mol. Struct. THEOCHEM 1989, 184, 247–260. [Google Scholar] [CrossRef]
- Aurbach, D.; Markovsky, B.; Shechter, A.; Ein-Eli, Y.; Cohen, H. A Comparative Study of Synthetic Graphite and Li Electrodes in Electrolyte Solutions Based on Ethylene Carbonate-Dimethyl Carbonate Mixtures. J. Electrochem. Soc. 2019, 143, 3809–3820. [Google Scholar] [CrossRef]
- Cao, Q.; Geng, X.; Wang, H.; Wang, P.; Liu, A.; Lan, Y.; Peng, Q. A review of current development of graphene mechanics. Crystals 2018, 8, 357. [Google Scholar] [CrossRef] [Green Version]
- Deng, B.; Hou, J.; Zhu, H.; Liu, S.; Liu, E.; Shi, Y.; Peng, Q. The normal-auxeticity mechanical phase transition in graphene. 2D Mater. 2017, 4, 021020. [Google Scholar] [CrossRef]
- Hou, J.; Deng, B.; Zhu, H.; Lan, Y.; Shi, Y.; De, S.; Liu, L.; Chakraborty, P.; Gao, F.; Peng, Q. Magic auxeticity angle of graphene. Carbon 2019, 149, 350–354. [Google Scholar] [CrossRef] [Green Version]
- Peng, Q.; Zamiri, A.R.; Ji, W.; De, S. Elastic properties of hybrid graphene/boron nitride monolayer. Acta Mech. 2012, 223, 2591–2596. [Google Scholar] [CrossRef]
- Peng, Q.; Liang, C.; Ji, W.; De, S. A theoretical analysis of the effect of the hydrogenation of graphene to graphane on its mechanical properties. Phys. Chem. Chem. Phys. 2013, 15, 2003–2011. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Chen, X.J.; Ji, W.; De, S. Chemically Tuning Mechanics of Graphene by BN. Adv. Eng. Mater. 2013, 15, 718–727. [Google Scholar] [CrossRef]
- Peng, Q.; Han, L.; Lian, J.; Wen, X.; Liu, S.; Chen, Z.; Koratkar, N.; De, S. Mechanical degradation of graphene by epoxidation: Insights from first-principles calculations. Phys. Chem. Chem. Phys. 2015, 17, 19484–19490. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Peng, Q.; Dai, Y.; Qian, Z.; Liu, S. Temperature dependence of Raman spectra of graphene on copper foil substrate. J. Mater. Sci. Mater. Electron. 2016, 27, 3888–3893. [Google Scholar] [CrossRef]
- Shi, T.; Peng, Q.; Bai, Z.; Gao, F.; Jovanovic, I. Proton irradiation of graphene: Insights from atomistic modeling. Nanoscale 2019, 11, 20754–20765. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, S.; Xie, L.; Zhu, P.; Li, R.; Peng, Q. Grain size and hydroxyl-coverage dependent tribology of polycrystalline graphene. Nanotechnology 2019, 30, 385701. [Google Scholar] [CrossRef]
- Mackay, A.L. Essentials of Crystallography Oxford Blackwell Scientific Publications 1986. viii+435 pp. Price: Cloth £25·00, limp £12·95; McKie, D., McKie, C., Eds.; Blackwell Science: Oxford, UK, 1988; Volume 52. [Google Scholar]
- Jia, X.; Hofmann, M.; Meunier, V.; Sumpter, B.G.; Campos-Delgado, J.; Romo-Herrera, J.M.; Son, H.; Hsieh, Y.P.; Reina, A.; Kong, J.; et al. Controlled formation of sharp zigzag and armchair edges in graphitic nanoribbons. Science 2009, 323, 1701–1705. [Google Scholar] [CrossRef]
- Enoki, T.; Kobayashi, Y.; Fukui, K.I. Electronic structures of graphene edges and nanographene. Int. Rev. Phys. Chem. 2007, 26, 609–645. [Google Scholar] [CrossRef]
- Nakada, K.; Fujita, M.; Dresselhaus, G.; Dresselhaus, M.S. Edge state in graphene ribbons: Nanometer size effect and edge shape dependence. Phys. Rev. B Condens. Matter Mater. Phys. 1996, 54, 17954–17961. [Google Scholar] [CrossRef] [Green Version]
- Zaghib, K.; Nadeau, G.; Kinoshita, K. Influence of edge and basal plane sites on the electrochemical behavior of flake-like natural graphite for Li-ion batteries. J. Power Sources 2001, 97–98, 97–103. [Google Scholar] [CrossRef]
- Shkrob, I.A.; Zhu, Y.; Marin, T.W.; Abraham, D. Reduction of carbonate electrolytes and the formation of solid-electrolyte interface (SEI) in lithium-ion batteries. 1. Spectroscopic observations of radical intermediates generated in one-electron reduction of carbonates. J. Phys. Chem. C 2013, 117, 19255–19269. [Google Scholar] [CrossRef]
Figure 1.
(a) Atomic structure of EC molecule determined by the DFT method. In the figure, red, yellow, blue balls represent oxygen, carbon, hydrogen atoms, respectively. (b) The most energetically favorable atomic structure of CH2CH2OCO2Li. In the figure, red, yellow, blue, and white balls represent oxygen, carbon, hydrogen, and lithium atoms, respectively. (c) gauche, (d) cis, (e) trans, (f) twisted cis, (g) twisted trans configuration of (CH2OCO2Li)2. (h) The two arrows form the lattice vectors for the primitive unit cell on the basal plane of graphite. (i) The side-view of atomic structure of the bulk graphite. (j) The schematic plot of basal surface, armchair edge (dotted line), and zigzag edge (dashed line).
Figure 1.
(a) Atomic structure of EC molecule determined by the DFT method. In the figure, red, yellow, blue balls represent oxygen, carbon, hydrogen atoms, respectively. (b) The most energetically favorable atomic structure of CH2CH2OCO2Li. In the figure, red, yellow, blue, and white balls represent oxygen, carbon, hydrogen, and lithium atoms, respectively. (c) gauche, (d) cis, (e) trans, (f) twisted cis, (g) twisted trans configuration of (CH2OCO2Li)2. (h) The two arrows form the lattice vectors for the primitive unit cell on the basal plane of graphite. (i) The side-view of atomic structure of the bulk graphite. (j) The schematic plot of basal surface, armchair edge (dotted line), and zigzag edge (dashed line).

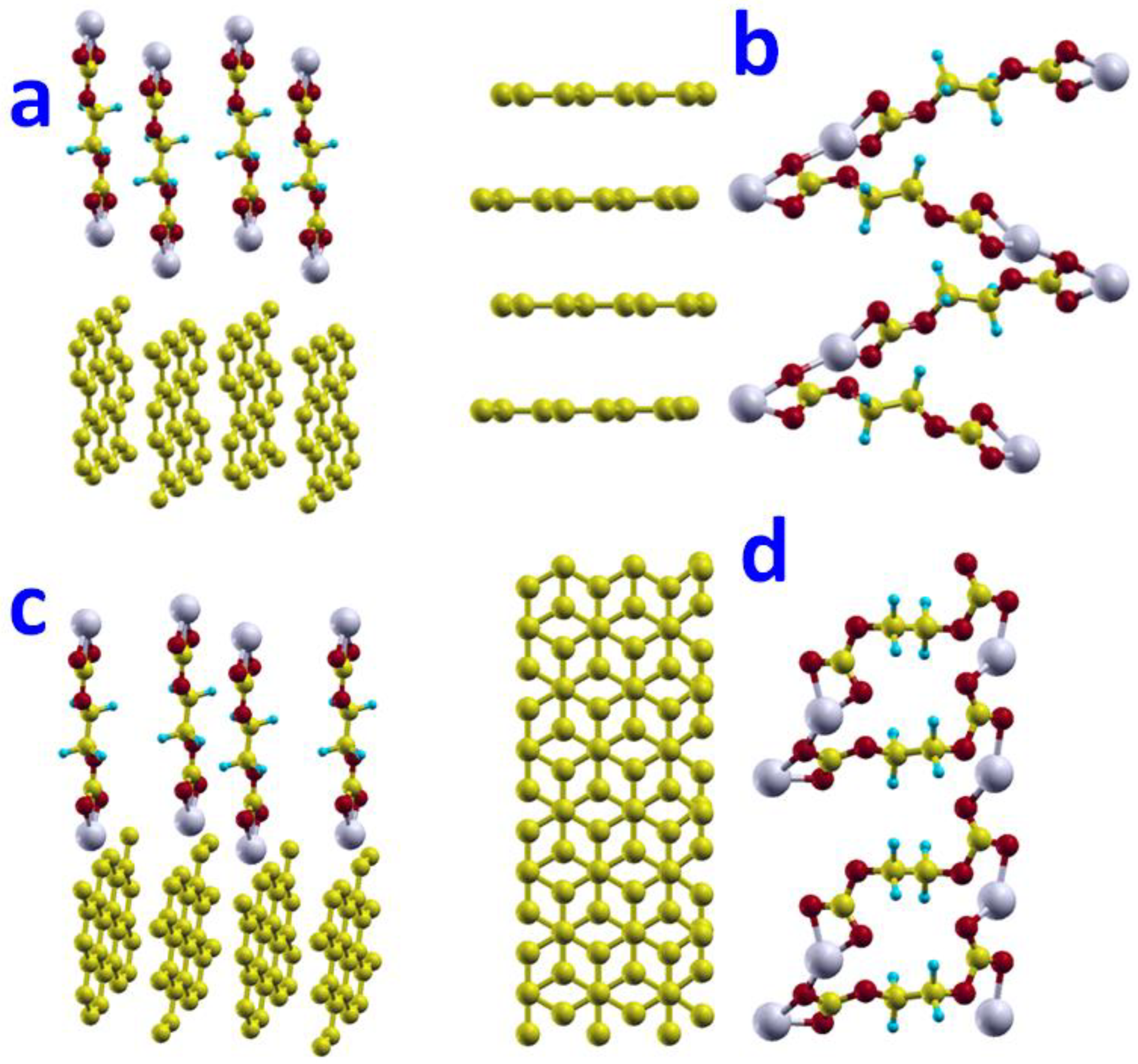
Figure 2.
Adsorptions of CH2CH2OCO2Li on the graphite surface with five orientations, ordered by their adsorption energy from lowest (a) to highest (c). Configuration (a) has the lowest total energy and strongest adsorption with adsorption energy of −0.24 eV on basal surface. Configuration (b) with the C1-O2-O3 plane parallel to the basal plane surface. Adsorptions of CH2CH2OCO2Li on the graphite edge surfaces (d) zigzag top-view (e) zigzag side-view and (f) armchair top-view (g) armchair side-view.
Figure 2.
Adsorptions of CH2CH2OCO2Li on the graphite surface with five orientations, ordered by their adsorption energy from lowest (a) to highest (c). Configuration (a) has the lowest total energy and strongest adsorption with adsorption energy of −0.24 eV on basal surface. Configuration (b) with the C1-O2-O3 plane parallel to the basal plane surface. Adsorptions of CH2CH2OCO2Li on the graphite edge surfaces (d) zigzag top-view (e) zigzag side-view and (f) armchair top-view (g) armchair side-view.
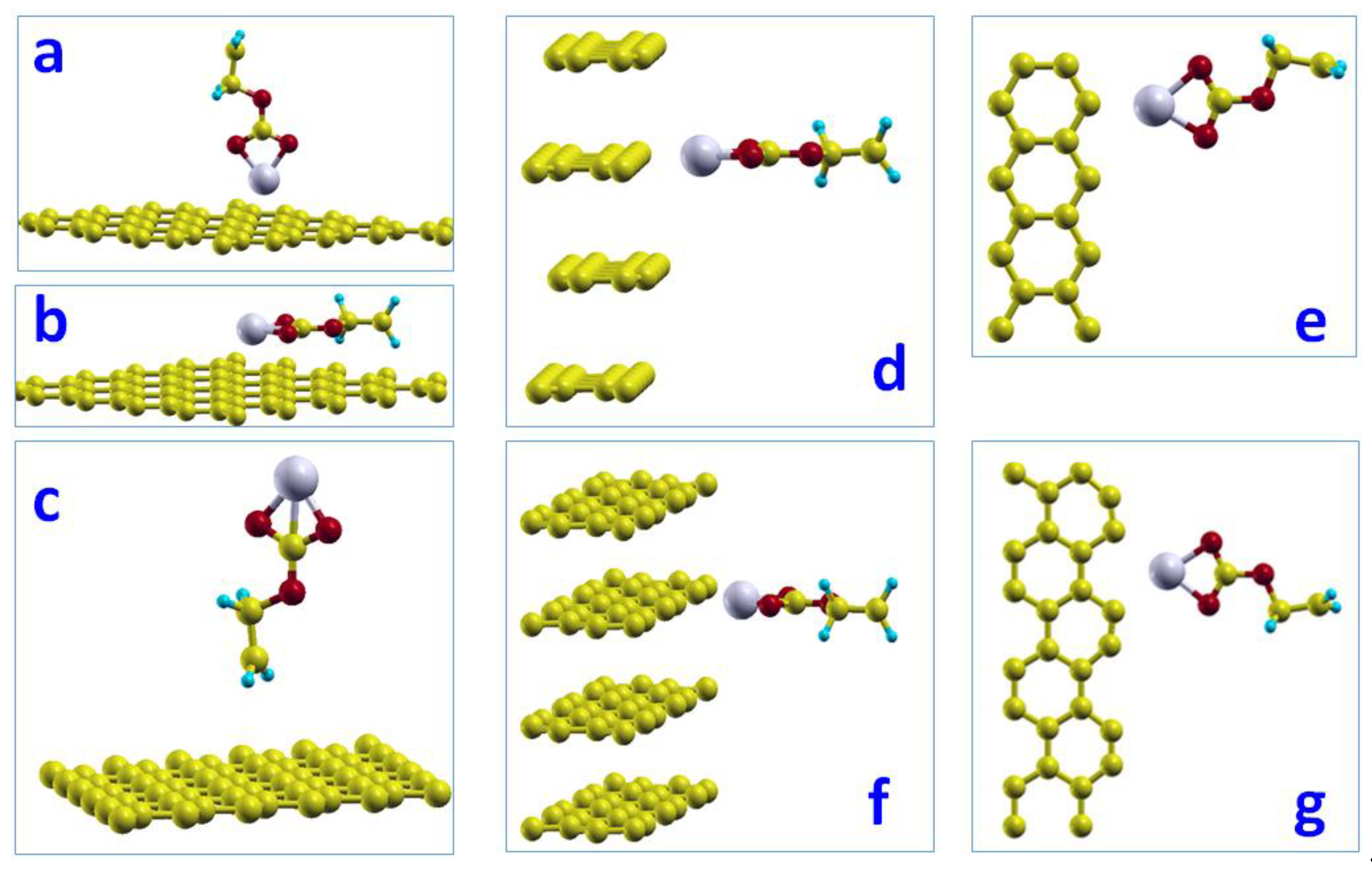
Figure 3.
Adsorption of (CH2OCO2Li)2 on graphite basal plane surface: (a) gauche where two lithium atoms are on Hollow-Hollow sites. (b) gauche where two lithium atoms are on Hollow-Top sites. (c) gauche where two lithium atoms are on Hollow-Bridge sites. (d) trans configuration.
Figure 3.
Adsorption of (CH2OCO2Li)2 on graphite basal plane surface: (a) gauche where two lithium atoms are on Hollow-Hollow sites. (b) gauche where two lithium atoms are on Hollow-Top sites. (c) gauche where two lithium atoms are on Hollow-Bridge sites. (d) trans configuration.

Figure 4.
Adsorptions of (CH2OCO2Li)2 on the graphite edge surfaces: (a) zigzag surface side view; (b) zigzag surface top view; (c) armchair surface side view; (d) armchair surface top view.
Figure 4.
Adsorptions of (CH2OCO2Li)2 on the graphite edge surfaces: (a) zigzag surface side view; (b) zigzag surface top view; (c) armchair surface side view; (d) armchair surface top view.
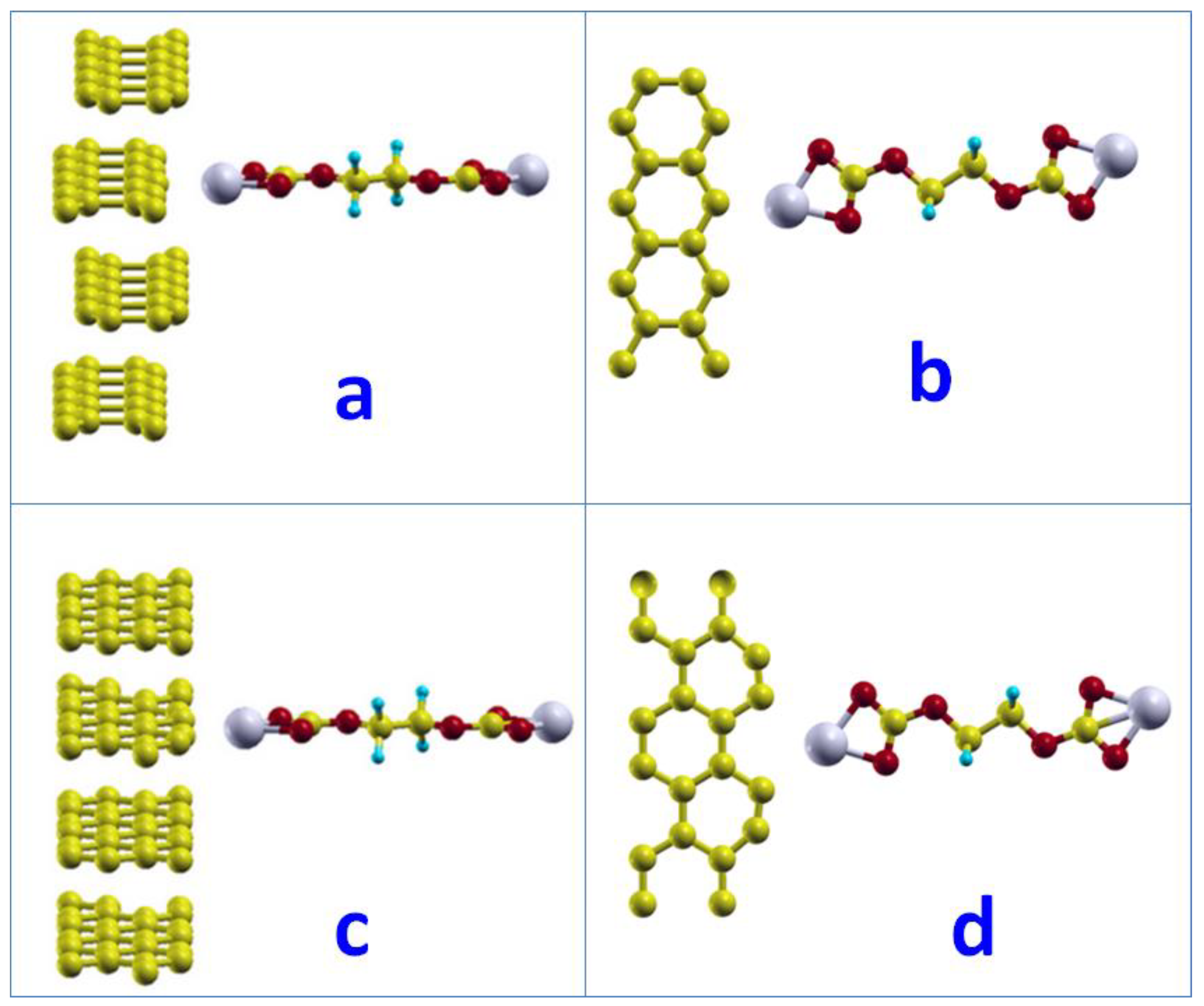
Figure 5.
The network of (CH2OCO2Li)2 molecules on basal surface of graphite: (a) initial structure (b) final structure.
Figure 5.
The network of (CH2OCO2Li)2 molecules on basal surface of graphite: (a) initial structure (b) final structure.
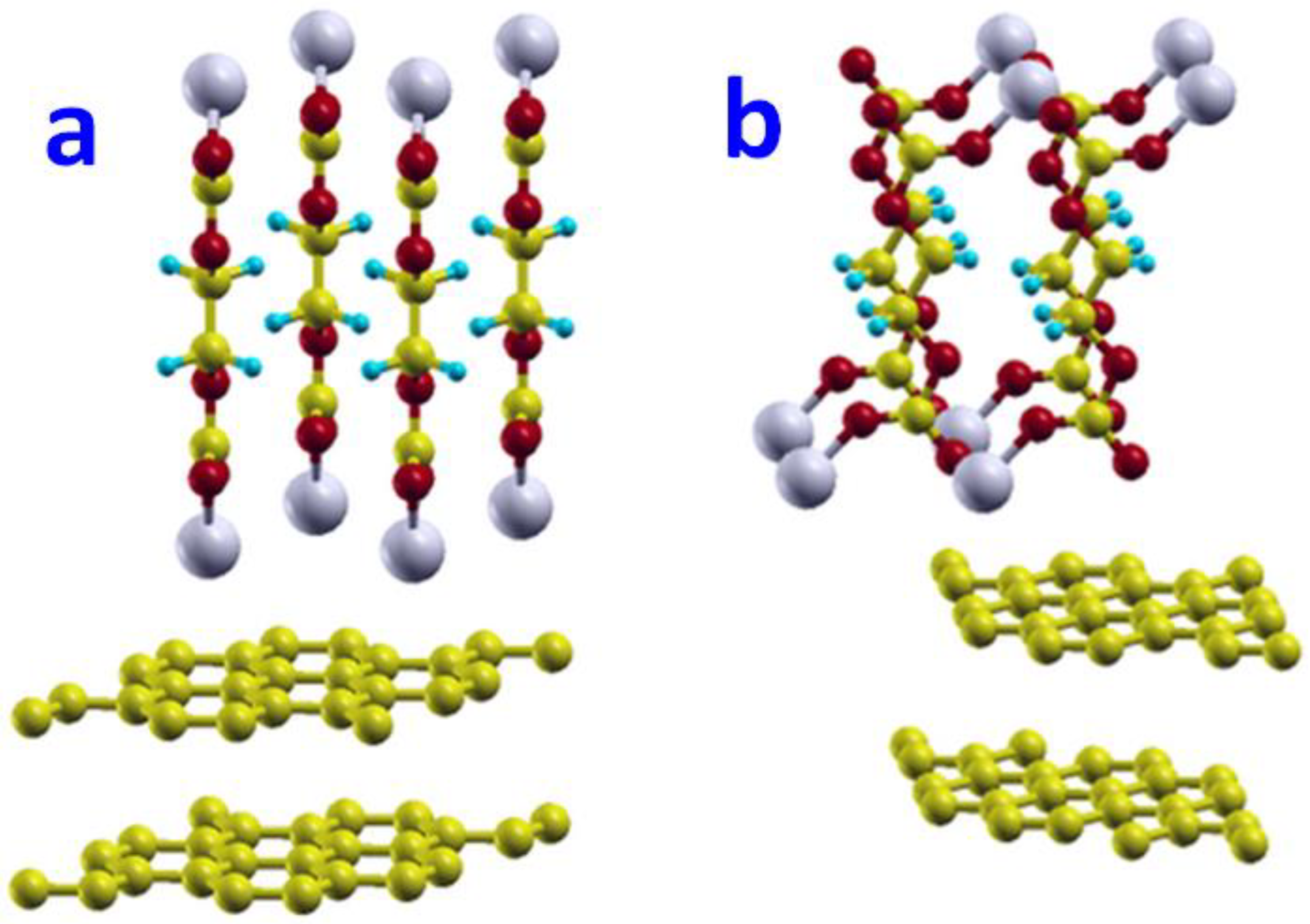
Figure 6.
The network of (CH2OCO2Li)2 on the zigzag (a,b) and armchair (c,d) edge surface. Initial structure (a) and final structure (b) of network of (CH2OCO2Li)2 on zigzag edge surface. Initial structure (c) and final structure (d) of network of (CH2OCO2Li)2 on armchair edge surface.
Figure 6.
The network of (CH2OCO2Li)2 on the zigzag (a,b) and armchair (c,d) edge surface. Initial structure (a) and final structure (b) of network of (CH2OCO2Li)2 on zigzag edge surface. Initial structure (c) and final structure (d) of network of (CH2OCO2Li)2 on armchair edge surface.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The results of the adsorption energies of CH2CH2OCO2Li and (CH2OCO2Li)2 on graphite basal, zigzag edge, armchair edge surfaces (units in eV).
Table 1.
The results of the adsorption energies of CH2CH2OCO2Li and (CH2OCO2Li)2 on graphite basal, zigzag edge, armchair edge surfaces (units in eV).
| Basal Surface | Zigzag Edge Surface | Armchair Edge Surface | |
|---|---|---|---|
| CH2CH2OCO2Li | −0.24 | −0.91 | −0.54 |
| (CH2OCO2Li)2 | −0.17 | −0.49 | −0.32 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Peng, Q. First-Principles Insights on the Formation Mechanism of Innermost Layers of Solid Electrolyte Interphases on Carbon Anodes for Lithium-Ion Batteries. Nanomaterials 2022, 12, 3654. https://doi.org/10.3390/nano12203654
AMA Style
Peng Q. First-Principles Insights on the Formation Mechanism of Innermost Layers of Solid Electrolyte Interphases on Carbon Anodes for Lithium-Ion Batteries. Nanomaterials. 2022; 12(20):3654. https://doi.org/10.3390/nano12203654
Chicago/Turabian StylePeng, Qing. 2022. "First-Principles Insights on the Formation Mechanism of Innermost Layers of Solid Electrolyte Interphases on Carbon Anodes for Lithium-Ion Batteries" Nanomaterials 12, no. 20: 3654. https://doi.org/10.3390/nano12203654
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.