
Activity Quantification of Fuel Cell Catalysts via Sequential Poisoning by Multiple Reaction Inhibitors

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
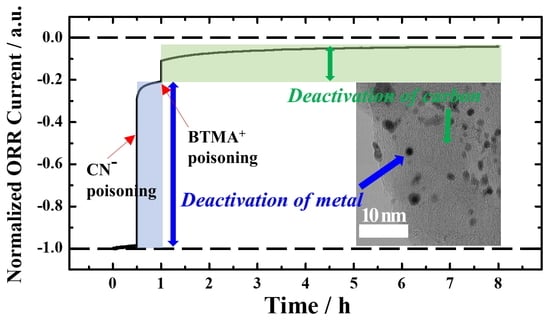
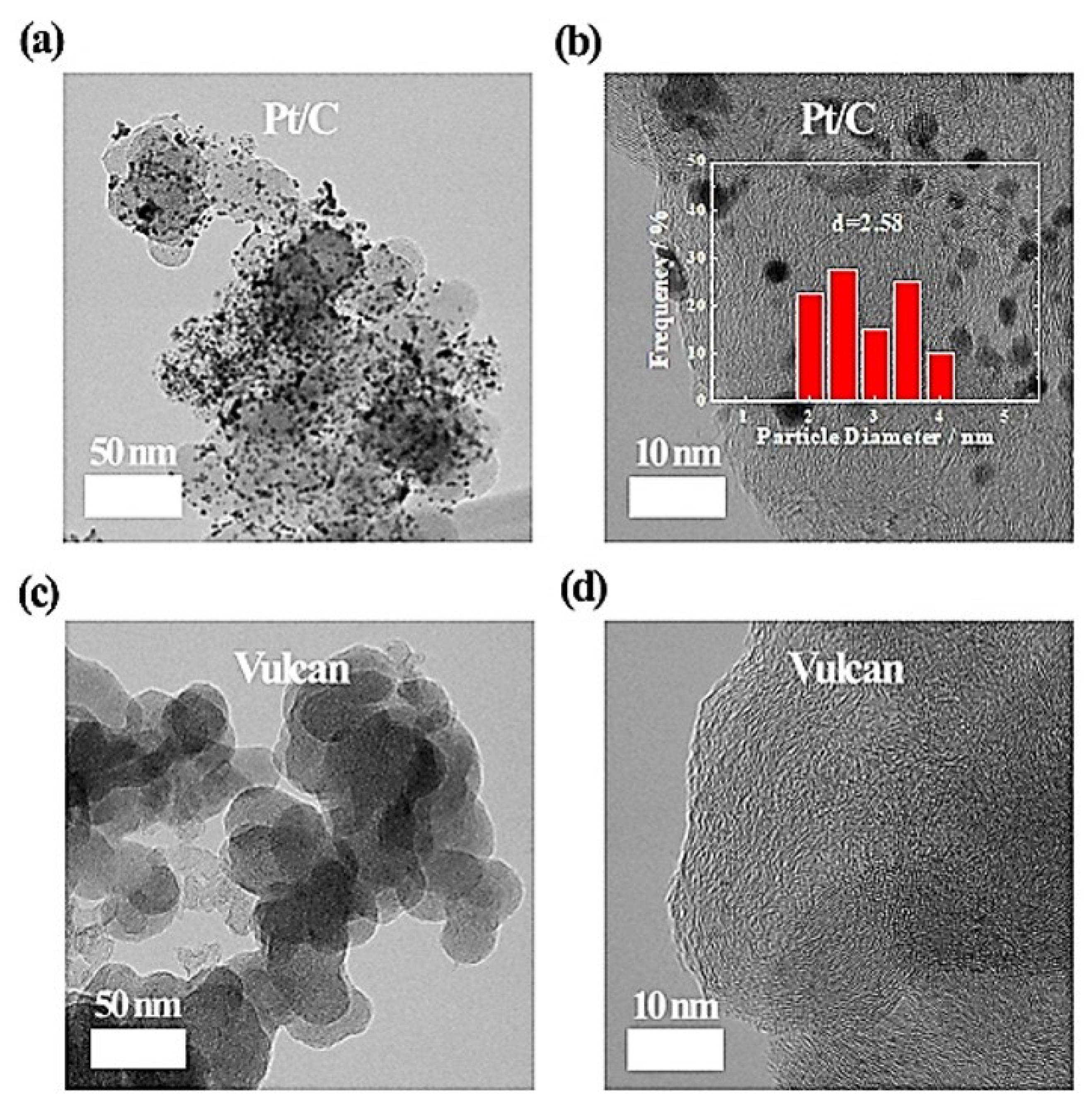
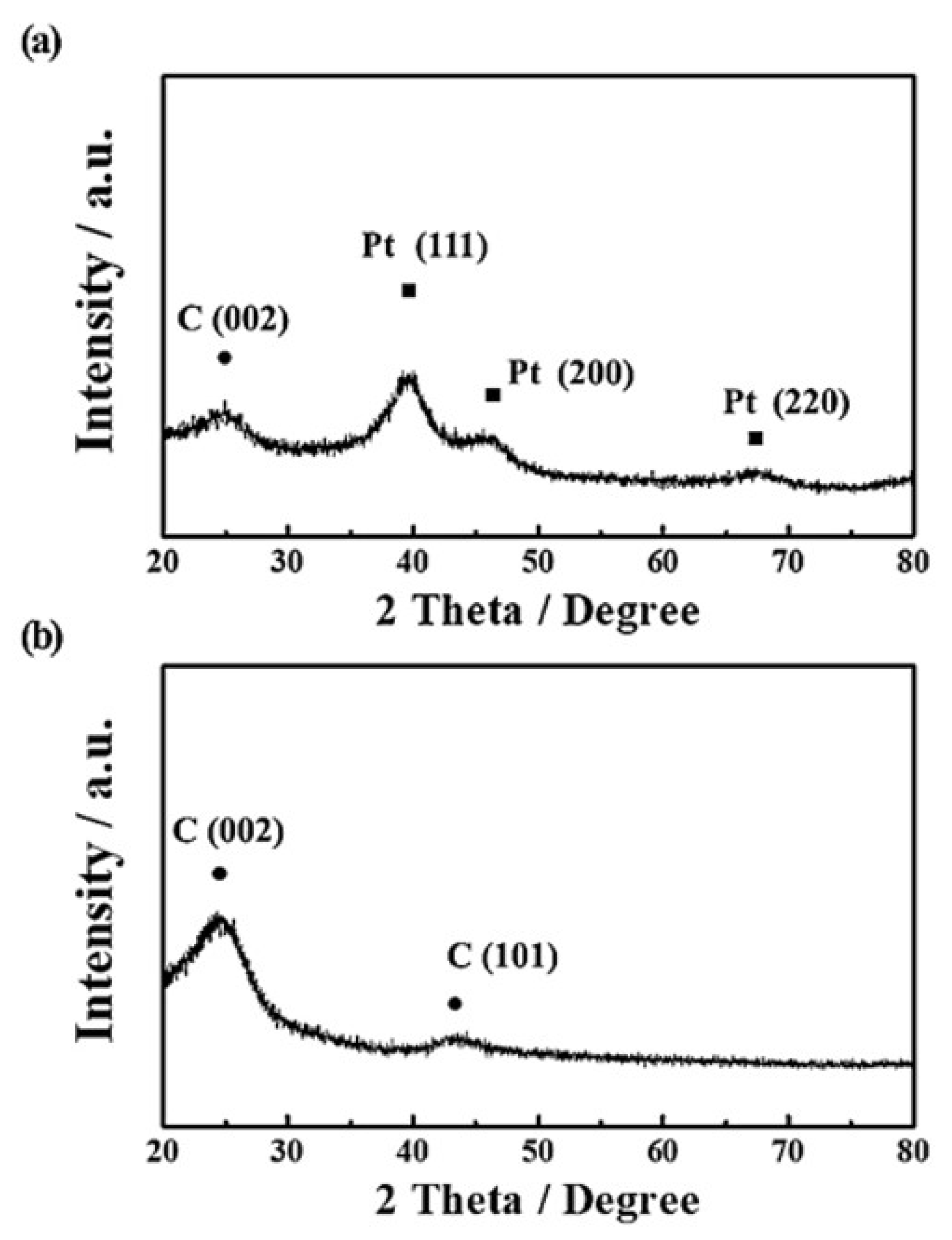
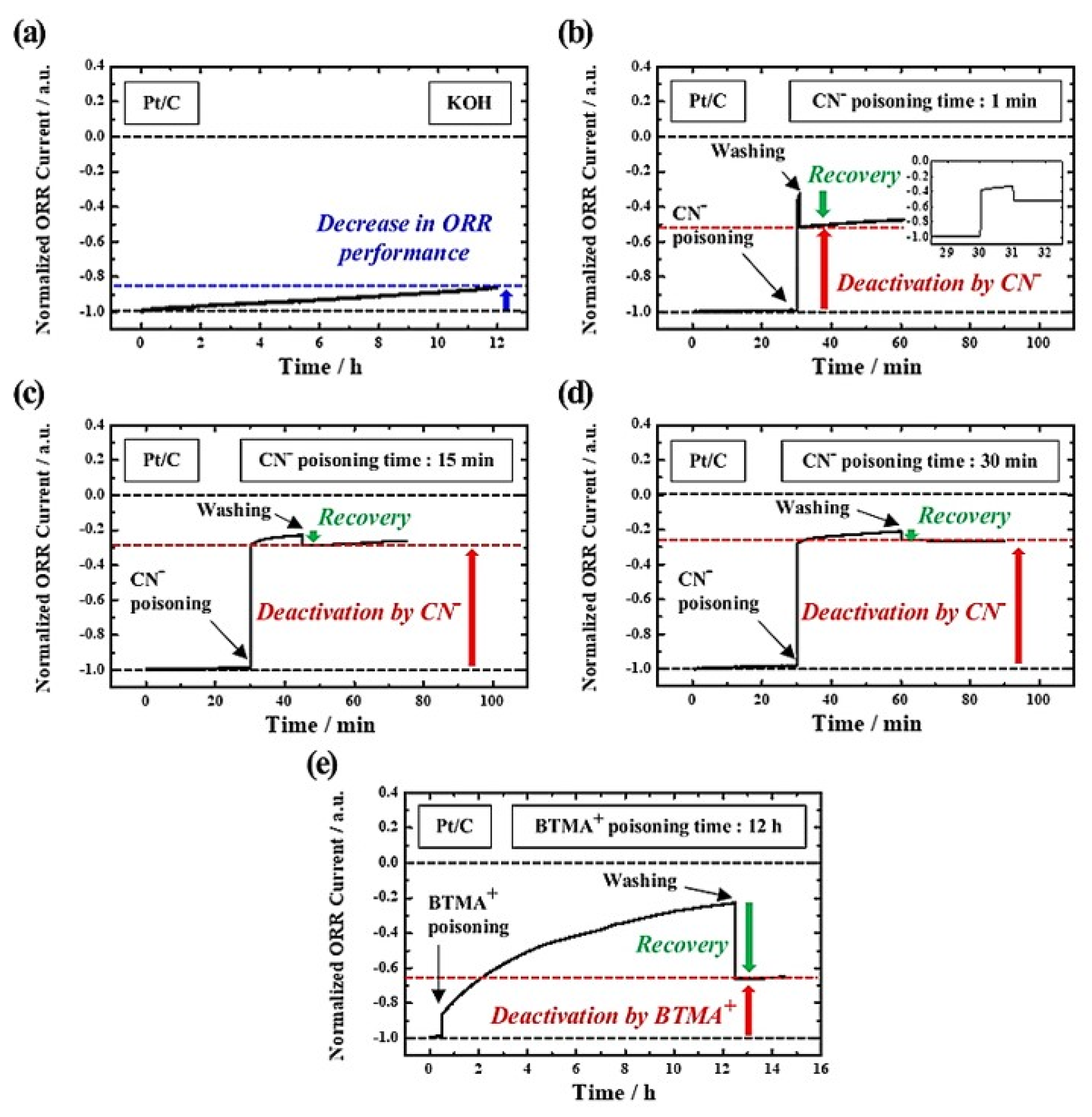
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sun, S.; Mitchell, S.; Li, J.; Lyu, P.; Wu, X.; Ramírez, J.P.; Lu, J. Design of Local Atomic Environments in Single-Atom Electrocatalysts for Renewable Energy Conversions. Adv. Mater. 2021, 33, 2003075. [Google Scholar] [CrossRef]
- Ferriday, T.B.; Middleton, P.H. Alkaline fuel cell technology. Int. J. Hydrogen Energy 2021, 46, 18489–18510. [Google Scholar] [CrossRef]
- Fan, L.; Tu, Z.; Chan, S.H. Recent development of hydrogen and fuel cell technologies: A review. Energy Rep. 2021, 7, 8421–8446. [Google Scholar] [CrossRef]
- Sharma, M.; Jung, N.; Yoo, S.J. Toward High-Performance Pt-Based Nanocatalysts for Oxygen Reduction Reaction through Organic−Inorganic Hybrid Concepts. Chem. Mater. 2018, 30, 2–24. [Google Scholar] [CrossRef]
- Shin, J.; Min, J.; Kim, Y.; Lee, J.H.; Chai, G.; Jung, N. Facile Strategy for Mass Production of Pt Catalysts for Polymer Electrolyte Membrane Fuel Cells Using Low-Energy Electron Beam. Nanomaterials 2020, 10, 2216. [Google Scholar] [CrossRef] [PubMed]
- Ko, K.; Min, J.; Kim, Y.; Hong, M.W.; Jeffery, A.A.; Chougule, S.S.; Yi, K.B.; Jung, N. Carbon Shell-Encapsulated Metal Alloy Catalysts with Pt-Rich Surfaces for Selective Hydrogen Oxidation Reaction. ChemElectroChem 2022, 9, e202200342. [Google Scholar] [CrossRef]
- Jang, J.; Sharma, M.; Choi, D.; Kang, Y.S.; Kim, Y.; Min, J.; Sung, H.; Jung, N.; Yoo, S.J. Boosting Fuel Cell Durability under Shut-Down/Start-Up Conditions Using a Hydrogen Oxidation-Selective Metal−Carbon Hybrid Core−Shell Catalyst. ACS Appl. Mater. Interfaces 2019, 11, 27735–27742. [Google Scholar] [CrossRef]
- Kim, Y.; Jeffery, A.A.; Min, J.; Jung, N. Modulating Catalytic Activity and Durability of PtFe Alloy Catalysts for Oxygen Reduction Reaction Through Controlled Carbon Shell Formation. Nanomaterials 2019, 9, 1491. [Google Scholar] [CrossRef] [Green Version]
- Min, J.; Kim, S.; Jeffry, A.A.; Shin, H.; Kang, Y.S.; Kim, Y.; Jang, J.; Lee, S.; Park, S.; Park, G.; et al. A paradigm shift in CO tolerant catalyst design for fuel cells via introducing defect-controlled carbon molecular sieve layers. Mater. Today Energy 2022, 29, 101124. [Google Scholar] [CrossRef]
- Sharma, M.; Jang, J.H.; Shin, D.Y.; Kwon, J.A.; Lim, D.H.; Choi, D.Y.; Sung, H.K.; Jang, J.H.; Lee, S.Y.; Lee, L.Y.; et al. Work function-tailored graphene via transition metal encapsulation as highly active and durable catalyst for oxygen reduction reaction. Energy Environ. Sci. 2019, 12, 2200–2211. [Google Scholar] [CrossRef]
- Chowdhury, S.R.; Ray, A.; Chougule, S.S.; Min, J.; Jeffery, A.A.; Ko, K.; Kim, Y.; Das, S.; Jung, N. Mixed Spinel Ni−Co Oxides: An Efficient Bifunctional Oxygen Electrocatalyst for Sustainable Energy Application. ACS Appl. Energy Mater. 2022, 5, 4421–4430. [Google Scholar] [CrossRef]
- Jang, J.H.; Jeffery, A.A.; Min, J.; Jung, N.; Yoo, S.J. Emerging carbon shell-encapsulated metal nanocatalysts for fuel cell and water electrolysis. Nanoscale 2021, 13, 15116–15141. [Google Scholar] [CrossRef]
- Zhu, C.; Xu, Q.S.; Fu, S.; Wan, G.; Yang, C.; Yao, S.; Song, J.; Zhou, H.; Du, D.; Beckman, S.P.; et al. Hierarchically Porous M–N–C (M = Co and Fe) Single-Atom Electrocatalysts with Robust MNx Active Moieties Enable Enhanced ORR Performance. Adv. Energy Mater. 2018, 8, 1801956. [Google Scholar] [CrossRef]
- Hossen, M.M.; Artyushkova, K.; Atanassov, P.; Serov, A. Synthesis and characterization of high performing Fe-N-C catalyst foroxygen reduction reaction (ORR) in Alkaline Exchange Membrane Fuel Cells. J. Power Sources 2018, 375, 214–221. [Google Scholar] [CrossRef]
- Liu, X.; Zheng, L.; Han, C.; Zong, H.; Yang, G.; Lin, S.; Kumar, A.; Jadhav, A.R.; Tran, N.Q.; Hwang, Y.; et al. Identifying the Activity Origin of a Cobalt Single-Atom Catalyst for Hydrogen Evolution Using Supervised Learning. Adv. Funct. Mater. 2021, 31, 2100547. [Google Scholar] [CrossRef]
- Kumar, A.; Vashistha, V.K.; Das, D.K.; Ibraheem, S.; Yasin, G.; Iqbal, R.; Nguyen, T.A.K.; Gupta, R.; Islam, R. M-N-C-based single-atom catalysts for H2, O2 & CO2 electrocatalysis: Activity descriptors, active sites identification, challenges and prospects. Fuel 2021, 304, 121420. [Google Scholar]
- Shah, S.S.A.; Najam, T.; Bashir, M.S.; Javed, M.S.; Rahman, A.; Luque, R.; Bao, S.J. Identification of Catalytic Active Sites for Durable Proton Exchange Membrane Fuel Cell: Catalytic Degradation and Poisoning Perspectives. Small 2022, 18, 2106279. [Google Scholar] [CrossRef]
- Zúñiga, C.; Candia-Onfray, C.; Venegas, R.; Muñozb, K.; Urra, J.; Sánchez-Arenillasc, M.F.; Marcoc, J.H.; Zagald, J.J.; Recio, F. Elucidating the mechanism of the oxygen reduction reaction for pyrolyzed Fe-N-C catalysts in basic media. Electrochem. Commun. 2019, 102, 78–82. [Google Scholar] [CrossRef]
- Guo, D.; Shibuya, R.; Akiba, C.; Saji, S.; Kondo, T.; Nakamura, J. Active sites of nitrogen-doped carbon materials for oxygen reduction reaction clarified using model catalysts. Electrochemistry 2016, 351, 6271. [Google Scholar] [CrossRef]
- Jain, D.; Zhang, Q.; Gustin, V.; Hightower, J.; Co, S.G.A.C.; Miller, J.T.; Asthagirl, A.; Ozkan, U.S. Experimental and DFT Investigation into Chloride Poisoning Effects on Nitrogen-Coordinated Iron−Carbon (FeNC) Catalysts for Oxygen Reduction Reaction. J. Phys. Chem. C 2020, 124, 10324–10335. [Google Scholar] [CrossRef]
- Zhang, X.; Lu, Z.; Yang, Z. Single non-noble-metal cobalt atom stabilized by pyridinic vacancygraphene: An efficient catalyst for CO oxidation. J. Mol. Catal. A Chem. 2016, 417, 28–35. [Google Scholar] [CrossRef]
- Ficca, V.C.A.; Santoro, C.; D’Epifanio, A.; Licoccia, S.; Serov, A.; Atanassov, P.; Mecheri, B. Effect of Active Site Poisoning on Iron-Nitrogen-Carbon Platinum-Group-Metal-Free Oxygen Reduction Reaction Catalysts Operating in Neutral Media: A Rotating Disk Electrode Study. ChemElectroChem 2020, 7, 3044–3055. [Google Scholar] [CrossRef]
- Thorum, M.S.; Hankett, J.M.; Gewirth, A.A. Poisoning the Oxygen Reduction Reaction on Carbon-Supported Fe and Cu Electrocatalysts: Evidence for Metal-Centered Activity. J. Phys. Chem. Lett. 2011, 2, 295–298. [Google Scholar] [CrossRef]
- Chen, Y.; Artyushkova, K.; Rojas-Carbonell, S.; Serov, A.; Matanovic, I.; Santoro, C.; Asset, T.; Atanassov, P. Inhibition of Surface Chemical Moieties by Tris(hydroxymethyl)aminomethane: A Key to Understanding Oxygen Reduction on Iron−Nitrogen−Carbon Catalysts. ACS Appl. Energy Mater. 2018, 1, 1942–1949. [Google Scholar] [CrossRef]
- Ge, X.; Sumboja, A.; Wuu, D.; An, T.; Li, B.; Thomas Goh, F.W.; Andy Hor, T.S.; Zong, Y.; Liu, Z. Oxygen Reduction in Alkaline Media: From Mechanisms to Recent Advances of Catalysts. ACS Catal. 2015, 5, 4643–4667. [Google Scholar] [CrossRef]
- Pana, Z.F.; Ana, L.; Zhaob, T.S.; Tang, Z.K. Advances and challenges in alkaline anion exchange membrane fuel cells. Prog. Energy Combust. Sci. 2018, 66, 141–175. [Google Scholar] [CrossRef]
- Malko, D.; Kucernak, A.; Lopes, T. In situ electrochemical quantification of active sites in Fe–N/C non-precious metal catalysts. Nat. Commun. 2016, 7, 13285. [Google Scholar] [CrossRef] [Green Version]
- Chung, M.W.; Choi, C.H. Carbon nanofibers as parent materials for a graphene-based Fe-N-C catalyst for the oxygen reduction reaction. Catal. Today 2017, 295, 125–131. [Google Scholar] [CrossRef]
- Tylus, U.; Jia, Q.; Strickland, K.; Ramaswamy, N.; Serov, A.; Atanassov, P.; Mukerjee, S. Elucidating Oxygen Reduction Active Sites in Pyrolyzed Metal-Nitrogen Coordinated Non-Precious-Metal Electrocatalyst Systems. J. Phys. Chem. C 2014, 118, 8999–9008. [Google Scholar] [CrossRef]
- Chung, M.W.; Chon, G.; Kim, H.; Jaouen, F.R.D.; Choi, C.H. Electrochemical Evidence for Two Sub-families of FeNxCy Moieties with Concentration-Dependent Cyanide Poisoning. ChemElectroChem 2018, 5, 1880–1885. [Google Scholar] [CrossRef]
- Maa, Y.; Wanga, H.; Ji, S.; Gohb, J.; Fengc, H.; Wang, R. Highly active Vulcan carbon composite for oxygen reduction reaction in alkaline medium. Electrochim. Acta 2014, 133, 391–398. [Google Scholar] [CrossRef]
- Carmo, M.; Santos, A.R.D.; Poco, J.G.R.; Linardi, M. Physical and electrochemical evaluation of commercial carbon black as electrocatalysts supports for DMFC applications. J. Power Sources 2007, 173, 860–866. [Google Scholar] [CrossRef]
- Jang, S.J.; Kang, Y.C.; Hyun, J.S.; Shin, T.H.; Lee, Y.W.; Roh, K.C. Hybrid Structure of TiO2-Graphitic Carbon as a Support of Pt Nanoparticles for Catalyzing Oxygen Reduction Reaction. Catalysts 2021, 11, 1196. [Google Scholar] [CrossRef]
- Maiyalagan, T.; Khan, F.N. Electrochemical oxidation of methanol on Pt/V2O5–C composite catalysts. Catal. Commun. 2009, 10, 433–436. [Google Scholar] [CrossRef]
- Benzie, J.W.; Bakhmutov, V.I.; Blumel, J. Benzene Adsorbed on Activated Carbon: A Comprehensive Solid- State Nuclear Magnetic Resonance Study of Interactions with the Pore Surface and Molecular Motions. J. Phys. Chem. C 2020, 124, 21532–21537. [Google Scholar] [CrossRef]
- Han, Y.; Quan, X.; Chen, S.; Zhao, H.; Cui, C.; Zhao, Y. Electrochemically enhanced adsorption of phenol on activated carbon fibers in basic aqueous solution. J. Colloid Interface Sci. 2006, 299, 766–771. [Google Scholar] [CrossRef]
- Dumana, O.; Ayrancib, E. Adsorptive removal of cationic surfactants from aqueous solutions onto high-area activated carbon cloth monitored by in situ UV spectroscopy. J. Hazard. Mater. 2010, 174, 359–367. [Google Scholar] [CrossRef]
- Friedrich, K.A.; Daum, W.; Kliinker, C.; Knabben, D.; Stimming, U.; Ibach, H. In-situ spectroscopy of cyanide vibrations on Pt(lll) and Pt(ll0) electrode surfaces: Potential dependencies and the influence of surface disorder. Surf. Sci. 1995, 335, 315–325. [Google Scholar] [CrossRef]
- Cuesta, A.; Escudero, M.; Lanova, B.; Baltruschat, H. Cyclic Voltammetry, FTIRS, and DEMS Study of the Electrooxidation of Carbon Monoxide, Formic Acid, and Methanol on Cyanide-Modified Pt(111) Electrodes. Langmuir 2009, 25, 6500–6507. [Google Scholar] [CrossRef]
- Escudero-Escribano, E.J.; Soldano, G.; Quaino, P.; Michoff, M.E.Z.; Leiva, E.P.M.; Schmickler, W.; Cuesta, A. Cyanide-modified Pt(1 1 1): Structure, stability and hydrogen adsorption. Electrochim. Acta 2012, 82, 524–533. [Google Scholar] [CrossRef] [Green Version]
- Ample, F.; Clotet, A.M.; Ricart, J. Structure and bonding mechanism of cyanide adsorbed on Pt(1 1 1). Surf. Sci. 2004, 558, 111–121. [Google Scholar] [CrossRef]
- Matanovic, I.; Chung, H.T.; Kim, Y.S. Benzene Adsorption: A Significant Inhibitor for the Hydrogen Oxidation Reaction in Alkaline Conditions. J. Phys. Chem. Lett. 2017, 8, 4918–4924. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, N.; Nakamura, M. Hotptsuyanagi. Active sites for the oxygen reduction reaction on the high indexplanes of Pt. ElectroChimica Acta 2013, 112, 899–904. [Google Scholar] [CrossRef]
- Perez-Alonso, F.J.; McCarthy, D.N.; Nierhoff, A.; Hernandez-Fernandez, P.; Strebel, C.; Stephens, I.E.L.; Nielsen, J.H.; Chorken dorff, I. The Effect of Size on the Oxygen Electroreduction Activity of Mass-Selected Platinum Nanoparticles. Angew. Chem. Int. Ed. 2012, 51, 4641–4643. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, E.; Taylor, S.; Rabis, A.; Levecque, P.; Conrad, O.; Kçtz, R.; Schmidt, T.J. The Effect of Platinum Nanoparticle Distribution on Oxygen Electroreduction Activity and Selectivity. ChemCatChem 2014, 6, 1410–1418. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.; Min, J.; Ko, K.; Sravani, B.; Chougule, S.S.; Choi, Y.; Choi, H.; Hong, S.; Jung, N. Activity Quantification of Fuel Cell Catalysts via Sequential Poisoning by Multiple Reaction Inhibitors. Nanomaterials 2022, 12, 3800. https://doi.org/10.3390/nano12213800
Kim Y, Min J, Ko K, Sravani B, Chougule SS, Choi Y, Choi H, Hong S, Jung N. Activity Quantification of Fuel Cell Catalysts via Sequential Poisoning by Multiple Reaction Inhibitors. Nanomaterials. 2022; 12(21):3800. https://doi.org/10.3390/nano12213800
Chicago/Turabian StyleKim, Yunjin, Jiho Min, Keonwoo Ko, Bathinapatla Sravani, Sourabh S. Chougule, Yoonseong Choi, Hyeonwoo Choi, SeoYeong Hong, and Namgee Jung. 2022. "Activity Quantification of Fuel Cell Catalysts via Sequential Poisoning by Multiple Reaction Inhibitors" Nanomaterials 12, no. 21: 3800. https://doi.org/10.3390/nano12213800