



Low-Temperature Predicted Structures of Ag2S (Silver Sulfide)
by
 , and
, and
Stanislav I. Sadovnikov
1,
Maksim G. Kostenko
1,
Aleksandr I. Gusev
1,* and
Aleksey V. Lukoyanov
2,3
1
Institute of Solid State Chemistry, Ural Branch of the Russian Academy of Sciences, 620990 Ekaterinburg, Russia
2
Mikheev Institute of Metal Physics, Ural Branch of the Russian Academy of Sciences, 620016 Ekaterinburg, Russia
3
Institute of Physics and Technology, Ural Federal University named after the first President of Russia B. N. Yeltsin, 620002 Ekaterinburg, Russia
*
Author to whom correspondence should be addressed.
Nanomaterials 2023, 13(19), 2638; https://doi.org/10.3390/nano13192638
Submission received: 21 August 2023
/
Revised: 18 September 2023
/
Accepted: 22 September 2023
/
Published: 25 September 2023
(This article belongs to the Topic Modeling, Fabrication, and Characterization of Semiconductor Materials and Devices)
Abstract
:Silver sulfide phases, such as body-centered cubic argentite and monoclinic acanthite, are widely known. Traditionally, acanthite is regarded as the only low-temperature phase of silver sulfide. However, the possible existence of other low-temperature phases of silver sulfide cannot be ruled out. Until now, there have been only a few suggestions about low-temperature Ag2S phases that differ from monoclinic acanthite. The lack of a uniform approach has hampered the prediction of such phases. In this work, the use of such an effective tool as an evolutionary algorithm for the first time made it possible to perform a broad search for the model Ag2S phases of silver sulfide, which are low-temperature with respect to cubic argentite. The possibility of forming Ag2S phases with cubic, tetragonal, orthorhombic, trigonal, monoclinic, and triclinic symmetry is considered. The calculation of the cohesion energy and the formation enthalpy show, for the first time, that the formation of low-symmetry Ag2S phases is energetically most favorable. The elastic stiffness constants cij of all predicted Ag2S phases are computed, and their mechanical stability is determined. The densities of the electronic states of the predicted Ag2S phases are calculated. The prediction of low-temperature Ag2S structures indicates the possibility of synthesizing new silver sulfide phases with improved properties.
1. Introduction
Silver sulfide has three phases: low-temperature monoclinic (space group P21/c) α-Ag2S (acanthite) exists at temperatures below 450 K, body-centered cubic (space group ) superionic β-Ag2S (argentite) exists in the temperature range of 452–860 K, and the high-temperature face-centered cubic γ-Ag2S phase is stable at T > 860 K [1,2]. When bcc β-Ag2S (argentite) is cooled below 450 K, a phase transition occurs with the formation of monoclinic α-Ag2S (acanthite) [3,4]. In cubic argentite (Ag2±δS) S with δ ≈ 0.002, both a slight deficit and a slight excess of silver can be observed. Bulk monoclinic α-Ag2S (acanthite) is stoichiometric. Silver sulfide nanoparticles with a size of less than ~50 nm is nonstoichiometric. Its composition is ~Ag1.93S due to vacant sites in the metal sublattice [2,5].
According to [2,6,7], the structure of α-Ag2S (acanthite) is a result of the distortion of the bcc sublattice of sulfur atoms (S) in the structure of β-Ag2S (argentite). Indeed, the unit cell of monoclinic (space group P21/c) α-Ag2S (acanthite) proposed in a previous study [6] has axes that are the combinations of the axes abcc, bbcc, and cbcc of the unit cell of bcc argentite.
The unusual physical and structural properties of the high-temperature phases, namely the enhanced ionic conductivity, the liquid-like behavior of silver sublattice [8,9], the uncertainty in the positions of silver atoms [10], and the alleged ordering, have always raised questions.
The transformation of “argentite–acanthite” is accompanied by a distortion of the bcc sublattice of S atoms to a monoclinic sublattice and displacements of sulfur and silver atoms [11]. The transformation of bcc argentite into low-temperature monoclinic acanthite can be considered a structural ordering [12,13]. It was noted in a previous study [12] devoted to the calculation and experimental determination of the Raman spectra of silver sulfide that the transformation of monoclinic silver sulfide α-Ag2S into superionic bcc argentite (β-Ag2S) is associated with disordering in the metal sublattice. As the temperature decreases below the transition temperature Ttrans, the S atoms, which occupied the sites of the bcc nonmetallic sublattice of argentite with equal probability, move to four sites of the monoclinic nonmetallic sublattice, leaving the remaining sites vacant (Figure 1). This relationship between the structures of α-Ag2S and β-Ag2S also raises questions regarding the existence of other ordered phases based on high-temperature β-Ag2S with the disordered silver sublattice.
This makes it possible to consider the formation of a monoclinic cell as an ordering in the bcc sublattice of sulfur atoms. Note that, in the bcc metal sublattice of argentite, upon transformation into acanthite, a peculiar ordering also occurs [14]. Indeed, silver atoms, which are randomly located with a probability of less than 0.1 in two cubic unit cells at 54 crystallographic positions of argentite, in acanthite occupy eight sites of the metal sublattice of a monoclinic unit cell with a probability of 1.
As shown earlier [13], the acanthite structure can be obtained from the argentite structure as a result of the disorder–order transition. In particular, an attempt to consider the variant of argentite ordering with the formation of a monoclinic (space group P21) Ag2S phase in which the Ag atoms are in a tetrahedral environment of four S atoms was made by Kashida et al. [15]. These authors considered three hypothetical models of structural ordering for the high-temperature argentite phase, in which cooperative ionic transport is possible. These models were only used for electronic structure calculations, and an evaluation of their thermodynamic stability has not been provided. We assumed that, in addition to acanthite, other phases of Ag2S can form in silver sulfide, especially nanocrystalline, with a decrease in temperature.
The Open Quantum Materials Database (OQMD) [16] and Materials Project databases [17] contain many other hypothetical structures of Ag2S which, apparently, were simulated on the basis of structural similarity using silver chalcogenides or related systems. Both resources consider the monoclinic (space group P21) structure proposed by Kashida et al. [15] as the best low-temperature model for silver sulfide, rather than the experimental acanthite phase. Thus, the real number of structures related exactly to Ag2S, as well as the anticipated sequences of phase transitions, remains unknown.
Theoretical data on the structure of several model Ag2S phases with triclinic, monoclinic, and orthorhombic symmetry [18,19,20,21,22,23,24,25] were calculated earlier within the framework of materials projects using the ELATE code proposed in [26]. However, the data [15,18,19,20,21,22,23,24,25] are extremely limited, often contradictory, and not entirely reliable. There is no information at all in the literature about possible highly symmetrical cubic and tetragonal Ag2S phases. To fill the noted gap, in this study, phases with cubic, tetragonal, orthorhombic, trigonal, monoclinic, and triclinic symmetry were considered as possible model phases of silver sulfide. The prediction of possible model phases of silver sulfide, which have a structure different from that of monoclinic acanthite, is important for obtaining the sulfide heteronanostructures of the Ag2S/ZnS type with a controlled band gap [27]. The band gap of the possible model phases of silver sulfide can be different from that of acanthite, which will expand the potential application of Ag2S/ZnS heterostructures.
Interest in the discussed low-temperature Ag2S polymorphs is due to the possibility of their wide application. All forms of silver sulfide (Ag2S) have attracted much attention [2,7,28]. Monoclinic silver sulfide (α-Ag2S) is a semiconductor at temperatures <~420–450 K, and body-centered cubic sulfide (β-Ag2S) exhibits superionic conductivity at temperatures greater than 452 K. Nanocrystalline silver sulfide is a versatile semiconductor for use in various optoelectronic devices, such as photocells, photoconductors, and infrared sensors [2,29,30]. The use of nanocrystalline silver sulfide is promising for the creation of Ag2S/Ag heteronanostructures intended for use in memory devices and resistance switches. Their action is based on the transformation of α-Ag2S (acanthite) into β-Ag2S (argentite) and the formation of a conducting channel between silver (Ag) and superionic β-Ag2S (argentite) [31,32,33,34]. The band gap of the predicted low-temperature Ag2S phases may be different from that of acanthite, which will expand the potential applications of silver sulfide.
Indeed, silver sulfide (Ag2S), along with lead, zinc, copper, cadmium, and mercury sulfides, is among the most in-demand semiconductors. This is an excellent material for the fabrication of heterostructures [35], which can also be used in solar cells [36], IR detectors [29,37], resistive switches, and nonvolatile memory devices [31,32,33,34]. Besides, Ag2S is promising for solar energy conversion into electricity [38]. The application of semiconducting nanostructured silver sulfide in biology and medicine as a biosensor is based on the quantum size effects that influence the optical properties of the material. Semiconducting Ag2S/noble metal heterostructures are treated as candidate materials for application in photocatalysis [39]. In particular, this is due to the narrow band gap of silver sulfide (about 0.9 eV).
The main fields of application of silver sulfide include microelectronics, biosensorics, and catalysis.
High-performance atomic resistive switches are a promising type of nonvolatile memory devices; here, read/write operations are implemented involving ion exchange [40]. In the case of cation migration, silver and silver sulfide heteronanostructures are the most widely used [31,32,33,34]. The Ag2S/Ag heteronanostructures combine the sulfide (semiconductor or ionic conductor, depending on the structure) and silver (electronic conductor) [41].
The design of fluorescent labels (biolabels and biomarkers) based on Ag2S quantum dots for applications in biology and medicine seems to be quite promising [42,43,44,45]. Nanocrystalline silver sulfide and heteronanostructures based on this substance are treated as effective materials for catalysis and photocatalysis [39,46,47]. Nanocrystalline silver sulfide has also been considered an effective thermoelectric material [48]. Silver sulfide is of interest as a thermoelectric material, owing to the reversible transition between the monoclinic semiconducting (α-Ag2S) and cubic superionic (β-Ag2S) phases. This allows the thermoelectric effect in Ag2S to be realized near the phase transition temperature, with silver sulfide acting as thermoelement.
Thus, silver sulfide (Ag2S) has a lot of possible applications. The prediction of new low-temperature phases of silver sulfide (Ag2S) will considerably allow for the expansion of its potential use.
The model of ordering described in previous works [2,7] implied that the initial bcc sulfur sublattice of argentite should be distorted. However, one can suggest simpler ordering models which are only based on the redistribution and “freezing” of the silver atoms, without the change in the sulfur bcc sublattice.
In addition to the conceptual structural models for ordered silver sulfide phases, we intend to use the modern technique for crystal structure prediction. In order to eliminate the inconsistencies and incompleteness in the theoretical and experimental data, we performed an extensive theoretical study of the structural properties of silver sulfide. Using a modern technique for crystal structure prediction [49], we intended to find the correct candidates for the low-temperature phases of Ag2S to check whether there are phases competing with acanthite at zero temperature and pressure and to compare the found structures with those given in the Materials Project database. The details of the calculations and methods we used are described in Section 2. In Section 3, we analyze the stability, electronic structure, and mechanical properties of the predicted phases. We also provide an estimation of the elastic moduli and hardness of silver sulfide since the corresponding experimental data were not available in the literature.
For the energy characterization of the model Ag2S phases, their cohesion energies and formation enthalpies were calculated, and the calculated elastic stiffness constants cij were used to evaluate the mechanical stability of the suggested and predicted phases. The variation of the formation enthalpies ΔHf of some of the predicted Ag2S phases with a reduction in symmetry is shown as an example in Figure 2.
2. Computational Simulations
When modeling the Ag2S phases, it was taken into account as one of the conditions that the nearest distances between Ag atoms should be greater than the doubled atomic radius of silver, equal to 0.144–0.146 nm [50], i.e., more than 0.288–0.292 nm. In other words, the Ag atoms in the model phases of silver sulfide (Ag2S) can be located at a distance of at least 0.288–0.292 nm from each other.
Crystal structure predictions were performed using the evolutionary algorithm (EA) USPEX (Universal Structure Predictor: Evolutionary Xtallography) [51,52,53]. The simulations were carried out for a primitive unit cell that contained one to six Ag2S formula units. The optimization started from a set of randomly generated structures. The number of initial random structures varied from 20 to 40, depending on the number of atoms in the cell. The structures for the next generations were produced randomly (40%) and from the previous ones (60%) via a set of flexible physically motivated variation operators (60%) [51,52,53].
The total energies of the predicted model structures were calculated according to density functional theory [54] using the Perdew–Burke–Ernzenhof version of the generalized gradient approximation (GGA) [55] and the projector-augmented wave method [56] as implemented in the VASP (Vienna ab initio simulation package) code [57,58]. To find equilibrium unit cell parameters and atomic positions, we performed stepwise relaxation [49] using VASP-implemented conjugate-gradient and quasi-Newton RMM-DIIS algorithms [59]. The energies of the initial USPEX-generated structures were computed using a kinetic energy cutoff of 259 eV and uniform k-point meshes with a reciprocal space resolution of 2π × 0.12 Å−1. During the following relaxation steps, these values were gradually improved to 388 eV and 2π × 0.05 Å−1, respectively. The structures with minimal energy generated using USPEX were finally treated at a denser k-point grid with a resolution of 2π × 0.03 Å−1.
In order to estimate the thermodynamic stability of the silver sulfide (Ag2S) model phases with different crystal structures, we calculated their enthalpies of formation at T = 0 K and P = 0 GPa (Table 1). Metallic silver (Ag) (space group No.225-) and orthorhombic sulfur (S) (space group No.70-Fddd) were considered as initial reactants. The formation enthalpies ΔHf of the predicted Ag2S phases according to the reaction
were determined, taking into account the real number of atoms in the unit cells of Ag, S, and Ag2S. The unit cell of metallic silver contains four atoms of Ag, the unit cell of sulfur (S) contains 128 atoms of S, and the predicted Ag2S phase contains Z formula units. The number of Ag2S formula units coincides with the number of S atoms in the predicted phase. Taking this into account, the formation enthalpy ΔHf of the predicted Ag2S phase according to reaction (1) was found using the formula
where E is the energies of the predicted Ag2S phase, condensed metallic silver (Ag) with a cubic (space group ) structure, and the condensed sulfur (S) phase with an orthorhombic (space group Fddd) structure obtained via the DFT calculations. In Formula (2), NAg and NS are the numbers of these atoms in the unit cell of the predicted Ag2S phase; Z is the number of formula units in the predicted Ag2S phase. The values of NAg-cond, Ns-cond, EAg-cond, and Es-cond are given in Table S1 (Supplementary Material file). The division by Z normalizes the ΔHf values to an Ag2S formula unit (i.e., to an S atom).
2Ag + S = Ag2S
The cohesion energy Ecoh is an alternative parameter to characterize the energy advantage of the predicted phase. In contrast to the formation enthalpy, Ecoh shows the energy gain in the formation of a substance from individual atoms:
where Ephase is the energy of the ground state of the predicted Ag2S phase according to DFT calculations, and EAg-at and ES-at are the energies of individual Ag and S atoms calculated as the energies of simple cubic structures, where atoms are separated by a vacuum to exclude their interaction. The values EAg-at and ES-at of individual Ag and S atoms are given in Table S1 (see Supplementary Material file).
For example, the cubic (space group ) Ag2S structure is characterized by the following quantities: Ephase = −19.498 eV, NAg = 4, NS = 2, and Z = 2 (see Table 1), and EAg-cond = −10.863 eV, NAg-cond = 4, ES-cond = −528.168 eV, NS-cond = 128, EAg-at = −0.198 eV, and ES-at = −1.081 eV (see Table S1, Supplementary Materials). According to Equation (2), the formation enthalpy ΔHf of the cubic (space group ) Ag2S structure for these quantities is
According to Equation (3), the cohesion energy Ecoh of the cubic (space group ) Ag2S structure for these quantities is
ΔHf = [−19.498 − (−10.863·4/4) − (−528.168·2/128)]/4 = [−19.498 + 10.863 + 8.252]/4 = −0.382/2 = −0.191 eV.
Ecoh = [−19.498 − (−0.198·4) − (−1.081·2)]/2 = [−19.498 + 0.792 + 2.162]/2 = −16.544/2 = −8.272 eV.
It is these values of ΔHf and Ecoh that are given in Table 1 for the cubic (space group ) Ag2S structure.
The structures predicted using the USPEX code were compared with the experimental acanthite phase, as well as the ordered models. To ensure the accuracy of this comparison, we optimized the unit cell parameters and positions of atoms using the identical settings of the DFT calculations of their energies. The structural optimization stopped if the forces acting on the atoms were less than 10−3 eV·Å−1. The forces were calculated according to the Hellmann–Feynmann theorem.
The DOS diagrams were computed using the tetrahedron method with Blöchl corrections [60]. The band structure curves were built according to the k-points trajectories suggested using the SeeK-path tool [61]. The elastic properties and mechanical stability of the predicted model Ag2S structures were estimated based on elastic tensor coefficients cij computed using the finite difference method as implemented in the VASP code [56,57,58]. The mechanical stability of the model structures was determined using the necessary and sufficient criteria given in previous works [62,63].
3. Results and Discussion
3.1. Structures and Energies of the Predicted Ag2S (Silver Sulfide) Phases
The literature’s theoretical data on the structure and lattice parameters of some model Ag2S phases with triclinic, monoclinic, and orthorhombic symmetry are presented in the form of a database on the websites of the Materials Project, such as mp-32284 [18], mp-556225 [19], mp-1095694 [20], mp-32669 [21], mp-36216 [22], mp-31053 [23], mp-32791 [24], and mp-610517 [25], and others. The crystallographic data of these silver sulfide phases were calculated ab initio using the ELATE code, described in general form in a previous study [26].
The main energy characteristics of the model Ag2S phases predicted using the USPEX code are given in Table 1.
The crystallographic data for the considered cubic, tetragonal, and trigonal phases can be found in Tables S2–S4, respectively (see Supplementary Material file). The crystallographic data for the orthorhombic and monoclinic (space group P21/c) α-Ag2S (acanthite) structure before and after relaxation, the predicted monoclinic (space group P21/c) α-Ag2S structure, and the predicted triclinic (space group P1) Ag2S structure are given in Table 2, Table 3, Table 4 and Table 5.
The unit cells of the model cubic, tetragonal, and trigonal Ag2S phases, which can presumably form as alternative low-temperature phases along with the monoclinic acanthite (α-Ag2S), are shown in Figure 3a–c, respectively. All the model crystal structures were visualized using the VESTA software [64]. The translation vectors and atomic coordinates in the unit cells of these model Ag2S phases are given in Tables S2 to S4, respectively (see Supplementary Material file).
The formation enthalpies ΔHf of the model cubic phases of Ag2S with space groups and (see Figure 3a) are −0.191 and +3.573 eV/form.unit (see Table 1). It is clear that the formation of the model cubic (space group ) Ag2S phase is thermodynamically impossible, while the cubic (space group ) Ag2S structure is quite favorable in terms of the enthalpy of formation. This cubic phase is markedly more favorable than the unrelaxed acanthite structure, but it is somewhat inferior to the relaxed acanthite structure (see Table 1).
The model tetragonal structures of Ag2S with space groups and P4/mmm calculated using the USPEX code are shown in Figure 3b. The translation vectors and atomic coordinates in the unit cells of these tetragonal Ag2S phases are given in Table 3. The suggested tetragonal structures with space groups P4/mmm and have positive formation enthalpies of +1.313 and +0.394 eV/form.unit, respectively (see Table 1). Therefore, the formation of such tetragonal structures of silver sulfide is energetically impossible.
A similar conclusion should be drawn for the trigonal (space groups and ) structures with the formation enthalpies of +0.042 and +0.041 eV/form.unit, respectively (see Table 1). The unit cells of these trigonal structures of Ag2S are shown in Figure 3c, and the translation vectors and atomic coordinates for these trigonal Ag2S phases are given in Table S4 (see Supplementary Material file).
Figure 4 shows the model orthorhombic Ag2S structures with space groups Cmce and Cmcm. The translation vectors and atomic coordinates in the unit cells of these orthorhombic Ag2S phases are given in Table 2. The calculated formation enthalpies ΔHf of these structures are −0.219 and −0.199 eV/form.unit, respectively (see Table 1), so their formation is quite possible.
Using the USPEX code, monoclinic (space group P21/c) models of the α-Ag2S (acanthite) structure were calculated before and after relaxation (Figure 5). The initial relaxation was carried out in terms of energy, and then the structure was recalculated with more accurate convergence criteria. Translation vectors and atomic coordinates in the unit cells of the unrelaxed and relaxed monoclinic (space group P21/c) phases of silver sulfide with the α-Ag2S (acanthite) structure are given in Table 3.
The first point we should pay attention to is a rather uncommon behavior of the acanthite structure. Its structure, obtained using an X-ray diffraction experiment (see Figure 5a), differs significantly from the structure corresponding to its DFT energy minimum (see Figure 5b). Although, during structural relaxation, the symmetry was fixed, and both variants of acanthite are of the same space group, P21/c, they are somewhat different (see Figure 5). The result of structural optimization for acanthite (Figure 5b) does not depend on whether energies or forces were used as a minimization criterion.
The relaxed monoclinic (space group P21/c) model of acanthite (α-Ag2S) turned out to be the best in terms of the formation enthalpy. The formation enthalpy ΔHf of the unrelaxed acanthite structure is −0.033 eV/form.unit, while the enthalpy ΔHf of formation of the relaxed acanthite structure is much lower and amounts to −0.199 eV/form.unit (see Table 1).
We calculated the X-ray diffraction (XRD) patterns of unrelaxed and relaxed monoclinic (space group P21/c) α-Ag2S (acanthite) (Figure 6). The calculations were carried out under CuKα1,2- radiation with a Δ(2θ) = 0.01° step. Figure 6 shows sections of the X-ray diffraction patterns in the range of angles 2θ = 19–61°, where the most characteristic and intense diffraction reflections of these acanthite phases are present. The calculated XRD pattern of unrelaxed acanthite coincides with high accuracy with the experimental XRD patterns of coarse-grained monoclinic (space group P21/c) α-Ag2S (acanthite) [2,65]. After the relaxation of acanthite, the unit cell parameters and, accordingly, the coordinates of the Ag and S atoms somewhat changed (Table 6). As a result of atomic displacements, a more ordered arrangement of atoms, especially Ag atoms, was observed in the relaxed phase compared to unrelaxed acanthite (see Figure 5). In particular, Ag atoms are predominantly located in atomic planes perpendicular to the a and c axes of the unit cell. This arrangement of atoms led to a noticeable decrease in the number of diffraction reflections in the XRD pattern of relaxed acanthite, although some of the reflections coincide in position with the reflections of the unrelaxed phase (indices of coinciding reflections are shown in Figure 5). Relaxed acanthite clearly exhibits interplanar distances of ~0.390, ~0.335, ~0.277, ~0.230, and ~0.210 nm, which are also characteristic of the unrelaxed phase. Note that, in numerous experimental studies of silver sulfide summarized in [2,7], only unrelaxed acanthite was observed as a low-temperature phase, regardless of the method and conditions of synthesis.
The calculation of other model Ag2S structures showed that the monoclinic structure of acanthite (α-Ag2S) described in the literature [6,65] is not the only possible and most energetically favorable low-temperature phase of silver sulfide. Because of calculations, it was possible to find a monoclinic (space group P21/c) Ag2S phase (Figure 7, left) with lower energy in the ground state compared to relaxed acanthite. The translation vectors and atomic coordinates in the unit cells of this monoclinic (space group P21/c) phase of silver sulfide are given in Table 4. The formation enthalpy ΔHf of this monoclinic (space group P21/c) Ag2S phase is −0.219 eV/form.unit (see Table 1), i.e., one 0.02 eV/form.unit less than the enthalpy of relaxed acanthite.
As a result of the performed simulation of other possible Ag2S structures, it was possible to establish that the monoclinic phases of silver sulfide (Ag2S) are not the most energetically favorable low-temperature phases. The calculations allowed us to find the triclinic (space group P1) Ag2S phase (Figure 7, right). This Ag2S phase has the lowest cohesion energy Ecoh = −8.304 eV/form.unit and formation enthalpy ΔHf = −0.223 eV/form.unit (see Table 1) in the ground state at T = 0 K and P = 0 Pa.
Translation vectors and atomic coordinates in the unit cell of the triclinic (space group P1) Ag2S phase are given in Table 5.
As for the USPEX predicted structures, ten of them have formation enthalpies significantly lower compared to unrelaxed acanthite and can be considered alternative low-temperature Ag2S modifications of silver sulfide (see Table 1). The triclinic (space group P1) structure (see Figure 7, right) has the lowest formation enthalpy (−0.223 eV/form.unit) of all the considered modifications. The other nine phases, despite the variety in their symmetries, have very similar structures. Their different symmetry is a result of small variations in angles and/or bond lengths. The monoclinic (space group P21) Ag2S phase proposed by Kashida et al. [15] also has a similar structure. The formation enthalpy of this monoclinic (space group P21) Ag2S phase is −0.208 eV/form.unit (see Table 1), which is slightly lower than the ΔHf of relaxed acanthite. The lowest formation enthalpy of −0.219 eV/form.unit for these almost identical structures is reached for the orthorhombic (space group Cmce) Ag2S phase (see Figure 4). The other examined structures of Ag2S with space groups Cmcm, P21/m, and Cmc21 from the Materials Project database are merely variants of relaxed acanthite (see Figure 5b) with formation enthalpies ranging from −0.1993 to −0.1990 eV/form.unit.
We assessed the influence of residual forces on the standard deviation of the formation enthalpies of the considered Ag2S phases. In particular, when assessing the impact of the residual forces on the unrelaxed acanthite structure, we found that the absolute deviations in formation enthalpy did not exceed 0.00034 eV/(Ag2S form.unit) or 0.00085 eV·Å−1·atom−1 when changing the force threshold from 0.002 eV·Å−1 to 0.001 eV·Å−1. The relative deviation of the formation enthalpy does not exceed 0.1%. For smaller values of the limiting parameter, it was impossible to complete the relaxation of the structure due to computational limitations. The other considered phases were more susceptible to relaxation, so deviations in their energies are very likely to be within this 0.1% range. This amount of uncertainty is enough to rank the Ag2S structures by their stability. For instance, the difference in the formation enthalpies, when taking into account their uncertainties, for the two best structures (triclinic and orthorhombic with space groups P1 and Cmce) is about 2%. Thus, the deviations in the formation enthalpies of structures with different symmetries are very small, which makes it possible to distinguish between these structures.
The Ag-S bond lengths for all the predicted Ag2S structures are given in Table S5 (see Supplementary Material file). The Ag-S bond length is 0.232–0.252 nm, depending on the symmetry of the predicted Ag2S structure.
In high-temperature body-centered cubic β-Ag2S (argentite) at a temperature close to 453 K, the length of the bonds Ag1-S and Ag2-S are 0.24307 and 0.25691nm, respectively. The coordination numbers of the Ag1 (b positions) and Ag2 (j positions) atoms are 6 and 1, respectively. In the crystal lattice of α-Ag2S acanthite, the length of the bonds Ag1-S and Ag2-S are 0.25113 and 0.25475 nm, respectively (see Table S5, Supplementary Material). The coordination numbers of the Ag1 and Ag2 atoms are the same and equal to 4. Thus, when taking into account the temperature change in the lattice parameters of silver sulfide during the transition from argentite to acanthite, the Ag1-S and Ag2-S bond lengths increased, and the coordination numbers of the Ag atoms became the same. The coordination numbers of the silver atoms in the predicted Ag2S structures are also less than 6. The smallest coordination number of Ag atoms, equal to 1, was observed for Ag-S1 bonds with a length of 0.23883 nm in the cubic (space group ) Ag2S phase. Coordination numbers of Ag atoms equal to 2 were observed for Ag-S bonds with lengths of 0.23556 nm and 0.24306 nm in the cubic (space group ) and orthorhombic (space group Cmce) Ag2S phases, as well as for all Ag-S bonds in a predicted triclinic (space group P1) Ag2S phase. The coordination number of Ag atoms in the Ag-S bonds of tetragonal (space group ) and trigonal (space groups and ) silver sulfides is 3. A coordination number of Ag atoms equal to 3 was also observed in relaxed monoclinic (space group P21/c) acanthite. Ag atoms have a fourfold coordination environment by S atoms in unrelaxed monoclinic (space group P21/c) acanthite and tetragonal (space group P4/mmm) silver sulfide.
In the structures with the lowest formation enthalpies (triclinic and orthorhombic with space groups P1 and Cmce), the Ag-S bond lengths range from 0.2431 to 0.2496 nm. The Ag-S bond lengths of the relaxed structure of acanthite are somewhat outside these limits and demonstrate an increasing disproportion. One of the bonds has a length of 0.2524 nm, while another 0.2403 nm. This disproportion is considerably larger in the other proposed ordered structures. The biggest difference in bond lengths is reached for the tetragonal (space group ) and one of the cubic (space group ) models. Therefore, from the chemical point of view, the formation of such structures seems unreasonable. The cubic (space group ) structure after relaxation contains chemically isolated sulfur atoms. The nearest distance to the silver atom is 0.6545 nm. Presumably, this peculiarity makes the cubic (space group ) structure the most unfavorable of all the considered structural models of silver sulfide. Another suggested cubic (space group ) structure has no disproportion in bond lengths. Its nearest Ag-S distance (0.2356 nm) is slightly less than that of the best structures. Nevertheless, the formation energy of the cubic (space group ) ordered structure is close to that of acanthite. Therefore, of all the suggested cubic, tetragonal, and trigonal models, only the cubic (space group ) structure can be considered a candidate for a new phase of silver sulfide.
As a result of the simulations and analysis, we should conclude that acanthite is not the most energetically favorable low-temperature variant in the crystal structure of silver sulfide. The predictions based on the evolutionary algorithm allowed us to find two new Ag2S phases (see Figure 7) with lower formation energies.
3.2. Symmetry Analysis of the Predicted Structures
Crystallographic point and space groups are comprehensively described in monographs [66,67,68,69]. In particular, all point groups and all their symmetry elements are listed in a monograph [69] in accordance with the notation adopted in [67]: forty-eight elements of the full cubic symmetry group (Oh) are sequentially denoted from h1 to h48.
The point group (Oh) of bcc argentite (β-Ag2S) includes all 48 symmetry elements h1-h48 of the cubic group [66], i.e., Nh-arg = 48. The rotational reduction in symmetry Nrot is equal to the ratio of the number of symmetry elements of the argentite point group to the number Nh-phase of symmetry elements included in the point group of the Ag2S model structure under consideration, i.e., Nrot = Nh-arg/Nh-phase = 48/Nh-phase. The reduction in translational symmetry is equal to the relative change in unit cell volume during the transformation of argentite into the Ag2S model structure. During the transformation of one phase of silver sulfide (in our case, argentite) into another Ag2S phase, the decrease in the translational symmetry Ntr can also be estimated as the ratio of the Zphase value (the number of Ag2S formula units in the discussed model phase) to the number of Ag2S formula units in argentite (Zarg = 2), i.e., Ntr = Zphase/Zarg. However, the decrease in translational symmetry is more accurately estimated from the relative change in unit cell volumes during the considered transformation. The total symmetry reduction Ntot = Nrot × Ntr is the product of the rotational symmetry reduction and the translational symmetry reduction. For example, in the transition of argentite to monoclinic α-Ag2S (acanthite), which contains Zacanth = 4 Ag2S formula units and whose point group 2/m (C2h) includes four symmetry elements, h1, h4, h25, and h28 (Nh-acanth = 4), the total symmetry reduction Ntot = 24.
The decrease in the symmetry Ntot of the predicted model Ag2S phases with respect to argentite is given in Table 1. When using the example of silver sulfide structures with the same monoclinic (space group P21/c) symmetry, it is clearly seen that the largest decrease in symmetry leads to the formation of phases with the lowest formation enthalpy (see Table 1).
In general, calculations of the model structures of silver sulfide (Ag2S) using the USPEX code [51,52,53] showed that a reduction in the symmetry of Ag2S phases from cubic, tetragonal, and trigonal to orthorhombic, monoclinic, and especially triclinic crystallographic systems is accompanied by a decrease in their formation enthalpy ΔHf and the emergence of the most energetically favorable structures. This was not previously known. Indeed, the transformation of high-temperature cubic (space group ) β-Ag2S (argentite) into any model Ag2S structure will occur with a decrease in symmetry.
3.3. Electronic Structure
The density of electron states (DOS) calculations for the predicted Ag2S phases were performed per electron density functional theory (DFT) [54] using the tetrahedron method with Blöchl corrections [60]. The exchange-correlation potential was described using the PBE [55] version of the generalized gradient approximation (GGA). The band structure was built along the trajectories of k-points determined using the SeeK-path service [61]. The kinetic energy of plane waves did not exceed 388 eV.
All the considered low-temperature Ag2S phases are very similar in their electronic structures. Their band structures exhibit a band gap with a width of ~0.6 to ~1.5 eV, which indicates the semiconductor properties of the predicted Ag2S phases. For relaxed Ag2S structures, the band gap Eg varies from 1.02 to 1.16 eV and is close to the experimentally measured values of 0.9–1.1 eV [2,7] for nanostructured silver sulfide.
The band structure of unrelaxed and relaxed monoclinic (space group P21/c) α-Ag2S (acanthite) is shown in Figure 8. It is clearly seen that unrelaxed acanthite is a direct-gap semiconductor. Because of relaxation, the band structure changed somewhat, since the electron located near the bottom of the conduction band acquired a momentum that differed from the momentum of the electron located near the maximum of the valence band. Thus, relaxed acanthite becomes an indirect-gap semiconductor.
The densities of states of unrelaxed and relaxed monoclinic (space group P21/c) α-Ag2S (acanthite) are shown in Figure 9. The structure of unrelaxed acanthite, formed from X-ray diffraction data [46], has a much smaller band gap of 0.72 eV compared to Eg = 1.02 eV for relaxed acanthite (α-Ag2S). The states near the Fermi level are formed by Ag 4d orbitals, and the S 3p states are most pronounced in the energy ranges below ~0.5–2.0 and ~4.5–6.0 eV relative to the Fermi level. The edge of the valence band of monoclinic (space group P21/c) acanthite near the Fermi level has a slope and resembles experimental optical spectra [70,71,72]. The width of the valence band of acanthite is approximately equal to 4–6 eV, which coincides with the experimental data on the photoelectron spectroscopy of silver sulfide [13,73].
The predicted cubic (space group ) Ag2S structure was proposed as a derivative of the bcc (space group ) structure of β-Ag2S (argentite). The formation enthalpy ΔHf = −0.191 eV/(Ag2S form.unit) of this structure is one of the lowest among the predicted silver sulfide phases. Nevertheless, its electronic structure (see Figure 9c) differs markedly from that of other low-temperature Ag2S phases. The valence band of the cubic (space group ) Ag2S structure is split, and the band gap is 1.54 eV, i.e., about 50% more than that of other predicted Ag2S structures.
3.4. Elastic Properties and Hardness
The elastic properties of the model cubic, tetragonal, trigonal, orthorhombic, monoclinic, and triclinic Ag2S structures were estimated by calculating the coefficients of elasticity tensors. The found matrices (C) of elastic stiffness constants cij (S6)–(S17) of the predicted model phases of Ag2S (silver sulfide) are given in the Supplementary Material file.
The analytical relationships between compliance coefficients sij and stiffness coefficients cij for low-symmetry (trigonal, monoclinic, and triclinic) crystals are very cumbersome and complex. For this reason, in general cases, the elastic compliance constants sij are found by calculating the matrix (S) of the compliance constants, which is inverse to the matrix (C), i.e., (S) = (C)−1. We found the matrices (S) of the predicted Ag2S structures by calculating the inverse matrices using the software [74] for the calculation of inverse matrices.
The calculated elastic constants cij (see matrices (S6)–(S17)) were used to evaluate the mechanical stability of the predicted model Ag2S phases using the Born criteria [75,76] and the necessary and sufficient conditions for the elastic stability of various crystal systems described in studies [62,63]. The calculated elastic stiffness constants cij and compliance constants sij were also used to estimate the isotropic elastic moduli of some predicted Ag2S phases of silver sulfide.
A necessary but not sufficient condition for the mechanical stability of a crystal of any symmetry is the positiveness of all diagonal elements of the matrix of elastic stiffness constants, i.e., cii > 0 (i = 1–6).
The stability criteria for cubic crystals have the form cii > 0, c11 > c12, c44 > 0, and c11 + 2c12 > 0 [62,63,75]. These criteria are met for the cubic (space group and ) Ag2S model phases. Thus, the cubic (space group ) Ag2S phase is mechanically stable. However, the positive formation enthalpy of a cubic phase with a space group excludes the existence of such a phase.
The mechanical stability conditions for tetragonal crystals have the form c11 > |c12|, c44 > 0, c66 > 0, and (c11 + c12)c33 > [43,44]. The elastic stiffness constants of the model tetragonal (space group P4/mmm) Ag2S phase do not satisfy these conditions since c44 < 0 and c55 < 0 (see matrix (S9), Supplementary Material file), so this tetragonal (space group P4/mmm)) Ag2S phase is mechanically unstable. The elastic stiffness constants of the tetragonal (space group ) Ag2S phase also do not satisfy these conditions since c11 < c12 and (c11 + c12)c33 < (see matrix (S8), Supplementary Material file), so this tetragonal (space group ) Ag2S phase is also mechanically unstable.
The mechanical stability conditions for trigonal (rhombohedral) crystals have the form c11 > |c12|, c44 > 0, (c11 + c12)c33 > , and (c11 − c12)c44 > + . The absence in the elastic matrices of the trigonal (space groups and ) Ag2S phases of elastic stiffness constants equal to 0, and the presence of negative elastic stiffness constants where there should be zero constants (see matrices (S10) and (S11), Supplementary Material file), means that the mechanical stability conditions are not satisfied for these phases. Thus, the model trigonal (space groups and ) Ag2S phases are mechanically unstable.
Necessary and sufficient conditions for the mechanical stability of orthorhombic crystals have the form cii > 0 (i = 1–6), c11c22 > , and c11c22c33 + 2c12c13c23 − − − > 0 [62,63] and are satisfied for the orthorhombic (space groups Cmcm and Cmce) Ag2S phases.
For the model monoclinic (space group P21) Ag2S phase, and also for unrelaxed and relaxed monoclinic (space group P21/c) α-Ag2S (acanthite), the elastic stiffness constants cii > 0, c22 + c33 > 2c23, c11 + c22 + c33 > 2(c12 + c13 + c23), c33c55 > , and c44c66 > (see matrices (S14)–(S16) in Supplementary Material file) satisfy the necessary and sufficient conditions for mechanical stability. Thus, the monoclinic (space groups P21 and P21/c) Ag2S phases with such elastic stiffness constants are mechanically stable.
For the predicted triclinic (space group P1) Ag2S phase, the elastic stiffness constants cii > 0 (see matrix (S17), Supplementary Material file). With this in mind, we can assume that the predicted triclinic (pace group P1) Ag2S phase satisfies the conditions of mechanical stability and is mechanically stable.
Thus, the mechanical stability of all the predicted Ag2S phases was determined for the first time. Previously, there was no information on the mechanical stability of low-temperature Ag2S phases.
The hardness of silver sulfide on the Moh’s scale is 2.0–2.5. There is no information on the Vickers hardness (HV) of silver sulfide (Ag2S) in the accessible literature. The availability of calculated data on the constants of the elastic stiffness cij and elastic compliance sij of the considered model Ag2S phases makes it possible to find the isotropic elastic moduli and hardness of these phases. We evaluated the isotropic elastic moduli EH, GH, and BH and the hardness (HV) of several predicted Ag2S phases that have the lowest formation enthalpies.
Pugh [77] suggested the use of the bulk modulus (B) to shear modulus (G) ratio as a criterion for the brittle and ductile behavior of polycrystalline metals. The isotropic elastic moduli of polycrystalline materials can be averaged based on the upper (BV) and lower (BR) limit values of the bulk modulus, and the upper (GV) and lower (GR) values of the shear modulus. The values of BV, BR, BH, GV, GR, and GH for crystals with any symmetry are calculated via the Voigt–Reuss–Hill method [78] (see Supplementary Material file) using elastic stiffness constants cij and compliance constants sij. The Poisson’s ratio μ was calculated as μ = (3BH − 2GH)/[2(3BH + GH)]. The calculated isotropic elastic moduli for GH, BH, EH of several polycrystalline silver sulfides (Ag2S) with the lowest formation enthalpies are given in Table 6.
The isotropic elastic moduli GH = 9.8–10.1 and BH = 28.3–31.1 GPa of unrelaxed and relaxed monoclinic (space group P21/c) α-Ag2S (acanthite) (see Table 6) are in good agreement with the values GH = 8 and BH = 26 GPa of the monoclinic (space group P21/c) phase of silver sulfide given in the Materials Project’s mp-610517 [25]. The BH modulus of acanthite calculated by us is 28–31 GPa (see Table 6) and is quite close to the experimental estimate of B = 33.7 GPa [79] at 0 K, determined from data on the temperature dependences of the heat capacity and thermal expansion coefficient. The calculated isotropic moduli GH = 10.2 and BH = 28.4 GPa of the predicted orthorhombic (space group Cmcm) Ag2S (silver sulfide) are close to the values of GH and BH found for the same orthorhombic sulfide (Ag2S) in [22] and equal to 9 and 24 GPa, respectively.
Currently, the inverse Pugh’s ratio, k = G/B, is used to estimate the theoretical Vickers hardness (HV) of solids. Chen et al. [80] proposed describing the Vickers hardness (HV) of solids as a function of the inverse Pugh’s ratio k and shear modulus G and bulk modulus B:
However, later, Tian et al. [81] noted that there are no physical grounds for using the free negative term −3 in Formula (4) since this leads to negative hardness values for some ionic and other crystals. Therefore, Tian et al. [81] proposed another formula for estimating the hardness HV:
HV = 2(k2G)0.585 − 3.
HV = 0.92k1.37G0.708.
In Formulas (4) and (5), the isotropic elastic moduli BH and GH of polycrystalline materials are used to evaluate hardness.
Later, Mazhnik and Oganov [82] suggested using Young’s modulus E instead of BH and GH, and they proposed using the empirical formula
where γ0 = 0.096 is a dimensionless constant independent of the material, and χ(μ) is a function that depends on the value of the Poisson’s ratio μ and has the form
HV = γ0χ(μ)E,
The found isotropic elastic moduli GH, BH, and EH of polycrystalline silver sulfides (Ag2S), the predicted structures of which have the lowest enthalpies of formation, are presented in Table 6. The use of Formula (4) led to negative hardness values for some silver sulfides, so we calculated the hardness using Equations (5) and (6). The hardness HV of polycrystalline silver sulfides (Ag2S), calculated using Equation (5), is from 1.0 to 3.2 GPa (Table 6). The use of Equation (6) resulted in slightly lower values, ranging from 0.9 to 1.5 GPa (see Table 6). The estimated hardness of silver sulfides is higher than the hardness of metallic silver and such metals as Fe, Ni, and Ti, and it is close to the hardness of calcium carbonate and fluoride (~1 and ~2 GPa, respectively).
Thus, for the first time, the isotropic elastic moduli of the predicted polycrystalline Ag2S phases were determined, and the hardness HV of the predicted Ag2S phases with the lowest formation enthalpies was estimated.
4. Conclusions
In this work, we considered alternative structural models for low-temperature modifications of silver sulfide (Ag2S) in addition to the known monoclinic structure of acanthite. The proposed structures, like acanthite itself, can be represented as the results of the ordering of the high-temperature cubic argentite phase. The possibility of the formation of cubic, tetragonal, orthorhombic, trigonal, monoclinic, and triclinic Ag2S phases was considered. The calculation of the cohesion energy and the formation enthalpy showed that the formation of low-symmetry Ag2S phases is energetically the most favorable. The most favorable phase with the lowest formation enthalpy has triclinic (space group P1) symmetry. The elastic stiffness constants cij of all the predicted Ag2S phases were calculated, and their mechanical stability was determined.
For the considered and predicted phases, we have presented their optimized crystallographic data, electronic structure, elastic moduli, and hardness. It was established that the band structure of all the considered low-temperature model phases of silver sulfide has a band gap, which indicates their semiconductor properties.
The predicted alternative low-temperature phases of silver sulfide (Ag2S), especially nanocrystalline ones, can also have many possible applications. One can expect that these phases will be suitable for biological and medical applications as biomarkers, and the predicted low-temperature Ag2S phases can be used for the manufacture of such electronic devices as photovoltaic cells, photoconductors, and infrared radiation detectors.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/nano13192638/s1, Table S1: Energies EVASP of individual Ag and S atoms and the condensed metallic silver (Ag) and condensed sulfur (S) phase. Table S2: Model predicted cubic Ag2S structures. Table S3: Model predicted tetragonal Ag2S structures. Table S4: Model predicted trigonal Ag2S structures. Subsection “Matrices of elastic stiffness constants computed for model Ag2S structures” with matrices from (S5) to (S16). Subsection “The Voigt–Reuss–Hill averaging scheme” with equations from (S17a–d) to (S18).
Author Contributions
S.I.S. proposed the main idea and methodology of the research, wrote the original draft of the manuscript, and supervised the work; M.G.K. carried out the crystal structures predictions and the data analysis; A.I.G. wrote the manuscript with contributions from all the other co-authors and performed its review and editing; A.V.L. carried out the VASP calculations and formal data analysis. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Russian Science Foundation (grant no. 19-79-10101-П) through the Institute of Solid State Chemistry of the Ural Branch of the RAS. The computations were performed on the Uran supercomputer at IMM UB RAS. Crystal structures were visualized using the VESTA software.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sharma, R.C.; Chang, Y.A. The Ag-S (Silver-Sulfur) system. Bull. Alloy Phase Diagr. 1986, 7, 263–269. [Google Scholar] [CrossRef]
- Sadovnikov, S.I.; Rempel, A.A.; Gusev, A.I. Nanostructured Lead, Cadmium and Silver Sulfides: Structure, Nonstoichiometry and Properties; Springer International Publishing AG: Cham/Heidelberg, Germany, 2018; 331p. [Google Scholar]
- Thompson, W.T.; Flengas, S.N. Drop calorimetric measurements on some chlorides, sulfides, and binary melts. Can. J. Chem. 1971, 49, 1550–1563. [Google Scholar] [CrossRef]
- Grønvold, F.; Westrum, E.F. Silver(I) sulfide: Ag2S heat capacity from 5 to 1000 K, thermodynamic properties, and transitions. J. Chem. Therm. 1986, 18, 381–401. [Google Scholar] [CrossRef]
- Sadovnikov, S.I.; Gusev, A.I.; Rempel, A.A. Nonstoichiometry of nanocrystalline monoclinic silver sulfide. Phys. Chem. Chem. Phys. 2015, 17, 12466–12471. [Google Scholar] [CrossRef] [PubMed]
- Sadanaga, R.; Sueno, S. X-ray study on the α-β transition of Ag2S. Mineralog. J. Jpn. 1967, 5, 124–148. [Google Scholar] [CrossRef]
- Sadovnikov, S.I.; Gusev, A.I. Recent progress in nanostructured silver sulfide Ag2S: From synthesis and nonstoichiometry to properties. J. Mater. Chem. A 2017, 5, 17676–17704. [Google Scholar] [CrossRef]
- Van Doorselaer, M.K. Solid state properties and photographic activiti of crystalline Ag2S and (Ag, Au)2S-specks at the surface of silver halide crystalls. J. Photograph. Sci. 1987, 35, 42–52. [Google Scholar] [CrossRef]
- Simonnin, P.; Sassi, M.; Gilbert, B.; Charlet, L.; Rosso, K.M. Phase transition and liquid-like superionic conduction in Ag2S. J. Phys. Chem. C 2020, 124, 10150–10158. [Google Scholar] [CrossRef]
- Sadovnikov, S.I.; Gerasimov, E.Y. Direct TEM observation of the “acanthite α-Ag2S–argentite β-Ag2S” phase transition in a silver sulfide nanoparticle. Nanoscale Adv. 2019, 1, 1581–1588. [Google Scholar] [CrossRef]
- Sadovnikov, S.I.; Gusev, A.I.; Rempel, A.A. High-temperature scanning electron microscopy study of acanthite—Argentite phase transformation in nanocrystalline silver sulfide powder. Phys. Chem. Chem. Phys. 2015, 17, 20495–20501. [Google Scholar] [CrossRef]
- Alekperov, O.; Jahangirli, Z.; Paucar, R. First-principles lattice dynamics and Raman scattering in ionic conductor β-Ag2S. Phys. stat. Sol. 2016, 253, 2049–2055. [Google Scholar] [CrossRef]
- Sadovnikov, S.I.; Gusev, A.I. Acanthite-argentite transformation in silver sulfide as a disorder-order transition. JETP Lett. 2019, 109, 584–588. [Google Scholar] [CrossRef]
- Blanton, T.; Misture, S.; Dontula, N.; Zdzieszynski, S. In situ high-temperature X-ray diffraction characterization of silver sulfide, Ag2S. Powder Diff. 2011, 26, 110–118. [Google Scholar] [CrossRef]
- Kashida, S.; Watanabe, N.; Hasegawa, T.; Iida, H.; Mori, M.; Savrasov, S. Electronic structure of Ag2S, band calculation and photoelectron spectroscopy. Sol. State Ion. 2003, 158, 167–175. [Google Scholar] [CrossRef]
- Saal, J.E.; Kirklin, S.; Aykol, M.; Meredig, B.; Wolverton, C. Materials Design and Discovery with High-Throughput Density Functional Theory: The Open Quantum Materials Database (OQMD). JOM 2013, 65, 1501–1509. [Google Scholar] [CrossRef]
- Available online: https://next-gen.materialsproject.org (accessed on 20 August 2023).
- Available online: https://next-gen.materialsproject.org/materials/mp-32884 (accessed on 20 August 2023).
- Available online: https://next-gen.materialsproject.org/materials/mp-556225 (accessed on 20 August 2023).
- Available online: https://next-gen.materialsproject.org/materials/mp-1095694 (accessed on 20 August 2023).
- Available online: https://next-gen.materialsproject.org/materials/mp-32669 (accessed on 20 August 2023).
- Available online: https://next-gen.materialsproject.org/materials/mp-36216 (accessed on 20 August 2023).
- Available online: https://next-gen.materialsproject.org/materials/mp-31053 (accessed on 20 August 2023).
- Available online: https://next-gen.materialsproject.org/materials/mp-32791 (accessed on 20 August 2023).
- Available online: https://next-gen.materialsproject.org/materials/mp-610517 (accessed on 20 August 2023).
- Gaillac, R.; Pullumbi, P.; Coudert, F.-X. ELATE: An open-source online application for analysis and visualization of elastic tensors. J. Phys. Cond. Matter 2016, 28, 275201. [Google Scholar] [CrossRef]
- Murugadoss, G.; Jayavel, R.; Rajesh Kumar, M.; Thangamuthu, R. Synthesis, optical, photocatalytic, and electrochemical studies on Ag2S/ZnS and ZnS/Ag2S nanocomposites. Appl. Nanosci. 2016, 6, 503–510. [Google Scholar] [CrossRef]
- Tang, A.; Wang, Y.; Ye, H.; Zhou, C.; Yang, C.; Li, X.; Peng, H.; Zhang, F.; Hou, Y.; Teng, F. Controllable synthesis of silver and silver sulfide nanocrystals via selective cleavage of chemical bonds. Nanotechnology 2013, 24, 355602. [Google Scholar] [CrossRef]
- Du, N.; Zhang, H.; Sun, H.Z.; Yang, D.R. Sonochemical synthesis of amorphous long silver sulfide nanowires. Mater. Lett. 2007, 61, 235–238. [Google Scholar] [CrossRef]
- Liu, L.; Hu, S.; Dou, Y.-P.; Liu, T.; Lin, J.; Wang, Y. Nonlinear optical properties of near-infrared region Ag2S quantum dots pumped by nanosecond laser pulses. Beilst. J. Nanotechnol. 2015, 6, 1781–1787. [Google Scholar] [CrossRef]
- Terabe, K.; Hasegawa, T.; Nakayama, T.; Aono, M. Quantized conductance atomic switch. Nature 2005, 433, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.H.; Terabe, K.; Hasegawa, T.; Aono, M. Resistance switching of an individual Ag2S/Ag nanowire heterostructure. Nanotechnology 2007, 18, 485202. [Google Scholar] [CrossRef]
- Xu, Z.; Bando, Y.; Wang, W.; Bai, X.; Golberg, D. Real-time in situ HRTEM-resolved resistance switching of Ag2S Nanoscale ionic conductor. ACS Nano 2010, 4, 2515–2522. [Google Scholar] [CrossRef] [PubMed]
- Belov, A.N.; Pyatilova, O.V.; Vorobiev, M.I. Synthesis of Ag/Ag2S nanoclusters resistive switches for memory cells. Adv. Nanopart. 2014, 3, 1–4. [Google Scholar] [CrossRef]
- Shen, S.; Zhang, Y.; Liu, Y.; Peng, L.; Chen, X.; Wang, Q. Manganese-doped Ag2S-ZnS heteronanostructures. Chem. Mater. 2012, 24, 2407–2413. [Google Scholar] [CrossRef]
- Nasrallah, T.B.; Dlala, H.; Amlouk, M.; Belgacem, S.; Bernede, J.C. Some physical investigations on Ag2S thin films prepared by sequential thermal evaporation. Synth. Met. 2005, 151, 225–230. [Google Scholar] [CrossRef]
- Karashanova, D.; Nihtianova, D.; Starbova, K.; Starbov, N. Crystalline structure and phase composition of epitaxially grown Ag2S thin films. Sol. State Ion. 2004, 171, 269–275. [Google Scholar] [CrossRef]
- Jadhav, U.M.; Patel, S.N.; Patil, R.S. Synthesis of silver sulphide nanoparticles by modified chemical route for solar cell applications. Res. J. Chem. Sci. 2013, 3, 69–74. [Google Scholar]
- Yang, J.; Ying, J.Y. Nanocomposites of Ag2S and noble metals. Angew. Chem. Int. Ed. 2011, 50, 4637–4643. [Google Scholar] [CrossRef]
- Hasegawa, T.; Terabe, K.; Tsuruoka, T.; Aono, M. Atomic switch: Atom/ion movement controlled devices for beyond Von-Neumann computers. Adv. Mater. 2012, 24, 252–267. [Google Scholar] [CrossRef]
- Wang, D.; Liu, L.; Kim, Y.; Huang, Z.; Pantel, D.; Hesse, D.; Alexe, M. Fabrication and characterization of extended arrays of Ag2S/Ag nanodot resistive switches. Appl. Phys. Lett. 2011, 98, 243109. [Google Scholar] [CrossRef]
- Li, C.; Zhang, Y.; Wang, M.; Zhang, Y.; Chen, G.; Li, L.; Wu, D.; Wang, Q. In vivo real-time visualization of tissue blood flow and angiogenesis using Ag2S quantum dots in the NIR-II window. Biomaterials 2014, 35, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Lutz, C.; Hasegawa, T.; Chikyow, T. Ag2S atomic switch-based ‘tug of war’ for decision making. Nanoscale 2016, 8, 14031–14035. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Nie, S. Quantum dot-encoded mesoporous beads with hgh brightness and uniformity: Rapid readout using flow cytometry. Anal. Chem. 2004, 76, 2406–2410. [Google Scholar] [CrossRef]
- Ma, X.; Zhao, Y.; Jiang, X.; Liu, W.; Liu, S.; Tang, Z. Facile preparation of Ag2S/Ag semiconductor/metal heteronanostructures with remarkable antibacterial properties. ChemPhysChem 2012, 13, 2531–2535. [Google Scholar] [CrossRef]
- Chang, P.; Cheng, H.; Lin, W.; Li, X.; Zhao, F. A stable and active AgxS crystal preparation and its performance as photocatalyst. Chin. J. Catal. 2015, 36, 564–571. [Google Scholar] [CrossRef]
- Zhang, D.; Xu, G.; Chen, F. Hollow spheric Ag–Ag2S/TiO2 composite and its applicationfor photocatalytic reduction of Cr(VI). Appl. Surf. Sci. 2015, 351, 962–968. [Google Scholar] [CrossRef]
- Xiao, C.; Xu, J.; Li, K.; Feng, J.; Yang, J.; Xie, Y. Superionic phase transition in silver chalcogenide nanocrystals realizing optimized thermoelectric performance. J. Amer. Chem. Soc. 2012, 134, 4287–4293. [Google Scholar] [CrossRef]
- Universal Structure Predictor: Evolutionary Xtallography. Manual. Version 9.4.4. Available online: http://uspex-team.org (accessed on 20 August 2023).
- Etris, S.F. Silver and Silver Alloys. In Kirk-Othmer Encyclopedia of Chemical Technology; John Wiley & Sons (Wiley): New York, NY, USA, 2001; Volume 4, pp. 761–803. [Google Scholar]
- Oganov, A.R.; Glass, C.W. Crystal structure prediction using ab initio evolutionary techniques: Principles and applications. J. Chem. Phys. 2006, 124, 244704. [Google Scholar] [CrossRef]
- Oganov, A.R.; Lyakhov, A.O.; Valle, M. How evolutionary crystal structure prediction works—And why. Accounts Chem. Res. 2011, 44, 227–237. [Google Scholar] [CrossRef]
- Lyakhov, A.O.; Oganov, A.R.; Stokes, H.T.; Zhu, Q. New developments in evolutionary structure prediction algorithm USPEX. Comp. Phys. Comm. 2013, 184, 1172–1182. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Vienna Ab-initio Simulation Package. VASP the GUIDE. 20 April 2016. Available online: http://cms.mpi.univie.ac.at/VASP/ (accessed on 20 August 2023).
- Blöchl, P.E.; Jepsen, O.; Andersen, O.K. Improved tetrahedron method for Brillouin-zone integrations. Phys. Rev. B 1994, 49, 16223–16233. [Google Scholar] [CrossRef]
- Hinuma, Y.; Pizzi, G.; Kumagai, Y.; Oba, F.; Tanaka, I. Band structure diagram paths based on crystallography. Comp. Mater. Sci. 2017, 128, 140–184. [Google Scholar] [CrossRef]
- Mouhat, P.; Coudert, F.-X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B 2014, 90, 224104. [Google Scholar] [CrossRef]
- Gusev, A.I.; Sadovnikov, S.I. Conditions of mechanical stability and elastic properties of crystal structures with different symmetry. Fiz. Tverd. Tela 2022, 64, 671–675. (In Russian) [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Sadovnikov, S.I.; Gusev, A.I.; Rempel, A.A. Artificial silver sulfide Ag2S: Crystal structure and particle size in deposited powders. Superlat. Microstr. 2015, 83, 35–47. [Google Scholar] [CrossRef]
- International Tables for X-ray Crystallography; Volume A1: Symmetry Relations between Space Groups; Wondratschek, H.; Müller, U. (Eds.) Kluwer Academic Publishers: Dordrecht, The Netherlands; Boston, NY, USA; London, UK, 2004; 731p. [Google Scholar]
- Kovalev, O.V. Representations of the Crystallographic Space Groups: Irreducible Representations, Induced Representations and Corepresentation, 2nd ed.; Gordon & Breach Science Publications: Yverdon, Switzerland; Paris, France; Berlin, Germany; London, UK; Tokyo, Japan; Amsterdam, The Netherlands, 1993; 390p. [Google Scholar]
- Flurry, R.L. Symmetry Groups: Theory and Chemical Applications; Prentice-Hall Inc.: Englewood Cliffs, NJ, USA, 1980; p. 356. [Google Scholar]
- Gusev, A.I.; Rempel, A.A.; Magerl, A.J. Disorder and Order in Strongly Nonstoichiometric Compounds: Transition Metal Carbides, Nitrides and Oxides; Springer: Berlin, Germany, 2001; 608p. [Google Scholar]
- Kryukov, A.I.; Stroyuk, O.L.; Zin’chuk, N.N.; Korzhak, A.V.; Kuchmii, S.Y. Optical and catalytical properties of Ag2S nanoparticles. J. Mol. Catal. A 2004, 221, 209–221. [Google Scholar] [CrossRef]
- Sadovnikov, S.I.; Kuznetsova, Y.V.; Rempel, A.A. Ag2S silver sulfide nanoparticles and colloidal solutions: Synthesis and properties. Nanostr. Nano-Object. 2016, 7, 81–91. [Google Scholar] [CrossRef]
- Sadovnikov, S.I.; Kozlova, E.A.; Gerasimov, E.Y.; Rempel, A.A.; Gusev, A.I. Enhanced photocatalytic hydrogen evolution from aqueous solutions on Ag2S/Ag heteronanostructure. Int. J. Hydrog. Energy 2017, 42, 25258–25266. [Google Scholar] [CrossRef]
- Liu, Q.; Pu, Y.; Zhao, Z.; Wang, J.; Wang, D. Synthesis of silver sulfide qantum dots via the liquid–liquid interface reaction in a rotating packed bed reactor. Transact. Tianjin Univ. 2020, 26, 273–282. [Google Scholar] [CrossRef]
- Available online: https://matrix.reshish.ru (accessed on 20 August 2023).
- Born, M. On the stability of crystal lattices. Math. Proc. Camb. Phil. Soc. 1940, 36, 160–172. [Google Scholar] [CrossRef]
- Born, M.; Huang, K. Dynamical Theory of Crystal Lattices; Oxford University Press: New York, NY, USA, 1998; 432p. [Google Scholar]
- Pugh, S.F. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Philos. Mag. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. A 1952, 65, 349–354. [Google Scholar] [CrossRef]
- Sadovnikov, S.I. Bulk modulus of coarse-crystalline and nanocrystalline silver sulfides. Phys. Solid State 2018, 69, 2437–2441. [Google Scholar] [CrossRef]
- Chen, X.-Q.; Niu, H.; Li, D.; Li, Y. Modeling hardness of polycrystalline materials and bulk metallic glasses. Intermet 2011, 19, 1275–1281. [Google Scholar] [CrossRef]
- Tian, Y.; Xu, B.; Zhao, Z. Microscopic theory of hardness and design of novel superhard crystals. Int. J. Refr. Metal Hard Mater. 2012, 33, 93–106. [Google Scholar] [CrossRef]
- Mazhnik, E.; Oganov, A.R. A model of hardness and fracture toughness of solids. J. Appl. Phys. 2019, 126, 125109. [Google Scholar] [CrossRef]
Figure 1.
Position of the monoclinic (space group P21/c) unit cell of α-Ag2S (acanthite) in the distorted bcc nonmetal sublattice of argentite. (○) and (●) are S atoms located beyond and inside (within) a monoclinic unit cell of α-Ag2S (acanthite), respectively; (●) are Ag atoms in a monoclinic unit cell of α-Ag2S.
Figure 1.
Position of the monoclinic (space group P21/c) unit cell of α-Ag2S (acanthite) in the distorted bcc nonmetal sublattice of argentite. (○) and (●) are S atoms located beyond and inside (within) a monoclinic unit cell of α-Ag2S (acanthite), respectively; (●) are Ag atoms in a monoclinic unit cell of α-Ag2S.
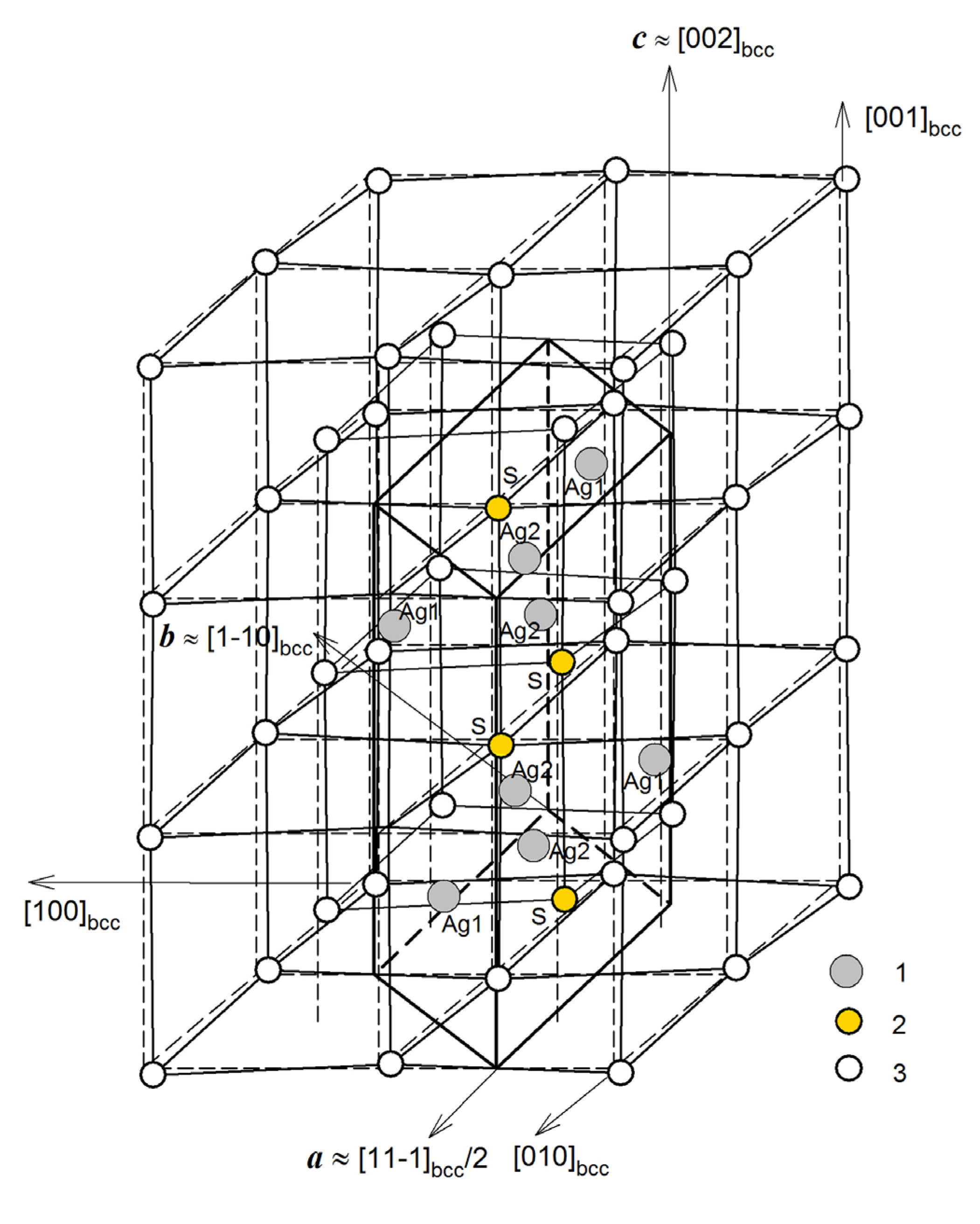
Figure 2.
Variation of the formation enthalpies Hf of some the predicted Ag2S phases with symmetry reduction.
Figure 2.
Variation of the formation enthalpies Hf of some the predicted Ag2S phases with symmetry reduction.
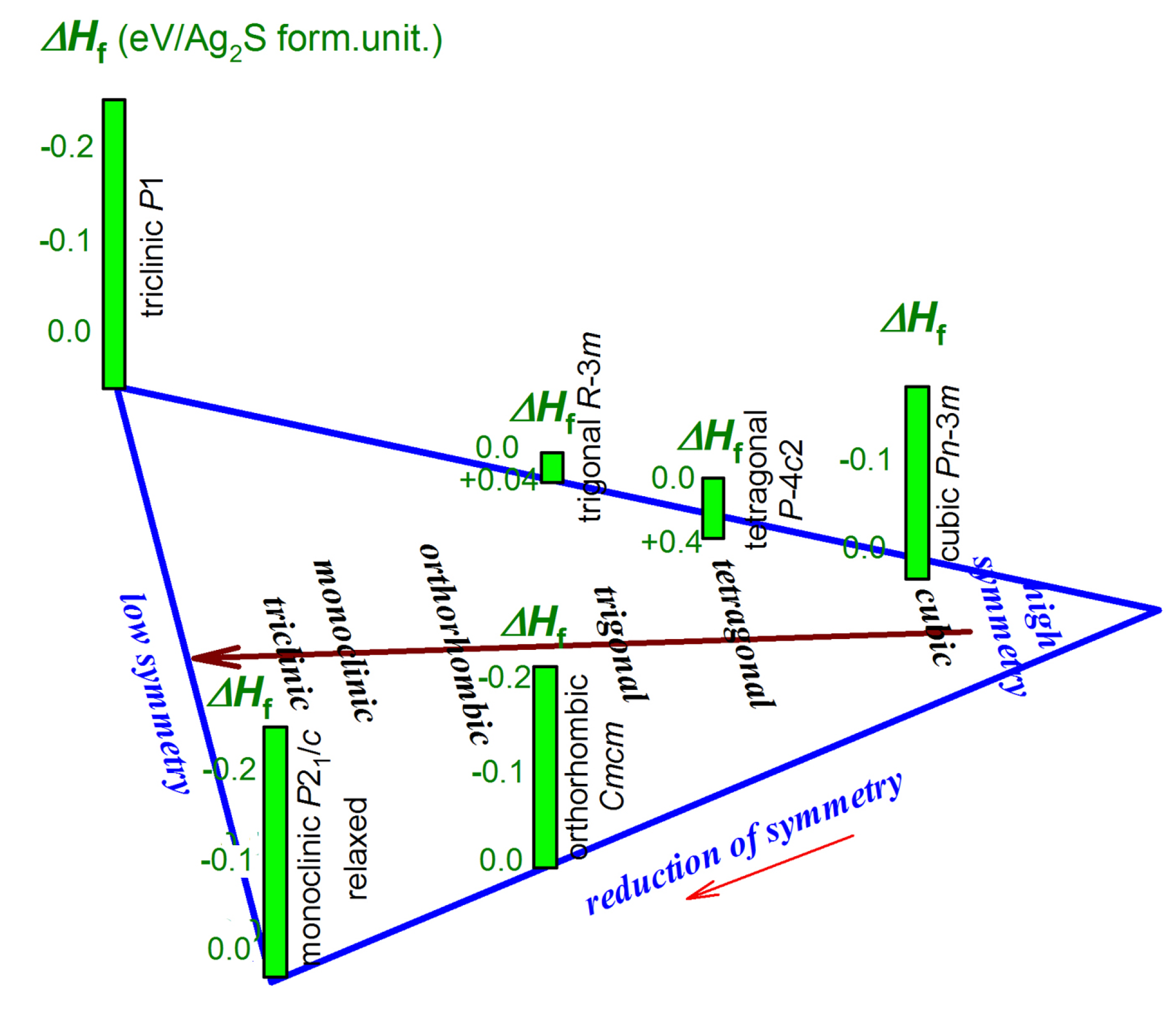
Figure 3.
Unit cells of possible ordered Ag2S structures derived from argentite: (a) model cubic Ag2S phases with space groups No.224- and No.227-, (b) model tetragonal Ag2S phases with space groups No.116- and No.123-P4/mmm, and (c) model trigonal Ag2S phases with space groups No.148- and No.166-.
Figure 3.
Unit cells of possible ordered Ag2S structures derived from argentite: (a) model cubic Ag2S phases with space groups No.224- and No.227-, (b) model tetragonal Ag2S phases with space groups No.116- and No.123-P4/mmm, and (c) model trigonal Ag2S phases with space groups No.148- and No.166-.
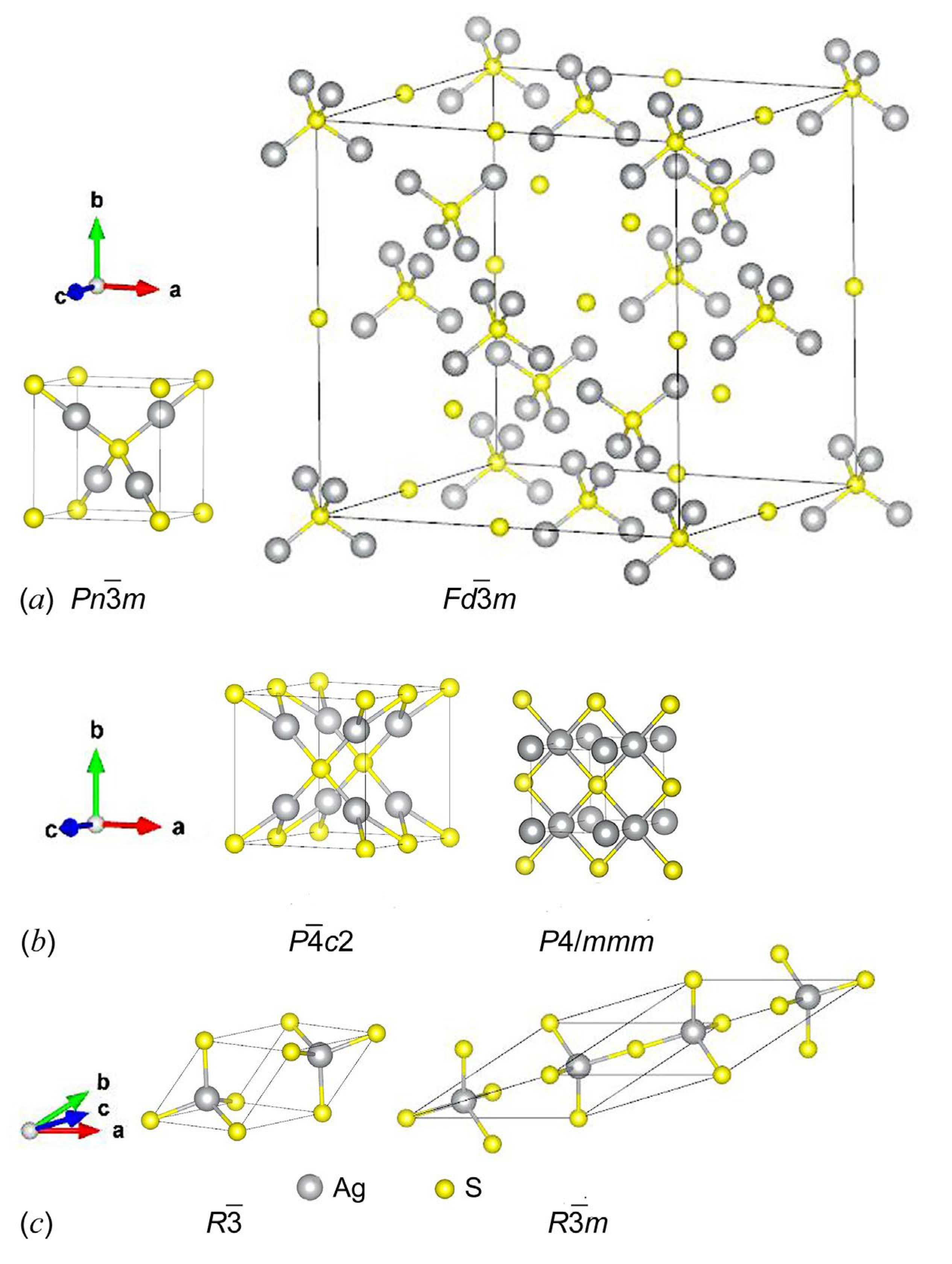
Figure 4.
Unit cells of model orthorhombic Ag2S phases with space groups No.64-Cmce and No.63-Cmcm.
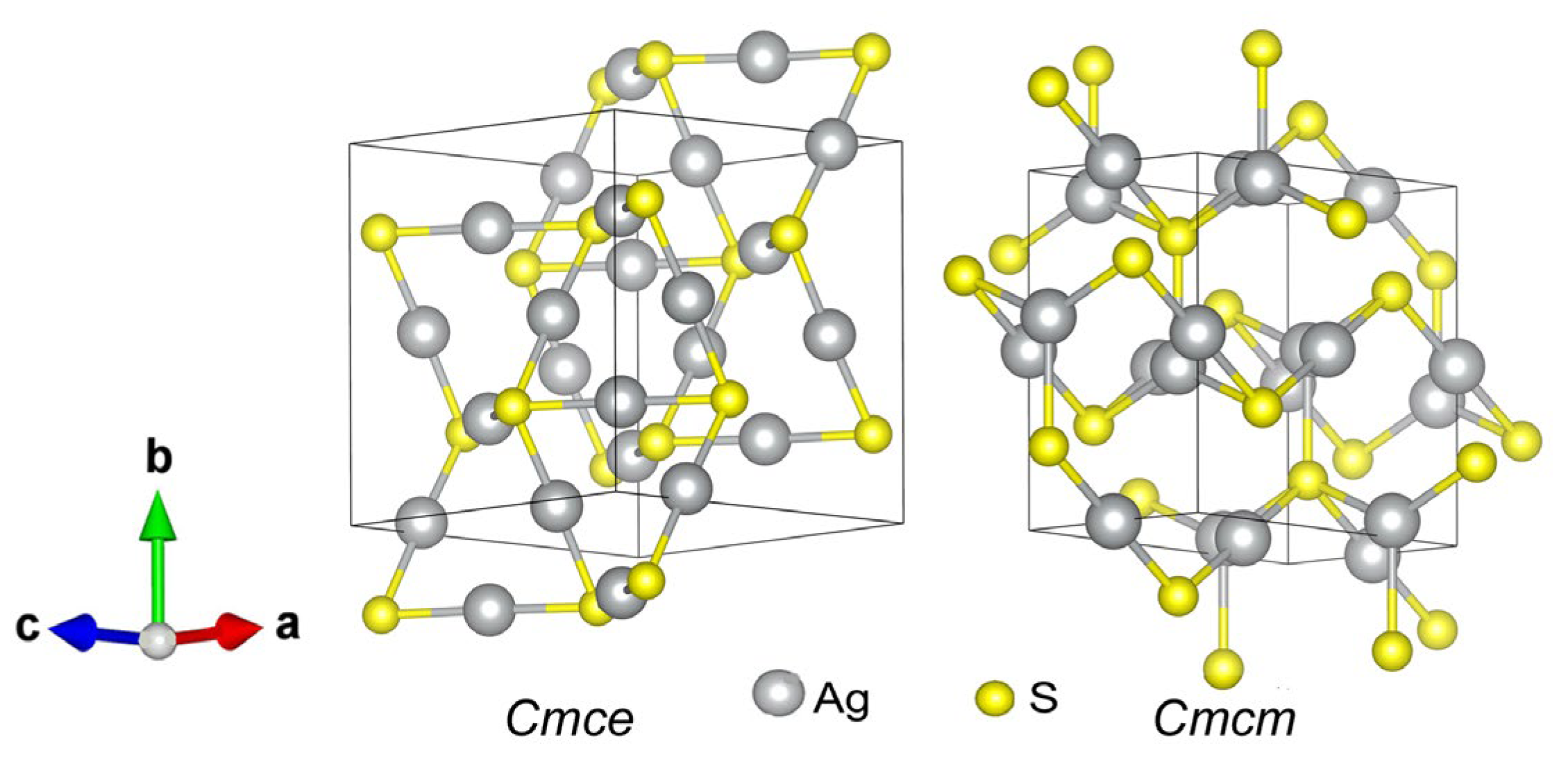
Figure 5.
Unit cells of monoclinic (space group P21/c) phases with α-Ag2S (acanthite) structure: (a) structure before relaxation according to diffraction experiment data [46], (b) structure after relaxation.
Figure 5.
Unit cells of monoclinic (space group P21/c) phases with α-Ag2S (acanthite) structure: (a) structure before relaxation according to diffraction experiment data [46], (b) structure after relaxation.
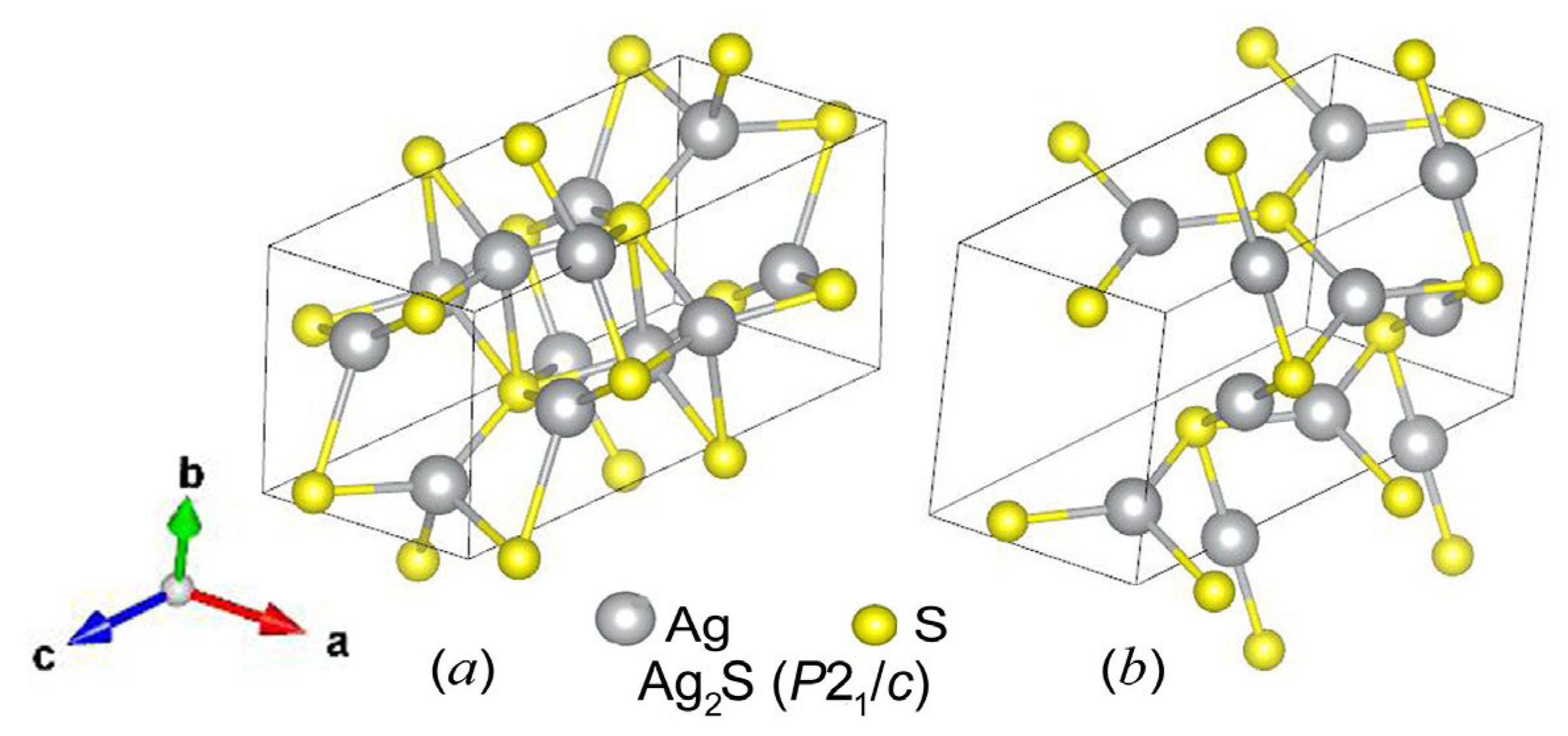
Figure 6.
Calculated XRD patterns (CuKα1,2 radiation) of monoclinic (space group P21/c) phases with an α-Ag2S (acanthite) structure: (a) unrelaxed structure, (b) relaxed structure. The theoretical XRD pattern of unrelaxed acanthite coincides with high accuracy with the experimental XRD pattern of coarse-crystalline monoclinic (space group P21/c) α-Ag2S (acanthite) [65].
Figure 6.
Calculated XRD patterns (CuKα1,2 radiation) of monoclinic (space group P21/c) phases with an α-Ag2S (acanthite) structure: (a) unrelaxed structure, (b) relaxed structure. The theoretical XRD pattern of unrelaxed acanthite coincides with high accuracy with the experimental XRD pattern of coarse-crystalline monoclinic (space group P21/c) α-Ag2S (acanthite) [65].
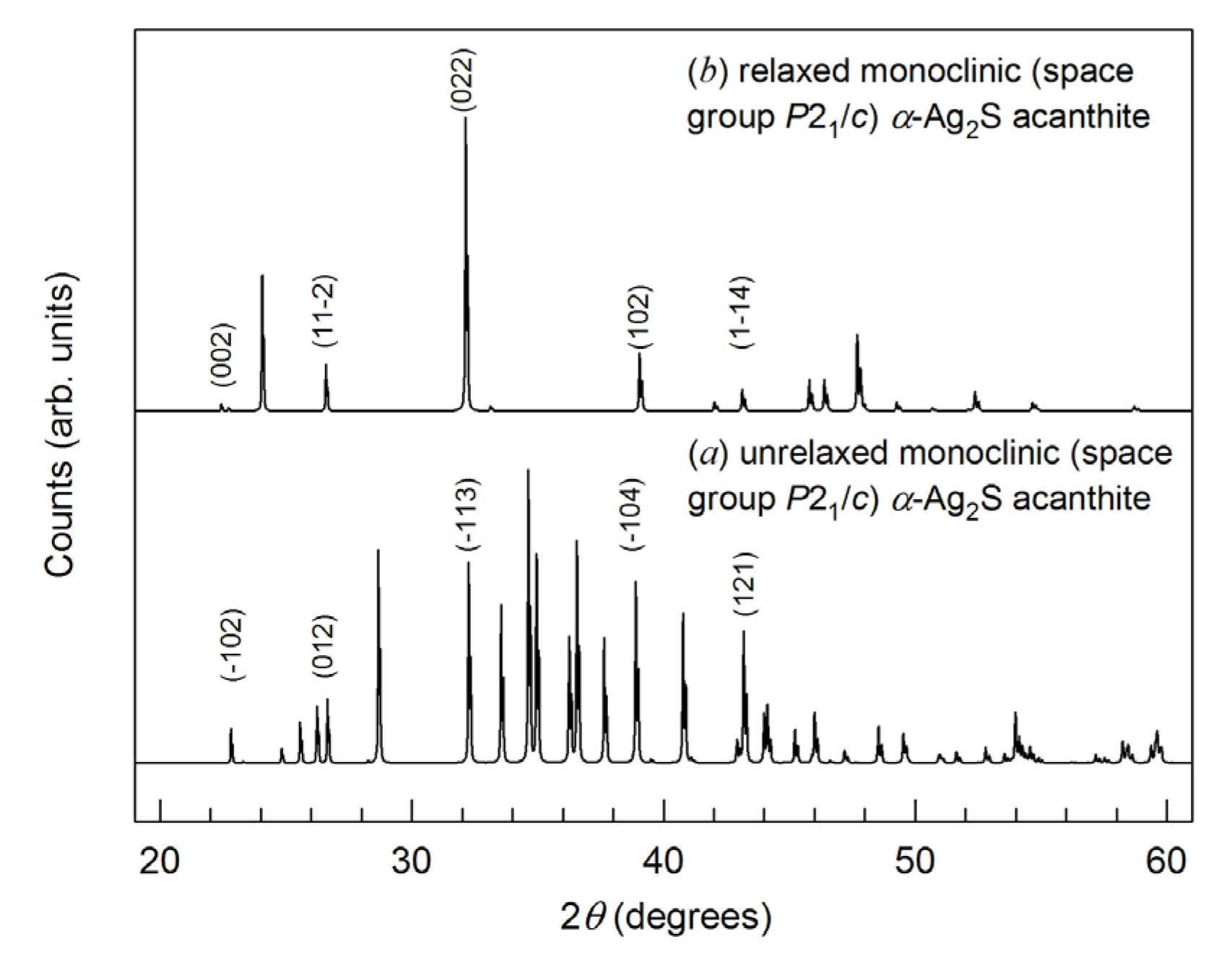
Figure 7.
Unit cell of the model monoclinic (space group P21/c) Ag2S phase (left), with the lowest formation enthalpy ΔHf = −0.219 eV/form.unit among the predicted monoclinic structures, and unit cell of the model triclinic (space group P1) Ag2S phase (right) with the lowest formation enthalpy ΔHf = −0.223 eV/form.unit.
Figure 7.
Unit cell of the model monoclinic (space group P21/c) Ag2S phase (left), with the lowest formation enthalpy ΔHf = −0.219 eV/form.unit among the predicted monoclinic structures, and unit cell of the model triclinic (space group P1) Ag2S phase (right) with the lowest formation enthalpy ΔHf = −0.223 eV/form.unit.
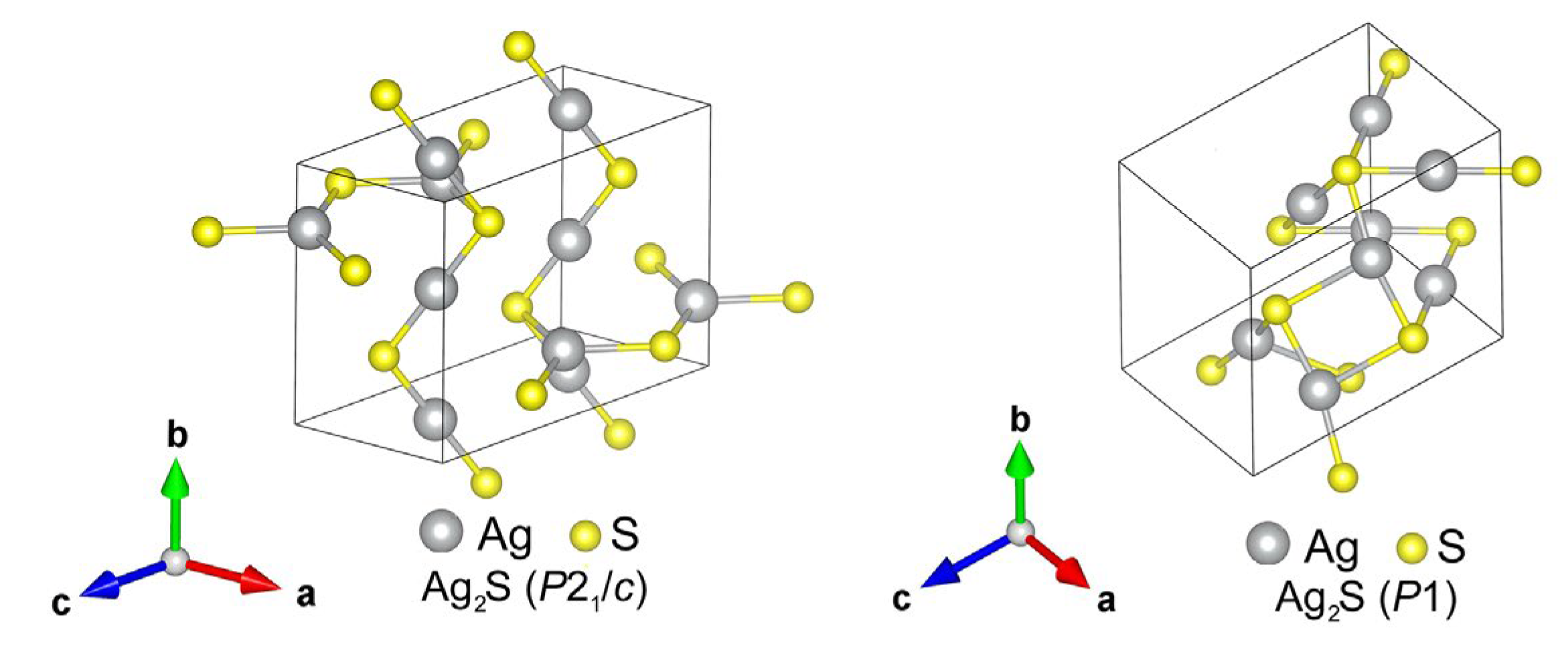
Figure 8.
Band structure of (a) unrelaxed and (b) relaxed monoclinic (space group P21/c) α-Ag2S (acanthite).
Figure 8.
Band structure of (a) unrelaxed and (b) relaxed monoclinic (space group P21/c) α-Ag2S (acanthite).
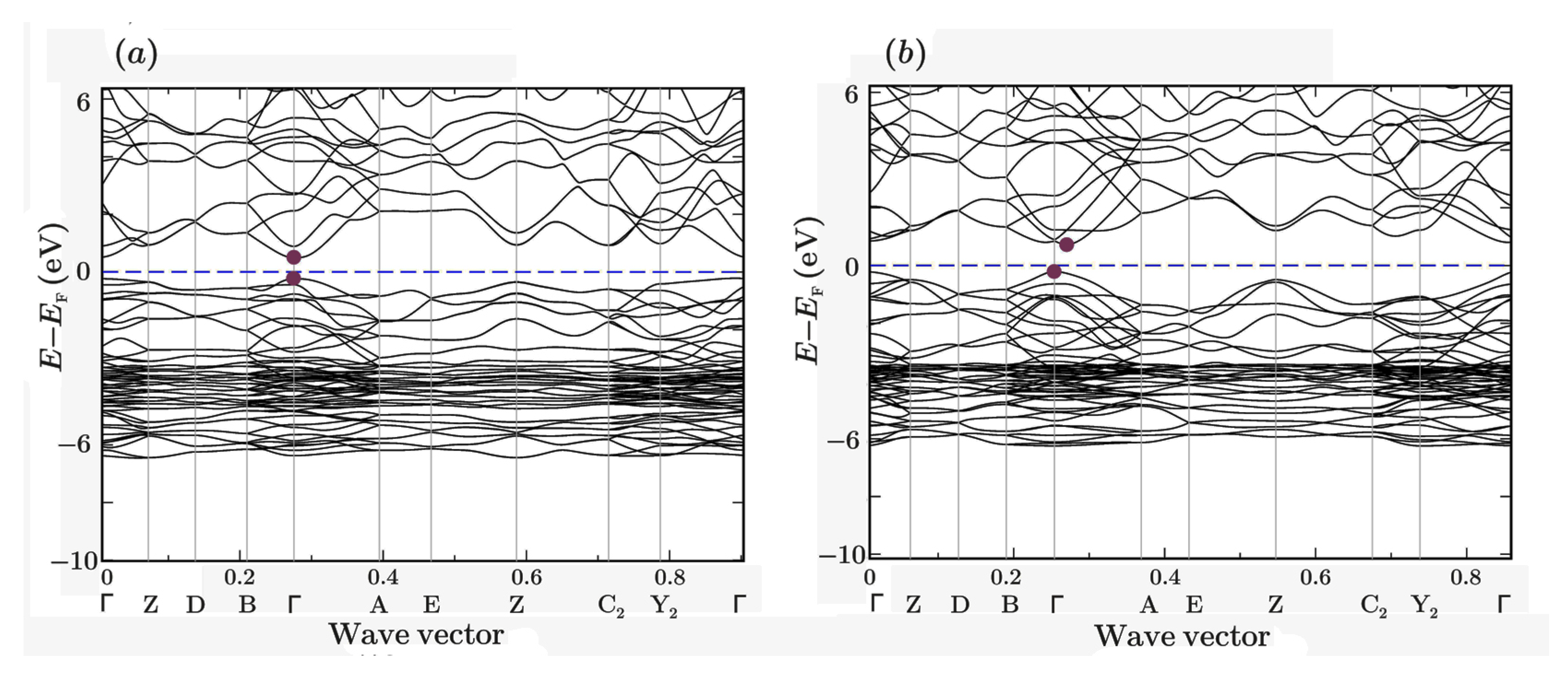
Figure 9.
Total and partial Ag 4d-, Ag 5s-, and S 3p-densities of electronic states (DOS) computed for (a) unrelaxed and (b) relaxed monoclinic (space group P21/c) α-Ag2S (acanthite) and (c) the proposed ordered cubic (space group ) Ag2S phase, which is a derivative of the argentite structure.
Figure 9.
Total and partial Ag 4d-, Ag 5s-, and S 3p-densities of electronic states (DOS) computed for (a) unrelaxed and (b) relaxed monoclinic (space group P21/c) α-Ag2S (acanthite) and (c) the proposed ordered cubic (space group ) Ag2S phase, which is a derivative of the argentite structure.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Calculated cohesion energy (Ecoh), formation enthalpy (ΔHf), rotational (Nrot), translational (Ntr), and total (Ntot) reductions of symmetry for predicted silver sulfide phases.
Table 1.
Calculated cohesion energy (Ecoh), formation enthalpy (ΔHf), rotational (Nrot), translational (Ntr), and total (Ntot) reductions of symmetry for predicted silver sulfide phases.
| Symmetry and Space Group | Number of Atoms | 1 Z | 2 Ephase, eV | E coh | 3 ΔHf | N rot | 4 Ntr | N tot | |
|---|---|---|---|---|---|---|---|---|---|
| N Ag | N S | eV/(Ag2S form.unit) | |||||||
| Cubic (No.224-) | 4 | 2 | 2 | −19.498 | −8.272 | −0.191 | 1 | 1.5 | 1.5 |
| Cubic (No.227-) | 32 | 16 | 16 | −95.749 | −4.507 | 3.573 | 1 | 8 | 8 |
| Tetragonal (No.116-) | 8 | 4 | 4 | −36.655 | −7.687 | 0.394 | 6 | 2 | 12 |
| Tetragonal (No.123-P4/mmm) | 2 | 1 | 1 | −8.2445 | −6.767 | 1.313 | 3 | 0.5 | 1.5 |
| Trigonal (No.148 -) | 2 | 1 | 1 | −9.516 | −8.039 | 0.042 | 8 | 0.5 | 4 |
| Trigonal (No.166 -) | 4 | 2 | 2 | −19.033 | −8.039 | 0.041 | 4 | 1 | 4 |
| Orthorhombic (No.64-Cmce) | 16 | 8 | 8 | −78.216 | −8.300 | −0.219 | 6 | 5 | 30 |
| Orthorhombic (No.63-Cmcm) | 8 | 4 | 4 | −39.027 | −8.280 | −0.199 | 6 | 2 | 12 |
| Orthorhombic (No.36-Cmc21) | 8 | 4 | 4 | −39.027 | −8.280 | −0.199 | 12 | 2 | 24 |
| Orthorhombic (No.19-P212121) | 8 | 4 | 4 | −38.384 | −8.119 | −0.038 | 12 | 2 | 24 |
| Monoclinic (No.11-P21/m) | 4 | 2 | 2 | −19.514 | −8.280 | −0.199 | 12 | 1 | 12 |
| Monoclinic (No.4-P21) | 4 | 2 | 2 | −19.532 | −8.289 | −0.208 | 24 | 1.5 | 36 |
| Monoclinic (No.14-P21/c) unrelaxed acanthite | 8 | 4 | 4 | −38.361 | −8.113 | −0.033 | 12 | 2 | 24 |
| Monoclinic (No.14-P21/c) relaxed acanthite | 8 | 4 | 4 | −39.028 | −8.280 | −0.199 | 12 | 2.5 | 30 |
| Monoclinic (No.14-P21/c) | 8 | 4 | 4 | −39.107 | −8.300 | −0.219 | 12 | 3 | 36 |
| Triclinic (No.1-P1) | 8 | 4 | 4 | −39.125 | −8.304 | −0.223 | 48 | 2 | 96 |
1 The number of Ag2S formula units in the predicted silver sulfide phase (Z coincides with the number of S atoms in the predicted phase); 2 Ephase: the energy of the predicted Ag2S structure obtained via the DFT calculation using the VASP code; 3 the estimated relative standard deviations of the formation enthalpies do not exceed 0.1%; 4 the reduction in translational symmetry is estimated as relative change in the unit cell volume of the predicted phase in comparison with the unit cell volume of argentite.
Table 2.
Model predicted orthorhombic Ag2S structures.
| Space Group | Atom | Position and Multiplicity | Atomic Coordinates in the Model Structures | ||
|---|---|---|---|---|---|
| x/a ≡ x/aorthorh | y/b ≡ y/borthorh | z/c ≡ z/corthorh | |||
| * No.63-Cmcm | Ag1 | 4(b) | 0 | 0.5 | 0 |
| Ag2 | 4(c) | 0 | 0.04657 | 0.25 | |
| S | 4(c) | 0 | 0.68496 | 0.25 | |
| ** No. 64-Cmce | Ag1 | 8(e) | 0.25 | 0.22162 | 0.25 |
| Ag2 | 8(f) | 0 | 0.02841 | 0.75496 | |
| S | 8(f) | 0 | 0.72452 | 0.39559 | |
| *** No.36-Cmc21 | Ag | 8(b) | 0 | 0.04705 | 0.06242 |
| S | 4(a) | 0 | 0.31480 | 0.56242 | |
| **** No.19-P212121 | Ag1 | 4(a) | 0.00985 | 0.77359 | 0.64205 |
| Ag2 | 4(a) | 0.15219 | 0.38829 | 0.45552 | |
| S | 4(a) | 0.12297 | 0.00356 | 0.34389 | |
* Parameters of the predicted unit cell (space group Cmcm): a = 0.461442 nm, b = 0.738306 nm, c = 0.791054 nm, V = 0.269500 nm3, and Z = 4; ** parameters of the predicted unit cell (space group Cmce): a = 0.832398 nm, b = 0.834841 nm, c = 0.862424 nm, V = 0.599315 nm3, and Z = 8; *** parameters of the predicted unit cell (space group Cmc21): a = 0.461617 nm, b = 0.737986 nm, c = 0.790727 nm, V = 0.269374 nm3, and Z = 4; **** parameters of the predicted unit cell (space group P212121): a = 0.438446 nm, b = 0.694766 nm, c = 0.733716 nm, V = 0.223502 nm3, and Z = 4.
Table 3.
Monoclinic (space group No.14-P21/c) α-Ag2S (acanthite) structures before relaxation [29] and after relaxation.
Table 3.
Monoclinic (space group No.14-P21/c) α-Ag2S (acanthite) structures before relaxation [29] and after relaxation.
| Monoclinic (Space Group P21/c) α-Ag2S | Atom | Position and Multiplicity | Atomic Coordinates in the Model Structures | ||
|---|---|---|---|---|---|
| x/a ≡ x/amon | y/b ≡ y/bmon | z/c ≡ z/cmon | |||
| * Unrelaxed unit cell [46] | Ag1 | 4(e) | 0.07157 | 0.48487 | 0.80943 |
| Ag2 | 4(e) | 0.27353 | 0.67586 | 0.56247 | |
| S | 4(e) | 0.4922 | 0.2341 | 0.13217 | |
| ** Relaxed unit cell | Ag1 | 4(e) | 0.04498 | 0.74996 | 0.47750 |
| Ag2 | 4(e) | 0.50004 | 0.00001 | 0.25002 | |
| S | 4(e) | 0.31581 | 0.25002 | 0.34210 | |
* Parameters of the unrelaxed unit cell (space group P21/c): a = 0.42264 nm, b = 0.69282 nm, c = 0.953171 nm, α = 90°, β = 125.554°, γ = 90°, V = 0.227068 nm3, and Z = 4; ** parameters of the relaxed unit cell (space group P21/c): a = 0.435628 nm, b = 0.791975, c = 0.871257 nm, α = 90°, β = 116.111°, γ = 90°, V = 0.269911 nm3, and Z = 4.
Table 4.
Model predicted monoclinic (space group No.14-P21/c) Ag2S structure.
| Atom | Position and Multiplicity | Atomic Coordinates in the Model Structures | ||
|---|---|---|---|---|
| x/a ≡ x/amon | y/b ≡ y/bmon | z/c ≡ z/cmon | ||
| Ag1 | 4(e) | 0.52641 | 0.24248 | 0.47333 |
| Ag2 | 4(e) | 0.02658 | 0.25188 | 0.47346 |
| S | 4(e) | 0.52441 | 0.39461 | 0.22983 |
Parameters of the unrelaxed unit cell (space group P21/c): a = 0.590253 nm, b = 0.863974 nm, c = 0.836796 nm, α = 90°, β = 135.1413°, γ= 90°, V = 0.301002437 nm3, and Z = 4.
Table 5.
Model triclinic (space group No.1–P1) Ag2S structure.
| Atom | Position and Multiplicity | Atomic Coordinates in the Model Structures | ||
|---|---|---|---|---|
| x/a ≡ x/atricl | y/b ≡ y/btricl | z/c ≡ z/ctricl | ||
| Ag1 | 1(a) | 0.86160 | 0.11768 | 0.65392 |
| Ag2 | 1(a) | 0.18251 | 0.45397 | 0.34767 |
| Ag3 | 1(a) | 0.68702 | 0.45957 | 0.35685 |
| Ag4 | 1(a) | 0.02431 | 0.56922 | 0.00840 |
| Ag5 | 1(a) | 0.52371 | 0.56411 | 0.00802 |
| Ag6 | 1(a) | 0.36410 | 0.10713 | 0.66484 |
| Ag7 | 1(a) | 0.00327 | 0.00177 | 0.00153 |
| Ag8 | 1(a) | 0.50720 | 0.00312 | 0.01042 |
| S1 | 1(a) | 0.62789 | 0.41309 | 0.70515 |
| S2 | 1(a) | 0.90766 | 0.15612 | 0.30911 |
| S3 | 1(a) | 0.11466 | 0.85369 | 0.70273 |
| S4 | 1(a) | 0.42086 | 0.71554 | 0.31158 |
Parameters of the triclinic unit cell (space group P1): a = 0.593602 nm, b = 0.706832 nm, c = 0.781798 nm, α = 116.4408°, β = 110.9258°, γ = 91.3246°. V = 0.267753731 nm3, and Z = 4.
Table 6.
Calculated bulk (BH) and shear (GH) elastic moduli (GPa), inverse Pugn’s ratio, k = GH/BH, Poisson’s ratio (μ), Young’s modulus (EH), and Vickers hardness (HV) (GPa) for polycrystalline predicted Ag2S (silver sulfides) with the lowest formation enthalpies.
Table 6.
Calculated bulk (BH) and shear (GH) elastic moduli (GPa), inverse Pugn’s ratio, k = GH/BH, Poisson’s ratio (μ), Young’s modulus (EH), and Vickers hardness (HV) (GPa) for polycrystalline predicted Ag2S (silver sulfides) with the lowest formation enthalpies.
| Symmetry, Space Group | G V | G R | G H | B V | B R | B H | k | μ | E H | H V | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Equation (5) | Equation (6) | ||||||||||
| Orthorhombic (Cmce) | 13.0 | 3.6 | 8.3 | 18.0 | 10.3 | 14.1 | 0.588 | 0.277 | 20.0 | 2.0 | 1.0 |
| Orthorhombic (Cmcm) | 11.8 | 8.6 | 10.2 | 34.9 | 21.9 | 28.4 | 0.360 | 0.359 | 25.9 | 1.2 | 1.5 |
| Monoclinic (P21) | 13.4 | −2.3 | 5.6 | 13.7 | −2.9 | 5.4 | 1.032 | 0.133 | 12.2 | 3.2 | 1.0 |
| Monoclinic (P21/c) unrelaxed acanthite | 11.6 | 8.6 | 10.1 | 34.3 | 27.9 | 31.1 | 0.326 | 0.371 | 25.9 | 1.0 | 1.5 |
| Monoclinic (P21/c) relaxed acanthite | 11.3 | 8.3 | 9.8 | 35.1 | 21.5 | 28.3 | 0.348 | 0.363 | 25.0 | 1.1 | 1.4 |
| Monoclinic (P21/c) | −0.8 | 12.4 | 5.8 | 25.3 | 61.3 | 43.3 | 0.134 | 0.445 | 15.5 | 0.2 | 0.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sadovnikov, S.I.; Kostenko, M.G.; Gusev, A.I.; Lukoyanov, A.V. Low-Temperature Predicted Structures of Ag2S (Silver Sulfide). Nanomaterials 2023, 13, 2638. https://doi.org/10.3390/nano13192638
AMA Style
Sadovnikov SI, Kostenko MG, Gusev AI, Lukoyanov AV. Low-Temperature Predicted Structures of Ag2S (Silver Sulfide). Nanomaterials. 2023; 13(19):2638. https://doi.org/10.3390/nano13192638
Chicago/Turabian StyleSadovnikov, Stanislav I., Maksim G. Kostenko, Aleksandr I. Gusev, and Aleksey V. Lukoyanov. 2023. "Low-Temperature Predicted Structures of Ag2S (Silver Sulfide)" Nanomaterials 13, no. 19: 2638. https://doi.org/10.3390/nano13192638
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.