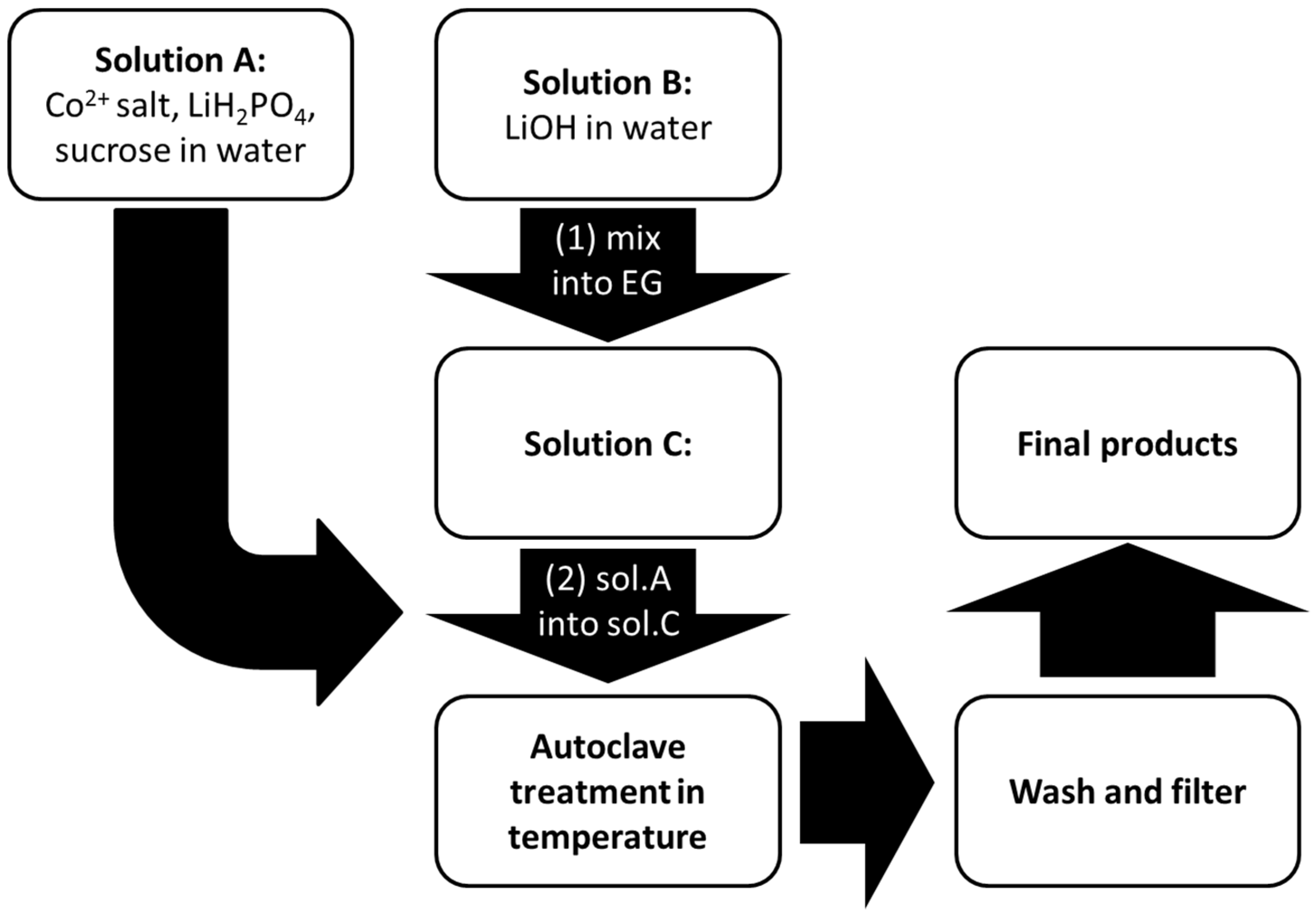
2.1. Synthesis of the LCP Materials
LCP materials have been synthesized by following a solvo-thermal route [
6]. A flowchart of the synthetic procedure is shown in the
Figure 1.
In the first step, two aqueous solutions containing LiH2PO4, sucrose (as a reducing agent to prevent possible oxidation of Co2+ to Co3+), and a Co2+ salt precursor (solution A), and LiOH (solution B) respectively, have been prepared under vigorous stirring. The amount of sucrose has always been set in respect to the cobalt precursor with a molar ratio Co2+:sucrose = 1:0.03. The relative amount of LiOH has been optimized in order to obtain a moderately-acidic solution to facilitate the precipitation of the olivine-phosphate. More details are discussed in the Results section. Subsequently the solution B has been dissolved in ethylene glycol (EG) to give solution C. The total H2O:EG volume ratio has been set to 1:2. Afterward, solution A has been added dropwise into solution C under stirring. The final slurry (solution D) has been poured into a Teflon vessel in a stainless steel autoclave. The final volume of the final solution has been set to 30 mL with a nominal [Co2+] concentration of 0.1 M. After sealing, the autoclave has been kept in an oven at 200–240 °C for few hours and then allowed to cool naturally to room temperature.
The resulting suspension has been filtered and the solid pinkish precipitate has been washed in deionized water and ethanol, and then oven-dried at 80 °C. Three different reaction temperatures (200 °C, 220 °C, and 240 °C) have been investigated and the reaction time optimized to each temperature condition (see next section). Four different Co2+ sources have been used: Co(NO3)2, CoSO4, Co(CH3COO)2, and CoCO3. A variant of this synthesis route has been developed in the case of LCP from carbonate. Due to the low Ksp (1 × 10−10 at 25 °C) of the cobalt (II) carbonate, the Co2+ precursor has been directly suspended in EG. Therefore, starting solutions are A (CoCO3 in EG), B (sucrose and LiH2PO4 in H2O), and C (LiOH in H2O). Solution B has been dropped into solution D, obtained by joining A and C.
Figure 1.
Schematic diagram of the synthesis route of LiCoPO4 (LCP).
Figure 1.
Schematic diagram of the synthesis route of LiCoPO4 (LCP).
2.2. Analysis of the Synthesis Conditions: Effect of the Solution Acidity
As mentioned above, the nature and ratio of the reagents, and in particular the amount of LiOH in the starting mixture, have been set in order to obtain a slightly acidic solution to facilitate the precipitation of the olivine phosphate. The precipitation of pure LCP samples from solutions at low temperature is a challenging task [
10]. Delacourt
et al. in reference [
10] calculated the solid-state repartition diagram in water as a function of pH and cobalt(II) concentration, thus highlighting an optimal pH region in aqueous solution for the precipitation of LCP (pH range 6.3–9.5). However their attempt to precipitate the pure LCP phase from water solutions failed due to the apparently unavoidable precipitation of competing phases, such as Co
3(PO
4)
2, Li
3PO
4, and Co(OH)
2 [
10]. Our case is even more complex due to the use of a mixed EG-H
2O solvent blend. In this view we performed an experimental screening of the [LiOH]/[LiH
2PO
4]/[Co
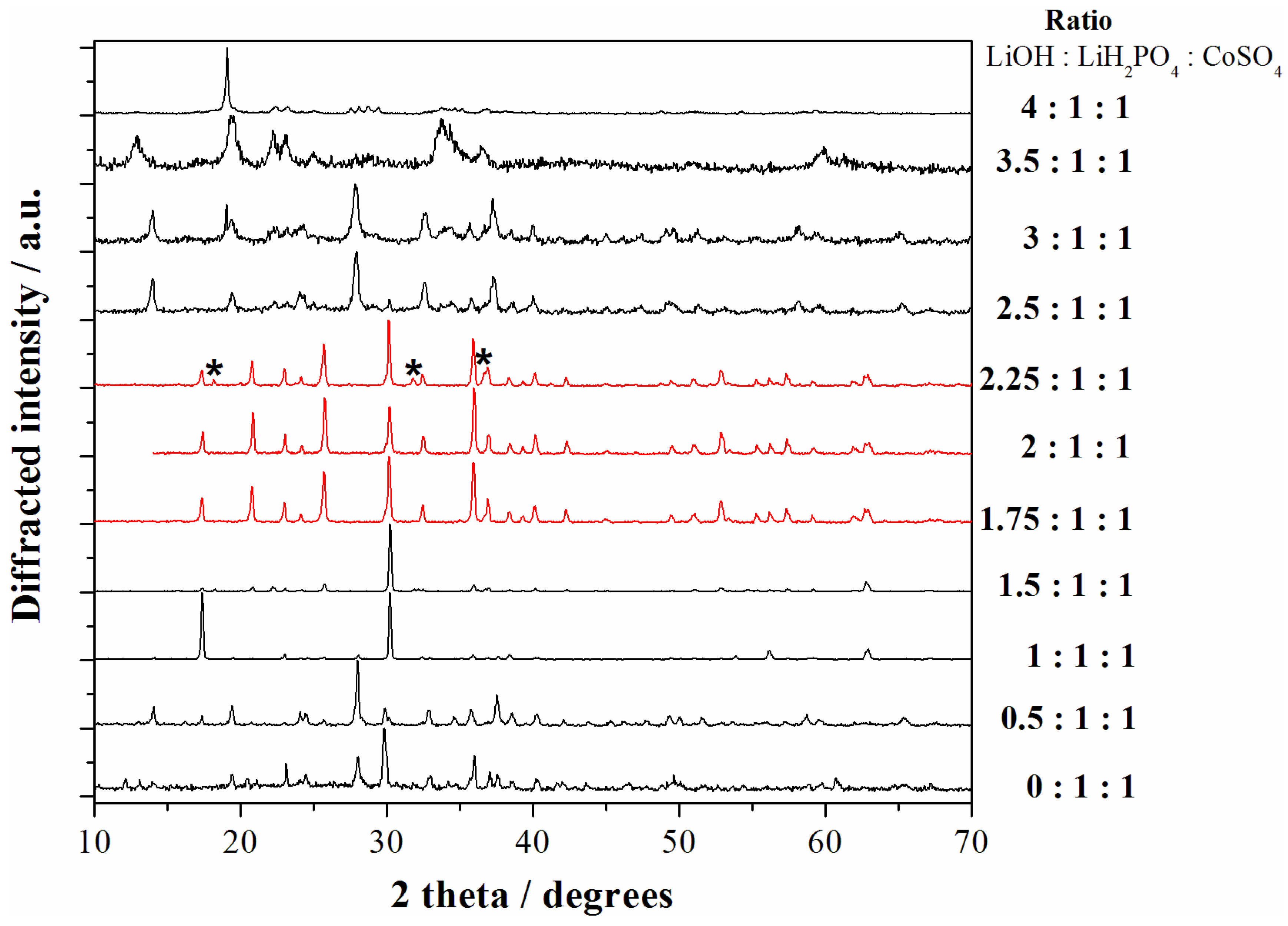
2+] ratio in the case of the cobalt sulfate salt in order to identify the most suitable range to obtain phase pure LCP particles. The X-ray diffraction (XRD) patterns of all the samples obtained starting from the reagent ratios LiOH:LiH
2PO
4:CoSO
4 ranging from 0:1:1 to 4:1:1 are shown in the
Figure 2.
Figure 2.
X-ray diffraction (XRD) patterns of all the samples obtained starting from the reagent ratios LiOH:LiH2PO4:CoSO4 ranging from 0:1:1 to 4:1:1. The red patterns correspond to the LCP reference diffractograms ((*) indicate peaks from impurities) whereas black patterns correspond to other reaction products. All materials have been obtained at 240 °C with solvo-thermal treatments of 5 h.
Figure 2.
X-ray diffraction (XRD) patterns of all the samples obtained starting from the reagent ratios LiOH:LiH2PO4:CoSO4 ranging from 0:1:1 to 4:1:1. The red patterns correspond to the LCP reference diffractograms ((*) indicate peaks from impurities) whereas black patterns correspond to other reaction products. All materials have been obtained at 240 °C with solvo-thermal treatments of 5 h.
Apparently the precipitation of the LCP phase (pure or almost pure) can be obtained in the LiOH:LiH
2PO
4:CoSO
4 ratios range within 1.75:1:1 and 2.25:1:1. Our results are therefore in agreement with the evaluation for water solution from Delancourt
et al. [
10]. In particular, neutral (reagents ratios 2:1:1) and moderately acidic pH (reagents ratio 1.75:1:1) lead to phase pure samples constituted by LCP without impurities. In fact the lack of unindexed peaks in the corresponding XRD patterns confirms the phase purity of these two samples and the absence of contaminant phases above 2%–3% in volume, which is the typically accepted detection limit for phase identification by powder XRD [
11]. All the other precipitation conditions lead to formation of other reaction products and have been, therefore, discarded.
On the other hand, from the point of view of the final elemental composition the two LCP-phase pure samples obtained in moderately acidic or neutral pH are not equivalent. In fact, whereas the material prepared starting from a reagent ratio LiOH:LiH2PO4:CoSO4 = 2:1:1 shows an experimental Li:Co ICP-AES ratio of 1.06:0.98, the material prepared in moderately acidic conditions (LiOH:LiH2PO4:CoSO4 = 1.75:1:1) has an almost stoichiometric LiCoPO4 experimental composition (Li:Co = 1.01:1.00). The difference in the elemental ratios between the samples prepared in neutral or moderately acidic conditions may imply the precipitation of minor contaminant phases in the first case, below the abovementioned detection limit of the XRD technique, or off-stoichiometric defects in the olivine lattice (see next section).
2.3. Analysis of the Synthesis Conditions: Effect of the Reaction Time
The evolution of the phase composition of the precipitates obtained from the solvo-thermal bath at 240 °C have been studied upon time for the reagent ratios LiOH:LiH
2PO
4:CoSO
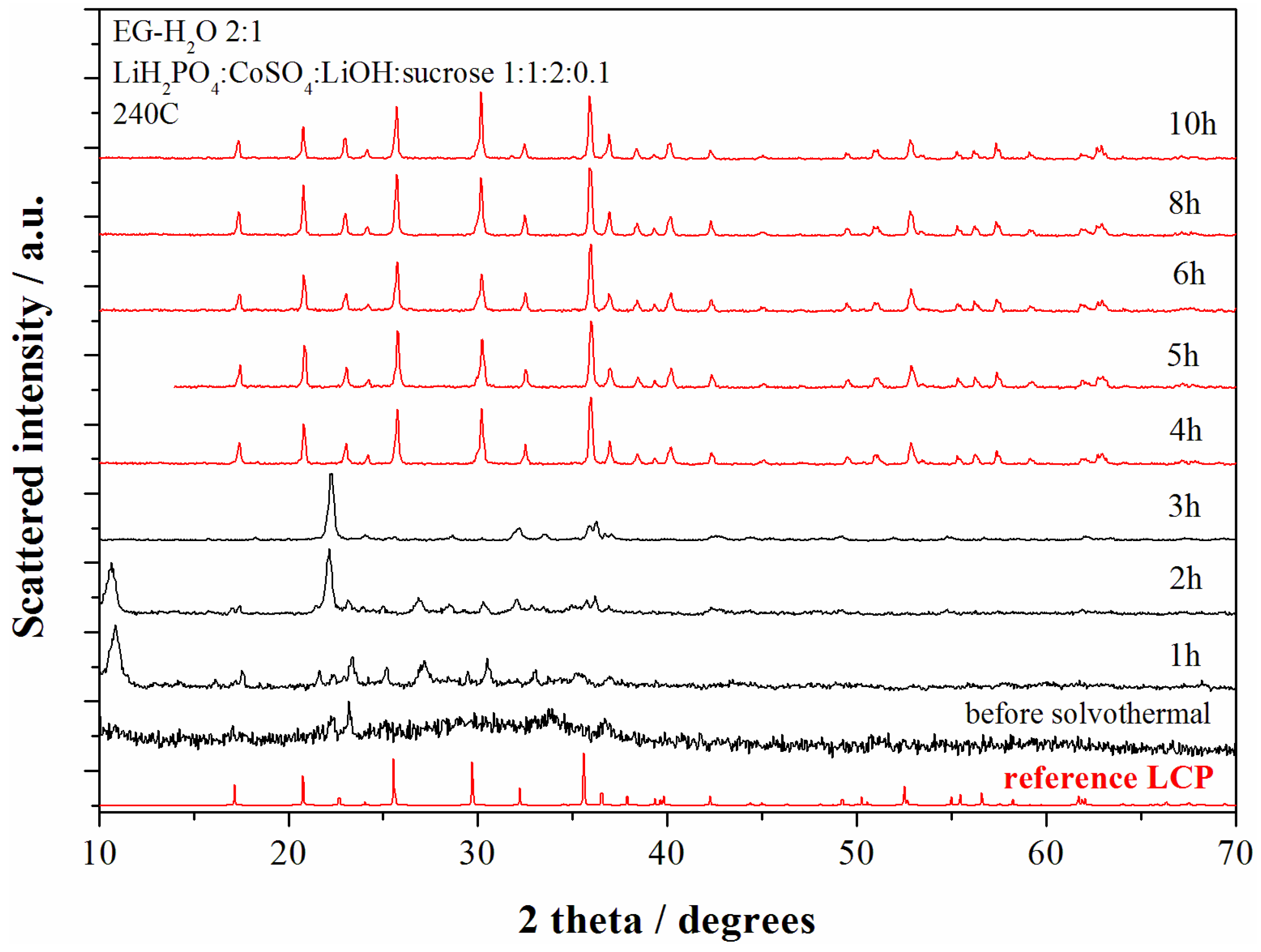
4 = 2:1:1 (neutral pH conditions). The XRD patterns of the materials obtained at various reaction time are shown in the
Figure 3.
Figure 3.
XRD patterns of the samples obtained starting from the reagent ratio LiOH:LiH2PO4:CoSO4 = 2:1:1 at 240 °C for different reaction time. The red patterns correspond to the LCP reference diffractograms whereas black patterns correspond to other reaction products.
Figure 3.
XRD patterns of the samples obtained starting from the reagent ratio LiOH:LiH2PO4:CoSO4 = 2:1:1 at 240 °C for different reaction time. The red patterns correspond to the LCP reference diffractograms whereas black patterns correspond to other reaction products.
The pristine product (before solvo-thermal treatment) obtained at the end of the starting solution mixing (violet precipitate) reveals an amorphous product with few broad unindexed peaks. After 1 h of solvo-thermal treatment Bragg reflections from crystalline phases start to become. After 4 h of reaction time at 240 °C the XRD pattern shows only reflections from the olivine LCP and no further structural evolution is observed up to 10 h of reaction.
The composition of the synthesized materials show a constant evolution with the reaction time. In fact the Li:Co ratio decreases monotonically from 1.85 (1 h), 1.37 (2 h), 1.15 (3 h), 1.10 (4 h), 1.08 (5 h), to 1.04 (10 h). The lithium excess in the LCP-pure samples (4–5–6–8–10 h) may suggest the presence of minor contaminants (below the XRD detection limit) or over-lithiation of the LCP olivine lattice.
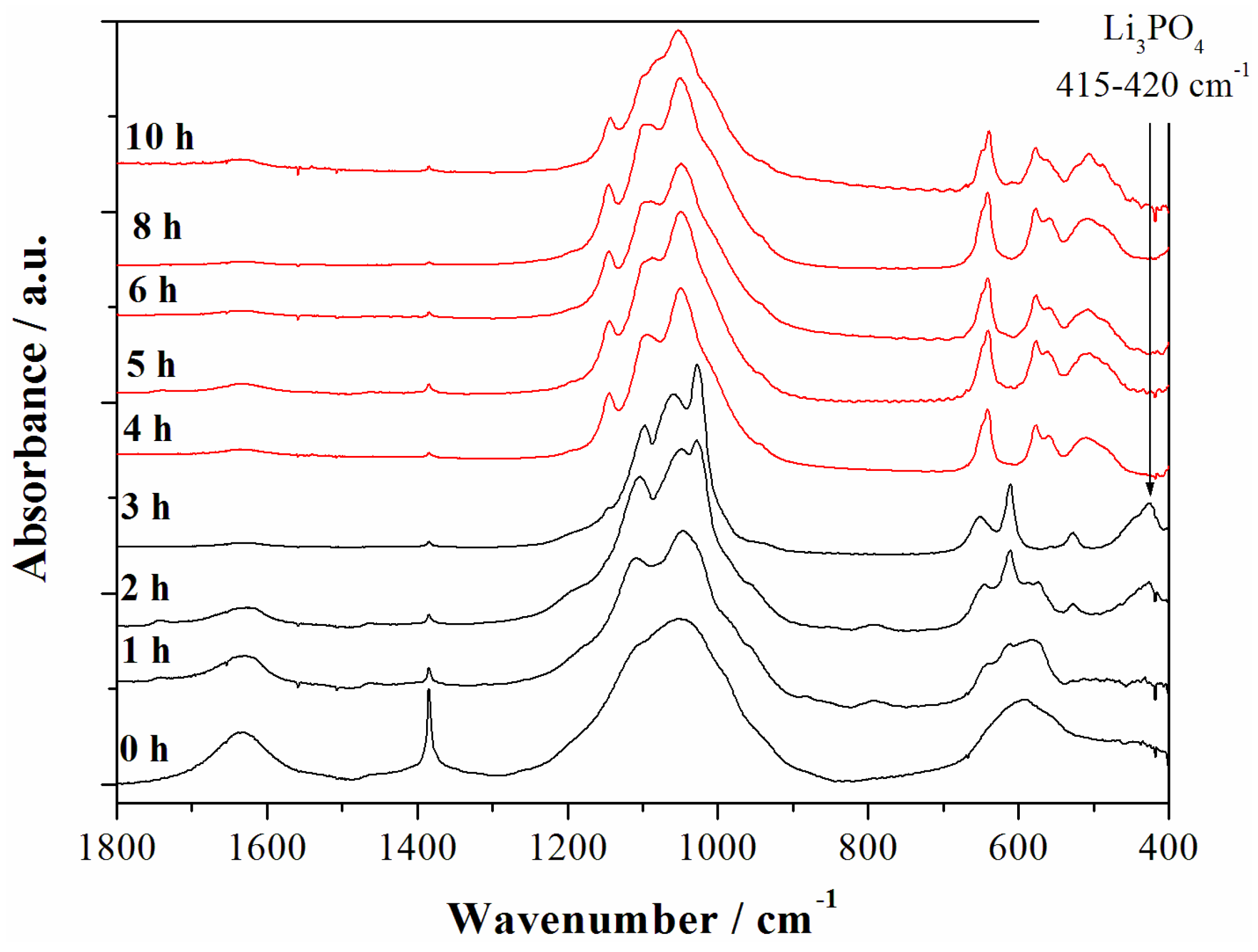
In order to check the possible precipitation of minor contaminant species in the synthesized materials the evolution of the Fast-Fourier Transform infrared spectroscopy (FTIR) spectra of the materials prepared at various reaction time have been studied as shown in the
Figure 4.
Figure 4.
Fast-Fourier Transform infrared spectroscopy (FTIR) spectra of the samples obtained starting from the reagent ratios LiOH:LiH
2PO
4:CoSO
4 = 2:1:1 at 240 °C for different reaction time. The red spectra correspond to the LCP reference one from reference [
6] whereas black patterns correspond to other reaction intermediates.
Figure 4.
Fast-Fourier Transform infrared spectroscopy (FTIR) spectra of the samples obtained starting from the reagent ratios LiOH:LiH
2PO
4:CoSO
4 = 2:1:1 at 240 °C for different reaction time. The red spectra correspond to the LCP reference one from reference [
6] whereas black patterns correspond to other reaction intermediates.
The strong water O–H bending mode at 1630–1640 cm
−1 is observed in the early synthesis stages (0–2 h) whereas it disappears almost completely after the formation of the LCP phase. The spectrum of the material after 4 h of reaction time matches perfectly the reference one for the phase pure LCP olivine lattice [
6] and remains almost unaltered for longer reaction time. In particular the intramolecular stretching modes of the PO
43− anions are observed at (
ν3) 1180, 1100, 1050, and (
ν1) 975 cm
−1, whereas the bending bands (
ν2,
ν4, and
ν2 +
ν4) are found at 640, 575, 550, 505, and 475 cm
−1. Additional bands are not observed in all LCP-pure samples (4, 5, 6, and 8–10 h). In particular, at 410–420 cm
−1 the typical fingerprint of the Li
3PO
4 phase is missing [
7], as well as absorption bands in the 650 cm
−1 to 940 cm
−1 region, where vibrations associated to other phosphate anions, such as P
2O
74− [
7], are located. In this view, considering the above reported results from the ICP-AES analysis for the trend of the Li:Co ratio upon reaction time, the absence of evident contaminations may be an indirect clue of the possible over-lithiation of the olivine lattice of samples prepared at 240 °C in neutral pH conditions. The excess of lithium incorporated in the material apparently decreases with the increase of the solvo-thermal reaction time.
Having established the absence of contaminant phases for all samples prepared at 240 °C after 4 h of reaction time and having measured the Li:Co ratios, it is possible to estimate a tentative stoichiometry for the corresponding LCP phase with the assumption of the electro-neutrality constraints between cations (Li
+, Co
2+) and anions (PO
43−) in the lattice. The resulting estimated stoichiometries are summarized in the
Table 1 together with the results of the Rietveld refinement fitting performances on the corresponding XRD patterns by the GSAS code [
12].
Cell constants are in agreement with literature values [
5]: lattice parameters are slightly expanded compared to both annealed samples (approximately +0.6%) and to single crystals (approximately +1.2%) [
5]. However, a clear structural evolution trend with the reaction time is missing, besides a slight increase of the overall cell volume for samples prepared for 8 h and 10 h. Similarly the structural disorder,
i.e., Li/Co anti-site defects [
4], occurs in all samples without a monotonic trend.
Table 1.
Estimated stoichiometries and Rietveld refinement results for the samples prepared at 240 °C in neutral pH conditions at various reaction time.
Table 1.
Estimated stoichiometries and Rietveld refinement results for the samples prepared at 240 °C in neutral pH conditions at various reaction time.
| Reaction Time | Estimated Stoichiometry | a/Å | b/Å | c/Å | V/Å3 ± 0.2 | %Me Disorder ± 0.6 | wRp |
|---|
| 4 h | Li1.06Co0.97PO4 | 10.211(1) | 5.924(2) | 4.705(3) | 284.6 | 3.1 | 7.3% |
| 5 h | Li1.05Co0.97PO4 | 10.208(2) | 5.923(2) | 4.704(3) | 284.4 | 2.7 | 8.0% |
| 6 h | Li1.05Co0.98PO4 | 10.210(3) | 5.924(2) | 4.705(4) | 284.6 | 2.6 | 6.5% |
| 8 h | Li1.03Co0.98PO4 | 10.214(2) | 5.928(1) | 4.705(2) | 284.9 | 2.8 | 6.3% |
| 10 h | Li1.03Co0.99PO4 | 10.216(3) | 5.925(4) | 4.706(1) | 284.9 | 2.7 | 7.6% |
Turning to the morphological evolution upon reaction time, the particle size smoothly evolves after the precipitation of a phase-pure LCP material (4 h). The comparison between the particles’ morphology of the materials obtained at 240 °C after 5 h and 10 h is shown in the
Figure 5.
A moderate increase in the size of the regular crystallites is observed with the increase of the reaction time. The LCP material obtained at 240 °C in 5 h is constituted by hexagonal sub-micrometric platelets with a thickness approximately below 200 nm, as estimated by the particle size routine using the ImageJ code [
13], whereas after 10 h particles are slightly larger and thicker but without remarkable changes in the overall particles shape and morphological homogeneity.
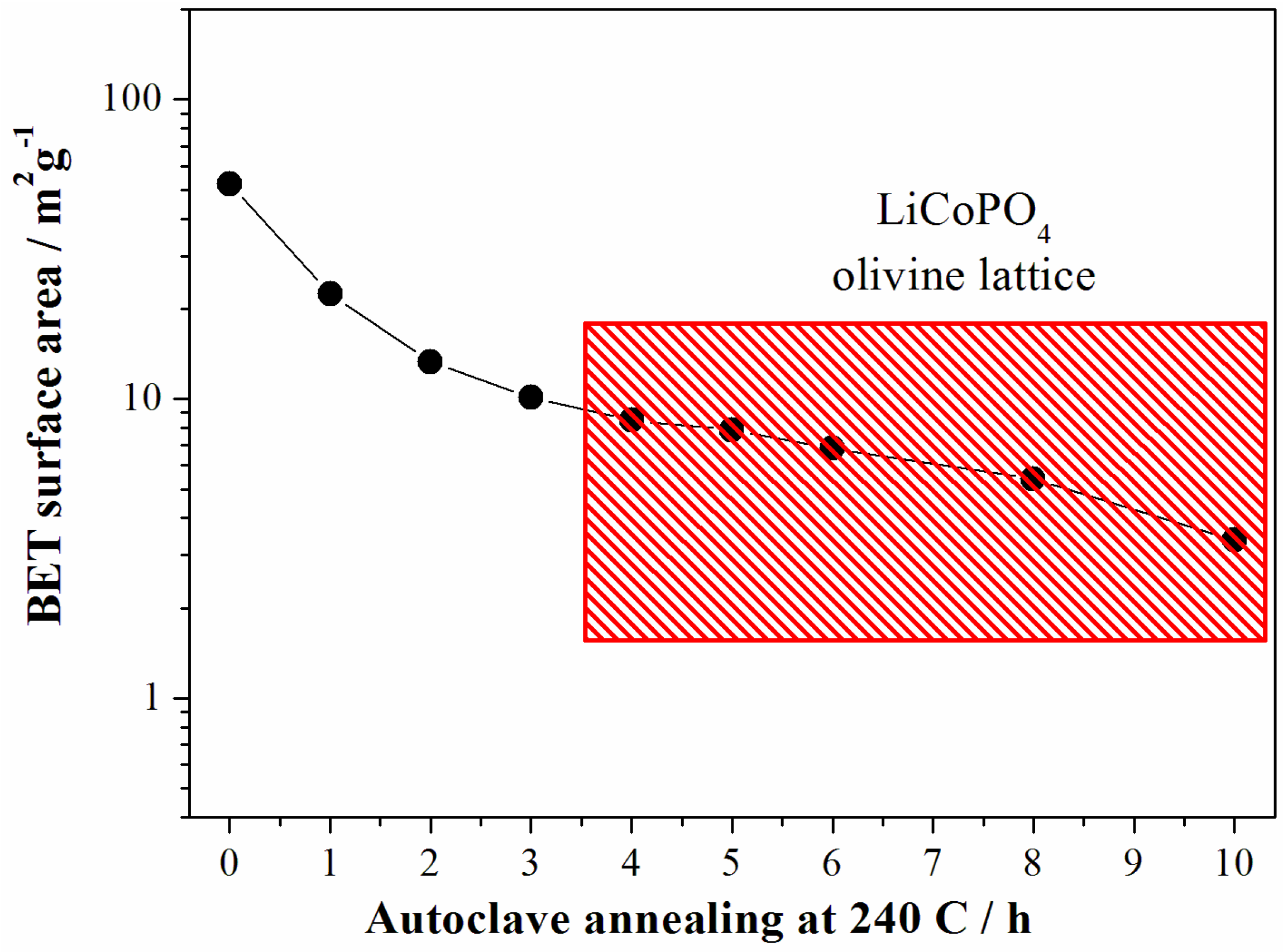
As expected the increase of the particle size for longer reaction time results in the decrease of the sample surface area as shown in the
Figure 6.
Figure 5.
Scanning electron microscopy (SEM) micrographs of the LCP phase-pure samples obtained at 240 °C from the reagent ratio LiOH:LiH2PO4:CoSO4 = 2:1:1 after a reaction time of 5 h ((a) panel) and 10 h ((b)panel).
Figure 5.
Scanning electron microscopy (SEM) micrographs of the LCP phase-pure samples obtained at 240 °C from the reagent ratio LiOH:LiH2PO4:CoSO4 = 2:1:1 after a reaction time of 5 h ((a) panel) and 10 h ((b)panel).
Figure 6.
Evolution of the surface area of the LCP samples prepared at 240 °C from the reagent ratio LiOH:LiH2PO4:CoSO4 = 2:1:1. Data from samples crystallized in a LCP olivine lattice falls into the red rectangle.
Figure 6.
Evolution of the surface area of the LCP samples prepared at 240 °C from the reagent ratio LiOH:LiH2PO4:CoSO4 = 2:1:1. Data from samples crystallized in a LCP olivine lattice falls into the red rectangle.
In summary, at 240 °C the LCP phase forms after 4 h of solvo-thermal treatment: the structures apparently do not evolve with longer reaction time, whereas the morphology of the particles continues to grow.
2.4. Analysis of the Synthesis Conditions: Effect of the Reaction Temperature
The effect of the temperature of the solvo-thermal bath has been tested by precipitating LCP-pure materials at three different temperatures,
i.e., 240, 220, and 200 °C, starting from the following reagent ratio: LiOH:LiH
2PO
4:CoSO
4 = 1.75:1:1. Details of the synthesis conditions are reported in the
Table 2. As expected, the adoption of different reaction temperatures implies different reaction kinetics. In this view, different solvo-thermal reaction time has been unavoidably optimized for each synthesis at different temperatures. The reaction time values reported in
Table 2 are the minimal reaction times to obtain a phase pure LCP material for each temperature.
Table 2.
Experimental conditions and structural refinements of samples prepared at different reaction temperatures in acidic pH conditions.
Table 2.
Experimental conditions and structural refinements of samples prepared at different reaction temperatures in acidic pH conditions.
| Reaction Temperature (°C) | Reaction Time (h) | Estimated Stoichiometry (from ICP Data) | a/Å | b/Å | c/Å | V/Å3 ± 0.3 | %Me Disorder ± 0.6 | wRp |
|---|
| 200 | 30 | Li1.01Co0.99PO4 | 10.205(5) | 5.923(4) | 4.700(3) | 284.1 | 1.6 | 8.5% |
| 220 | 15 | Li1.01Co1.00PO4 | 10.219(2) | 5.926(2) | 4.707(4) | 284.9 | 2.9 | 2.1% |
| 240 | 5 | Li0.99Co1.00PO4 | 10.212(3) | 5.922(3) | 4.705(1) | 284.5 | 2.5 | 7.7% |
The XRD patterns of the three materials obtained at 240, 220, and 200 °C show phase-pure materials with olivine structure without contaminants: patterns are very similar to those shown in the
Figure 3 (red lines) and have, therefore, been omitted to save space. The stoichiometries of the LCP phases for each sample derived from ICP-AES data are reported in the
Table 2 together with the Rietveld Refinment results from the corresponding XRD pattern.
All preparations have line composition that matches, within errors, the theoretical LCP Li:Co = 1:1 stoichiometry. Materials prepared at 220 and 240 °C are very similar from the point of view of the cell volume and cationic disorder, whereas the sample prepared at 200 °C shows a slight lattice contraction, which occurs with the expected parallel decrease of the disorder [
4].
The morphologies of the three materials are shown in the SEM micrographs reported in the
Figure 7. Apparently the particle size and shape moderately depend on the solvo-thermal temperature. All materials are constituted by regular prismatic particles. Samples prepared at 240 °C (
Figure 7c), are constituted by hexagonal-octagonal platelets, similar to those obtained at the same temperature but at neutral pH shown in the
Figure 5. However, the morphological homogeneity and uniformity of the particle sizes are reduced compared to neutral pH samples.
On the other hand syntheses carried out at lower temperatures (220 and 200 °C) show slightly larger prismatic particles (especially thicker platelets) compared to the thin, regular octagonal/hexagonal platelets prepared at 240 °C. This trend is confirmed by the measured BET surface areas being 7.9 ± 0.2, 5.1 ± 0.1 and 3.8 ± 0.5 for the materials prepared at 240, 220, and 200 °C, respectively. One may speculate that syntheses carried out at lower temperature produce a smaller number of starting crystalline seeds due to a possible kinetic bottleneck in the nucleation activation energy. However, the study of the LCP crystal growth thermodynamics and kinetics is beyond the scope of this communication and therefore our hypothesis is presented only as mere speculation.
Figure 7.
SEM micrographs of the materials prepared at (a) 200, (b) 220, and (c) 240 °C in acidic pH conditions.
Figure 7.
SEM micrographs of the materials prepared at (a) 200, (b) 220, and (c) 240 °C in acidic pH conditions.
2.5. Analysis of the Synthesis Conditions: Effect of the Cobalt Anion
In their work about the hydrothermal growth of LFP olivine lattice Lu
et al. [
14] observed that the presence of charged species dissolved into the reaction bath directly affects the olivine crystal growth. In fact the different affinity of various anions and cations towards different lattice surfaces of the LFP particles leads to an effective selectivity to drive the final prismatic morphology. In order to shed light on this effect the LCP materials have been synthesized starting from several cobalt salts. Four different Co
2+ sources have been used,
i.e., Co(NO
3)
2, CoSO
4, Co(CH
3COO)
2, and CoCO
3. The experimental conditions (reagent ratio, temperature, and reaction time) suitable to obtain the precipitation of pure LCP materials starting from the four different cobalt salts are summarized in
Table 3. It is to be noted that shorter reaction times lead to the incomplete formation of the LCP phase with presence in the final material of phase impurities (cobalt phosphate, lithium phosphate). Moreover, it is important to underline the very long reaction time required to obtain an LCP-phase pure sample starting from cobalt carbonate. This extended reaction time (100 h) is uncommon in solvo-thermal and hydrothermal synthesis but, in this case, the low solubility of both reagents (cobalt carbonate) and products (LCP) apparently slow down the kinetics of the reaction strongly, compared to the other three cases where soluble cobalt salts are used as precursors.
All of the materials prepared in the conditions reported in the
Table 3 are constituted by the pure LCP phase without contaminants: XRD patterns are very similar to those shown in the
Figure 2 and
Figure 3 (red lines) and have, therefore, been omitted to avoid redundancies. The results of the Rietveld refinements carried out on the XRD pattern of each sample are reported in the
Table 4.
Table 3.
Experimental conditions applied during solvo-thermal synthesis of the LCP samples starting from different cobalt salts (solvent volume 30 mL, reaction temperature 220 °C).
Table 3.
Experimental conditions applied during solvo-thermal synthesis of the LCP samples starting from different cobalt salts (solvent volume 30 mL, reaction temperature 220 °C).
| Co(II) Salt | Co Salt (10−3 mol) | LiH2PO4 (10−3 mol) | Sucrose (10−3 mol) | LiOH (10−3 mol) | Reaction Time (h) |
|---|
| Nitrate | 3 | 3 | 0.1 | 5.25 | 15 |
| Sulfate | 3 | 3 | 0.1 | 5.25 | 15 |
| Acetate | 3 | 3 | 0.1 | 3 | 30 |
| Carbonate | 3 | 3 | 0.1 | 0.25 | 100 |
Table 4.
Results of the structural refinements of samples prepared from different cobalt(II) salts.
Table 4.
Results of the structural refinements of samples prepared from different cobalt(II) salts.
| Co(II) Salt | Reaction Time (h) | Estimated Stoichiometry (from ICP Data) | a/Å | b/Å | c/Å | V/Å3 ± 0.3 | %Me Disorder ± 0.3 | wRp |
|---|
| Nitrate | 15 | Li1.00Co0.99PO4 | 10.205(2) | 5.915(1) | 4.700(1) | 283.8 | 1.9 | 8.6% |
| Sulfate | 15 | Li1.01Co1.00PO4 | 10.219(2) | 5.926(2) | 4.707(4) | 284.9 | 2.9 | 2.1% |
| Acetate | 30 | Li1.00Co1.00PO4 | 10.210(1) | 5.922(1) | 4.704(1) | 284.4 | 1.7 | 6.1% |
| Carbonate | 100 | Li1.00Co0.99PO4 | 10.202(1) | 5.915(1) | 4.699(1) | 283.6 | 1.7 | 7.7% |
The stoichiometries of all samples have been estimated from ICP-AES data and correspond to an almost ideal Li:Co = 1:1 stoichiometry within errors. On the other hand the structures of three out of four olivine lattices are very similar: only the sample synthesized from the sulphate salt shows larger cell parameters, and the corresponding expansion of the cell volume, together with an increase of the metal disorder between Li and Co sites.
Similarly, the morphologies of the four samples can be grouped, putting together materials prepared from nitrate/acetate/carbonate precursors opposite to the LCP material synthesized from CoSO
4. The SEM micrographs of the four samples are shown in the
Figure 8.
As already discussed the LCP sample prepared from cobalt sulfate is constituted by octagonal/hexagonal thin platelets. On the contrary the three materials prepared from nitrate/acetate/carbonate precursors show large isotropic prismatic crystallites with sizes above 1–2 mm in all the crystallographic directions. In particular, samples prepared from cobalt nitrate and carbonate are constituted by regular and well-formed prisms with hexagonal bases.
The LCP material prepared from cobalt acetate is also constituted by very large isotropic prisms, but with less clear prismatic regularities and with a significant morphological impurities (i.e., small irregular particles spread on the surfaces of the prisms).
These results are in very nice agreement with the findings of Lu
et al. [
14] for the growth of LFP crystals in hydrothermal conditions. As already discussed, these authors observed that different anions (sulfate and nitrate, in their case) have different absorption ability on different lattice planes of the same olivine crystals. This selective absorption apparently drives the crystal growth along specific crystal directions as an example by inhibiting or promoting the precipitation on the (101) plane [
14]. Additionally, in the case of the LCP preparation in solvo-thermal conditions, the use of sulfate salts apparently inhibit the crystal growth perpendicular to the (101) crystal facet, thus leading to the precipitation of octagonal/hexagonal platelets very similar to those observed by Lu
et al. for LFP crystals from sulfate precursors [
14].
Figure 8.
SEM micrographs of the prepared LiCoPO4 powders: (a) nitrate; (b) sulfate; (c) acetate; and (d) carbonate.
Figure 8.
SEM micrographs of the prepared LiCoPO4 powders: (a) nitrate; (b) sulfate; (c) acetate; and (d) carbonate.
In summary, our evidence confirms that the tuning of a crystal growth in a specific lattice direction or its limitation as well as the number of nucleation sites are strongly altered by the nature of the cobalt counter-anion in the precursor salt. As already mentioned, a similar effect has been reported by Lu
et al. [
14] for LFP materials and here we also confirm this trend for the LCP crystal growth in solvo-thermal conditions.
On the contrary, a simple effect of the nature of the cobalt salt precursors on the lattice ordering in the final LCP phase is not straightforward. In fact, although the choice of specific anions seem to drive crystallization with improved cation ordering, since the experimental synthesis conditions are necessarily different for different salts, it is not possible to draw a simple cause-effect rule.
2.6. Electrochemical Tests of the LCP-Pure Samples in Li Cells
In the previous sections a number of different preparation recipes have been reported to obtain phase-pure LCP materials with slightly different morphological identity and homogeneity, structural properties, and composition. A summary of the synthesized materials is presented in the
Table 5.
Table 5.
LCP phase-pure materials prepared with different solvo-thermal recipes.
Table 5.
LCP phase-pure materials prepared with different solvo-thermal recipes.
| Sample | LiOH/Co2+ Salt Ratio | Reaction Time (h) | Reaction Temperature (°C) | Co(II) Precursor |
|---|
| LCP-01 | 2:1 | 5 | 240 | Sulfate |
| LCP-02 | 1.75:1 | 5 | 240 | Sulfate |
| LCP-03 | 2:1 | 4 | 240 | Sulfate |
| LCP-04 | 2:1 | 6 | 240 | Sulfate |
| LCP-05 | 2:1 | 8 | 240 | Sulfate |
| LCP-06 | 2:1 | 10 | 240 | Sulfate |
| LCP-07 | 1.75:1 | 15 | 220 | Sulfate |
| LCP-08 | 1.75:1 | 30 | 200 | Sulfate |
| LCP-09 | 1.75:1 | 15 | 220 | Nitrate |
| LCP-10 | 1:1 | 30 | 220 | Acetate |
| LCP-11 | 0.083:1 | 100 | 220 | Carbonate |
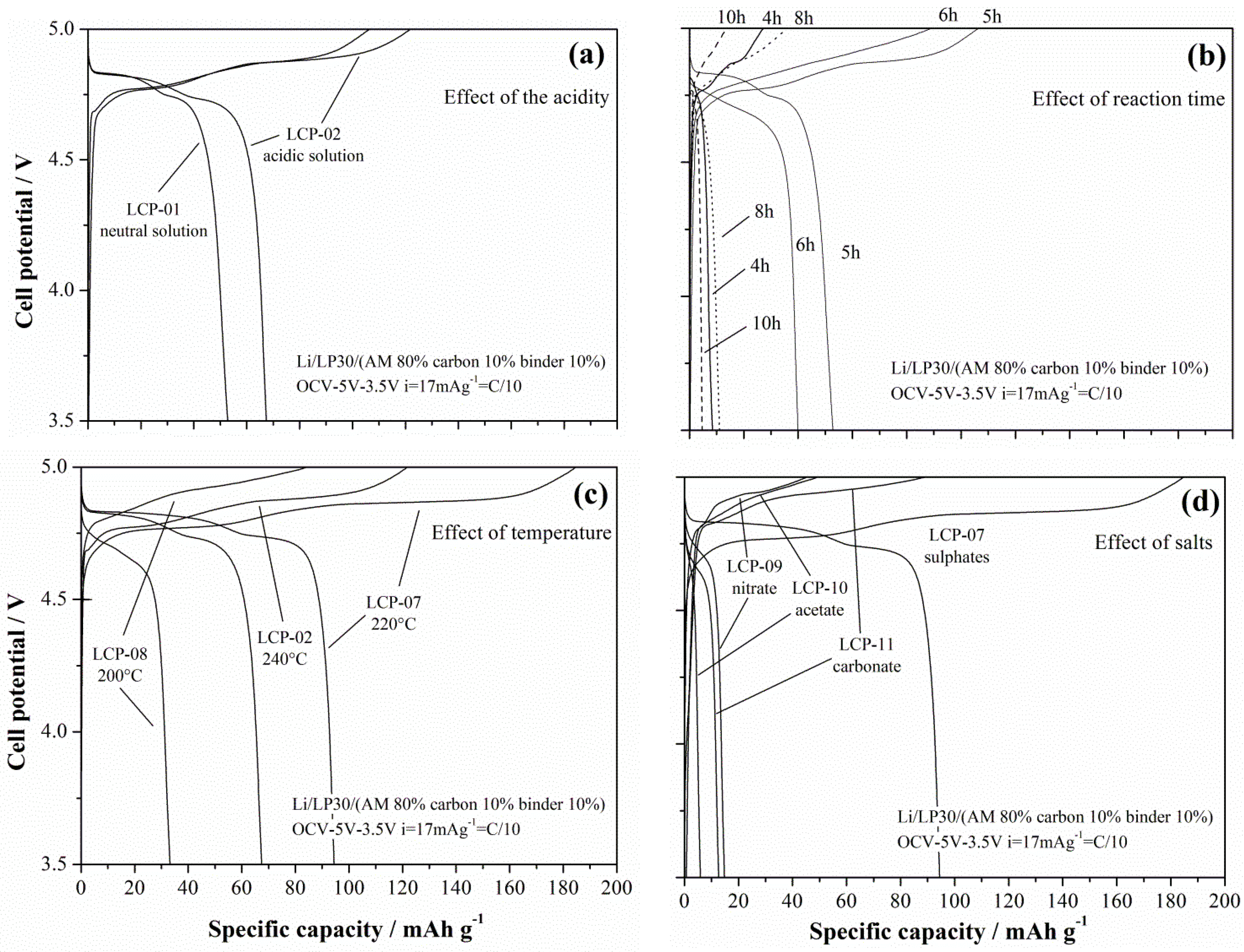
In this section the electrochemical performances of the 11 synthesized materials are compared in order to highlight the effect of the various preparation conditions on the ability to reversibly exchange lithium in a Li-cell.
The lithium de-insertion/insertion curves for galvanostatic cycling at 17 mA·g
−1 (C/10) are shown in
Figure 9a–d.
Figure 9.
Lithium de-insertion/insertion curves from galvanostatic cycling of the LCP materials.
Figure 9.
Lithium de-insertion/insertion curves from galvanostatic cycling of the LCP materials.
The LCP material reversibly cycles lithium ions at approximately 4.8 V
vs. Li: the oxidation/reduction steps show two plateaus in agreement with the reaction mechanism by Bramnik [
15]. Apparently, different synthesis conditions lead to different performances in lithium cells. In particular, acidic solvo-thermal solutions improves the performance, as well as assessed reaction times. Concerning this last effect, the occurrence of an optimal reaction time to achieve the best performances has also been observed by Lu
et al. [
14] for similar synthesis of the LFP olivine. Lu
et al. put this behavior in direct correlation with two competing effects,
i.e., the ordering of the olivine lattice and the crystal growth that improves and decreases the ability of reversibly-exchanged lithium, respectively. Furthermore, for the here-synthesized LCP materials, both of these parameters are apparently playing a similar role: in fact, the cationic disorder is estimated to reduce slightly after 5 h of synthesis without further signficant changes for longer reaction times, whereas the size of the prismatic particles increase, thus reducing the surface area of the material. These findings are also in agreement with the observation reported by Truong and coworkers in References [
16].
Additionally, the trend of the performances in lithium cells vs. the synthesis temperature show an interesting optimum value. In fact, the LCP-07 material that has been synthesized at 220 °C shows improved performance compared to both the materials prepared at higher (LCP-02 at 240 °C) and lower (LCP-08 at 200°C) temperatures. In this case a clear explanation of this trend is missing, since apparently the competition between the lattice ordering and the crystallite size cannot account for this peculiar behavior. One may speculate that, at different temperatures, the growth of the crystal facets with different hkl Miller index may be different, as well as the number of nucleation sites of the prismatic particles. Therefore, as a mere hypothesis, one may suggest an optimal reaction temperature may result from the balancing between the nucleation thermodynamics and crystal growth kinetics.
Turning to the effect of the cobalt salt precursor, apparently the selection of different anions has a massive impact on the ability of the resulting LCP materials to reversibly cycle lithium in cells. On the other hand, this behavior is expected due to the very large difference in morphology between the four compared materials. In fact the only LCP material with crystallite size below 1 micron is the only electrochemically active one, whereas all micro-sized samples show negligible performances, likely due to the much-extended diffusion paths within the LCP lattice for the lithium ions to be extracted or inserted in the structure. Lithium diffusion is a well-known limiting factor for olivine-based materials [
4,
5] in particular for LCP phases, thus the need of sub-micro-sized particles to achieve acceptable electrochemical performances.
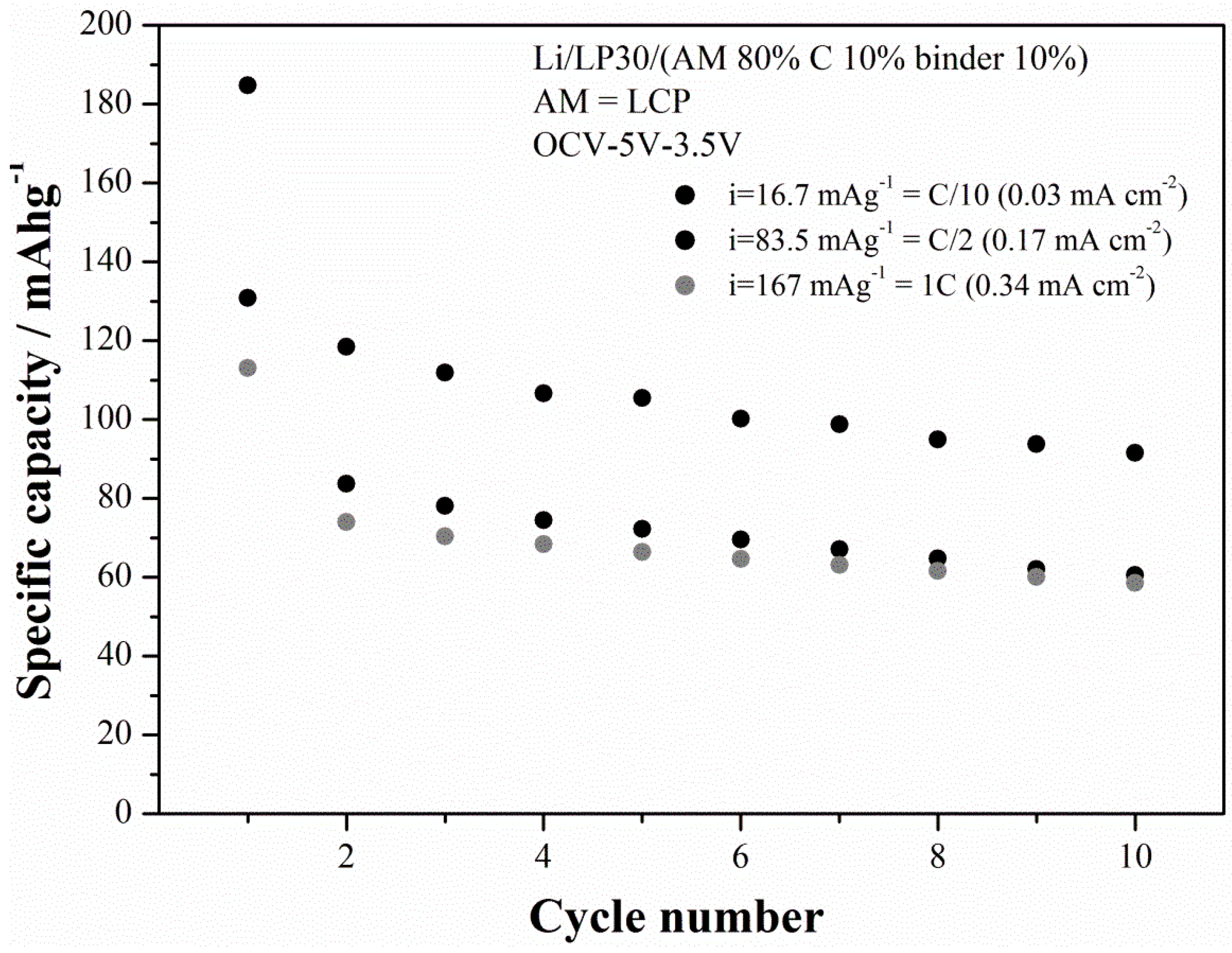
Among all the synthesized LCP materials the outperforming one is LCP-07 that has been prepared starting from cobalt sulfate in a moderately acidic solution treated at 220 °C for 15 h. The cycling performance at three different current rates is shown in the
Figure 10 for the LCP-07 material.
The capacity in the first charge at C/10 slightly exceeds the theoretical capacity (167 mAh·g
−1), thus suggesting the occurrence of the electrolyte decomposition at high voltage. In fact EC:DMC electrolytes with fluorinated salts are at the limits of their stability window and have been reported to decompose on LCP-based electrolyte even with the use of stabilization additives [
5,
17]. This hypothesis is also confirmed by the value of the coulombic efficiencies in the first cycle,
i.e., 51%, as well as by the fading trend of the specific capacities upon cycling. The last effect has been recently put in correlation with the occurrence on an apparently unavoidable increase of the cationic disorder within the olivine lattice [
18]. On the other hand, it is to be noted that, at C/10, the ability of the material to supply a specific capacity upon cycling approaches 90–100 mAh·g
−1 after 10 cycles. At higher current rates performance drops to approximately 1/3 of the theoretical performance. However the deterioration of the capacity performance at high rates is likely due to the poor electronic conductivity of this material [
5] and may be mitigated by the growth of carbon coatings or doping, similarly to the parent LFP-phase [
4,
19]. In this view this solvo-thermal synthetic route allows the preparation of a material with large room for improvements.
Figure 10.
Cycling performance (charge capacities) of the LCP-07 material prepared at 220 °C under moderately-acidic conditions for 15 h.
Figure 10.
Cycling performance (charge capacities) of the LCP-07 material prepared at 220 °C under moderately-acidic conditions for 15 h.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







