Layered-Expanded Mesostructured Silicas: Generalized Synthesis and Functionalization

 , ,
, ,  ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Synthesis
2.2. Chemicals
2.3. Preparative Procedure
2.4. Reactivity Assays by Chemical Exchange of the Surfactant
2.5. Physical Measurements
3. Results and Discussion
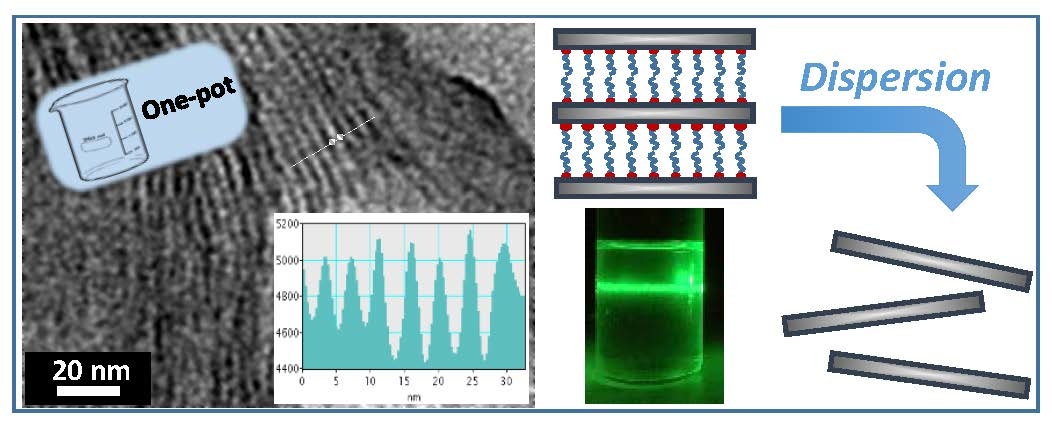
3.1. Synthesis Strategy
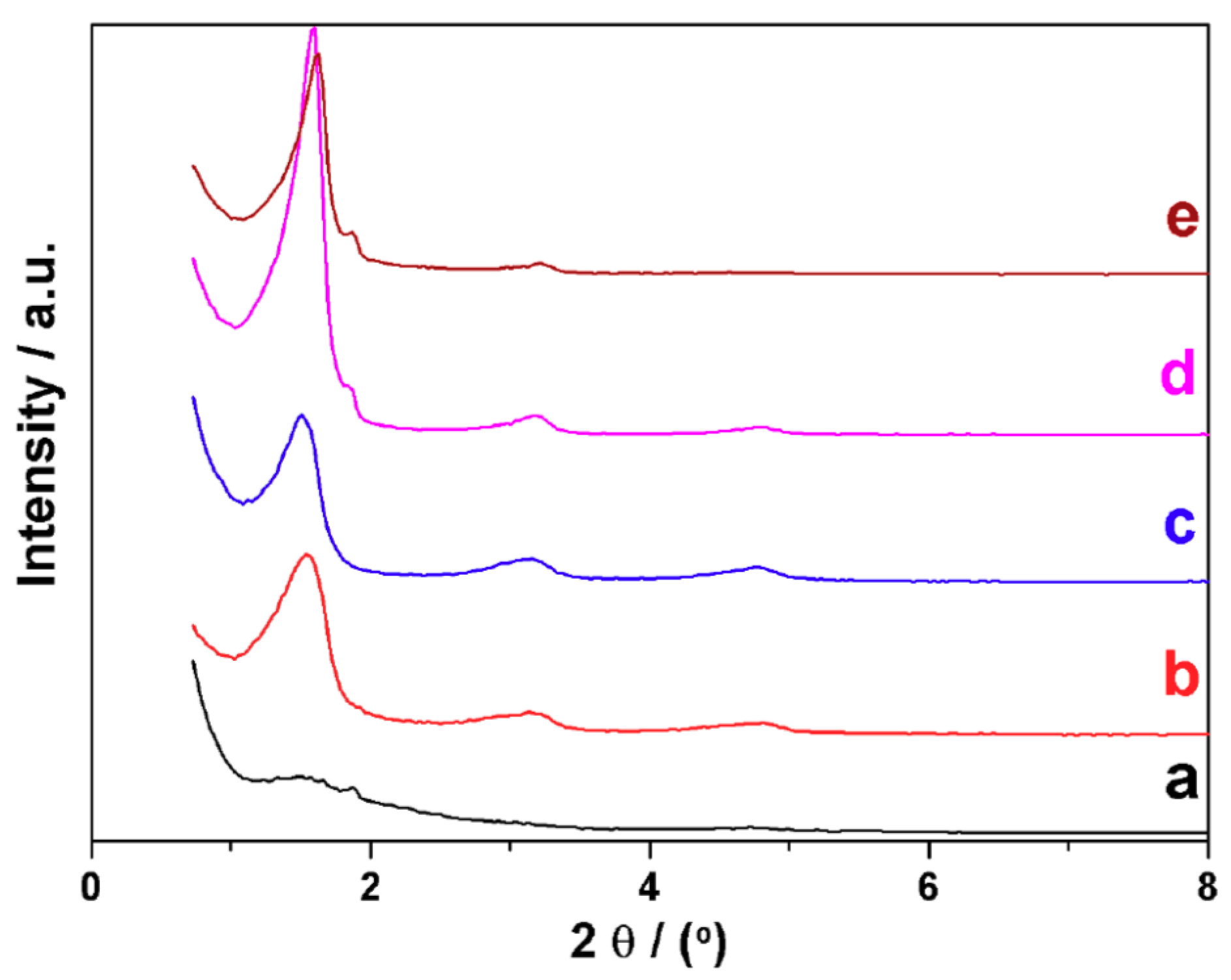
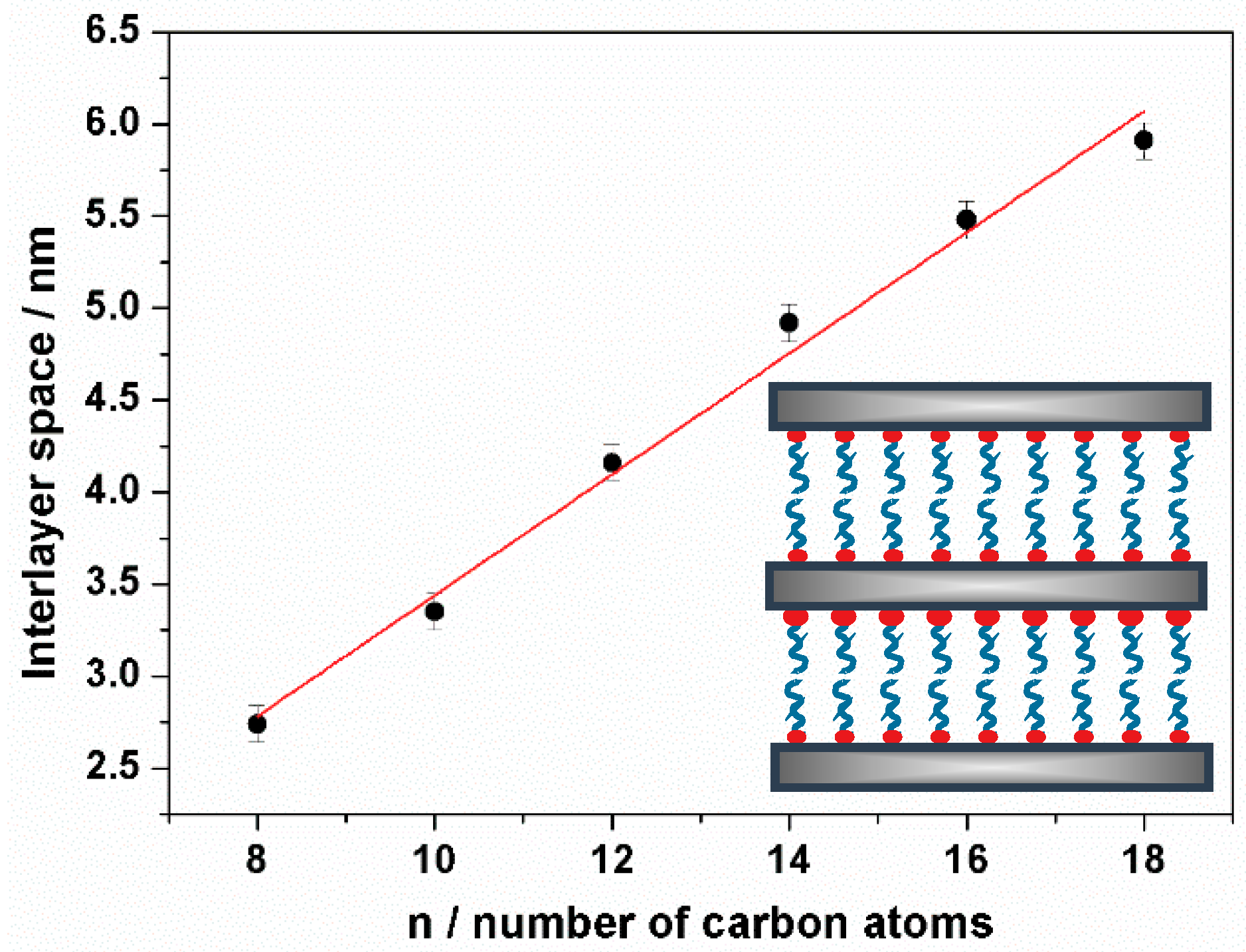
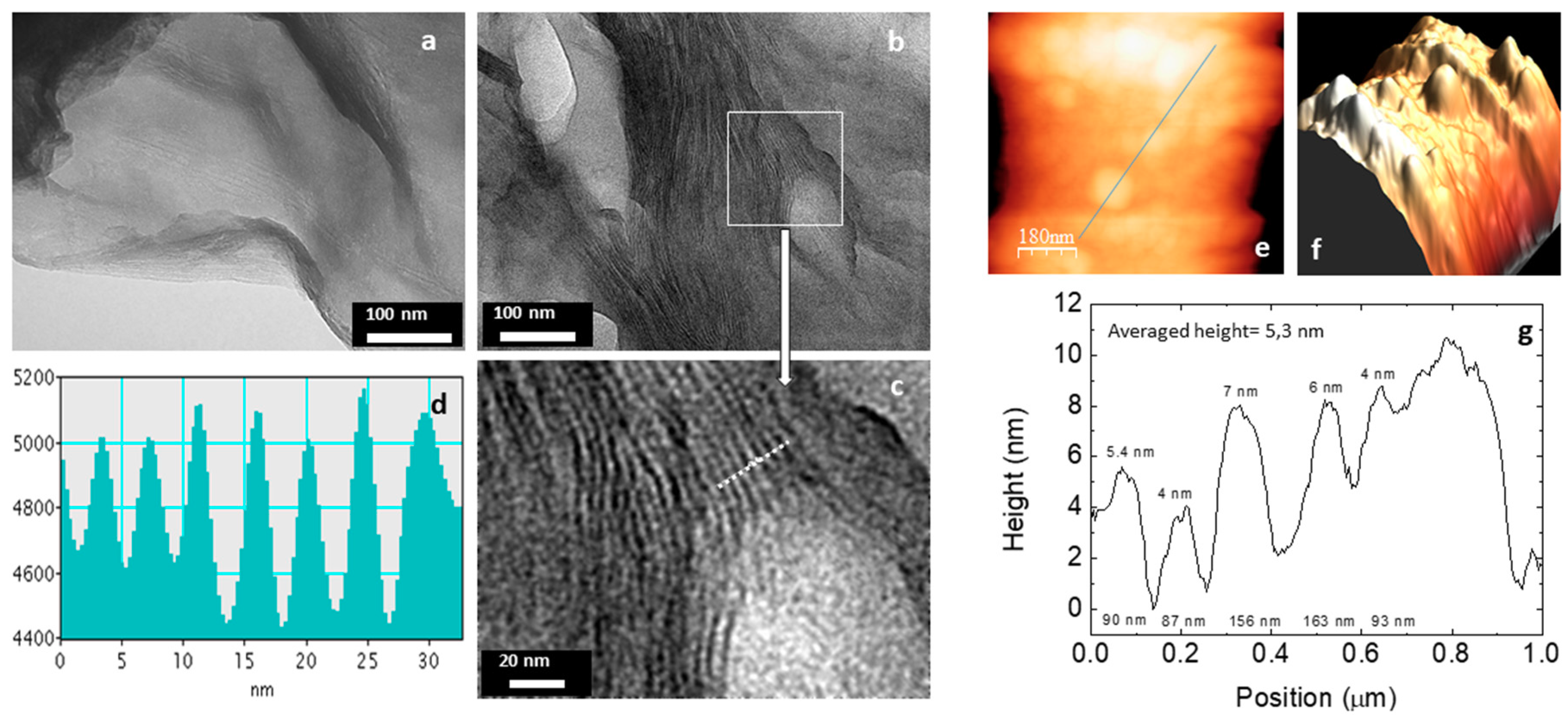
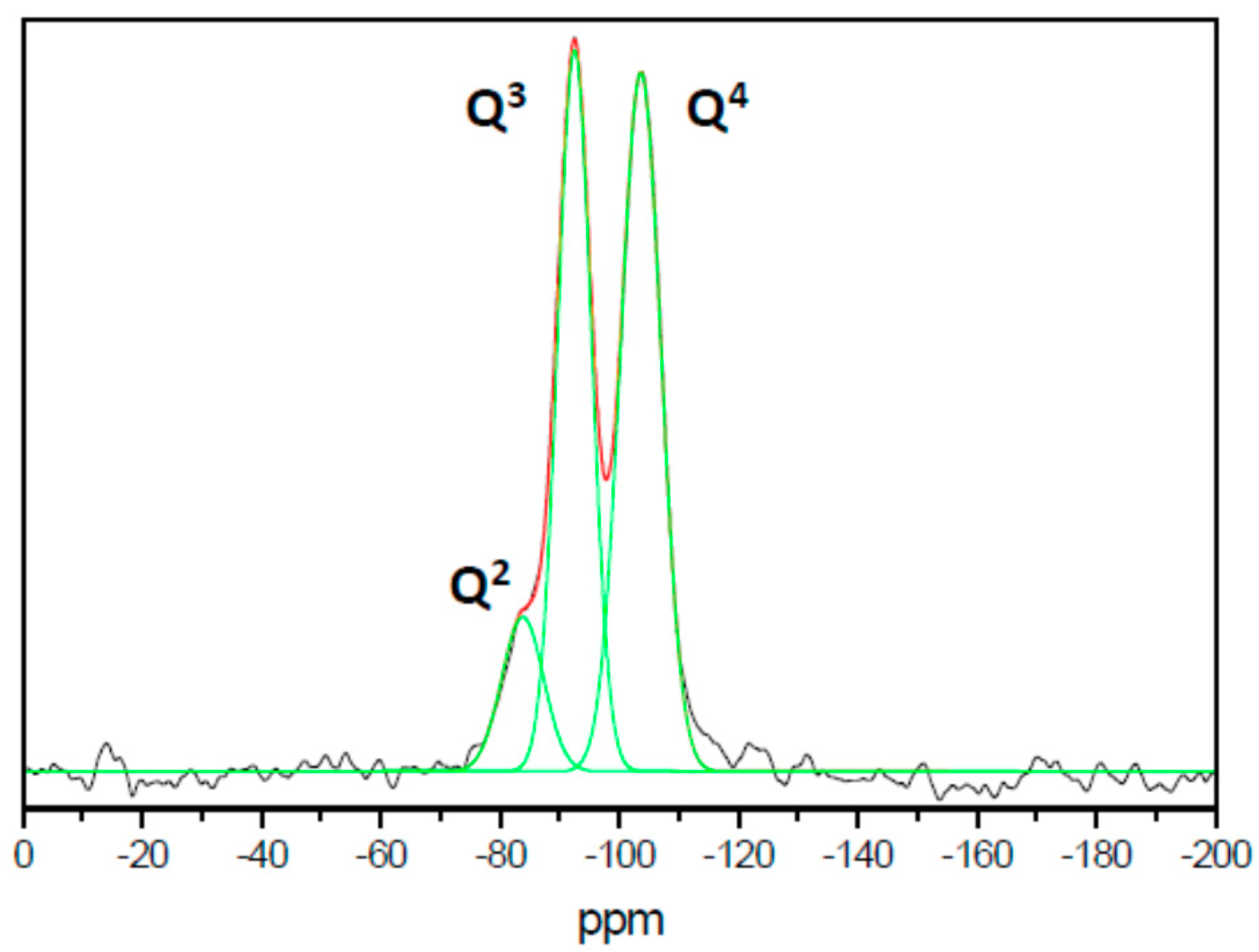
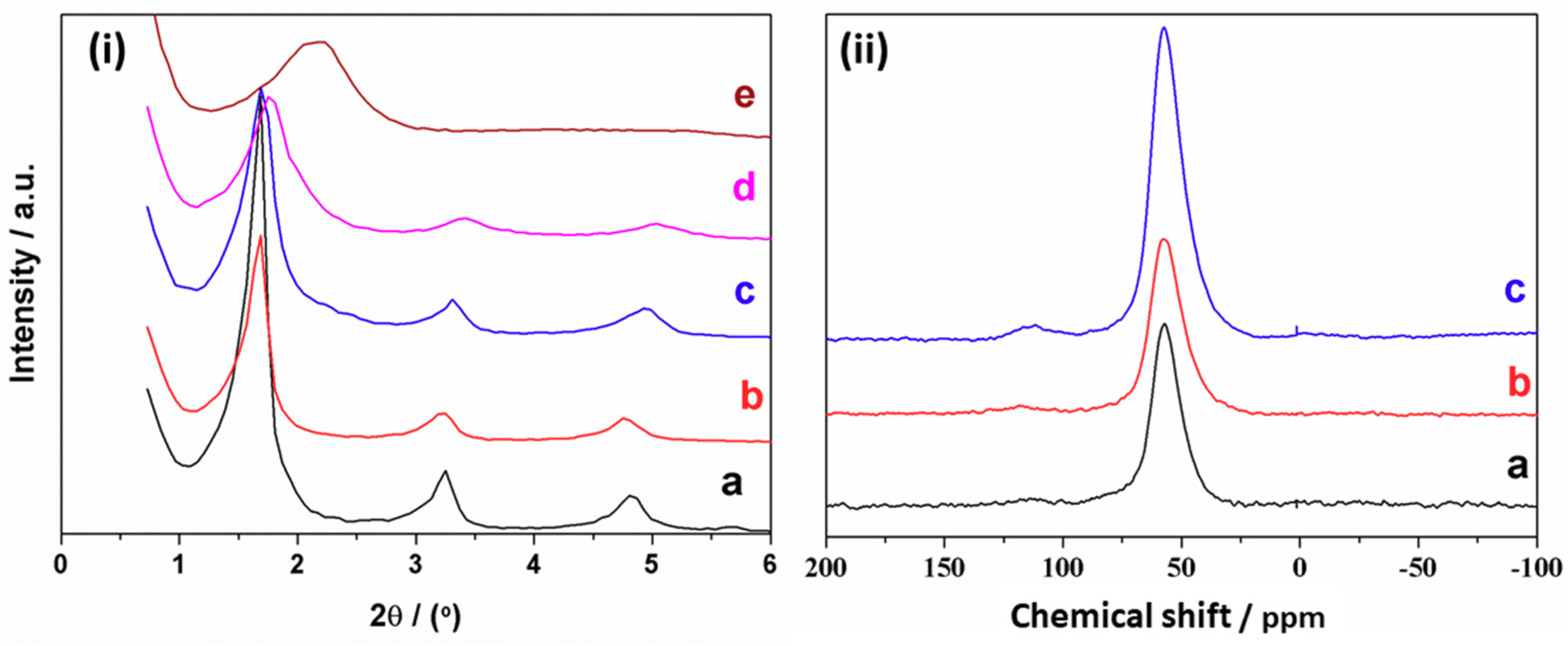
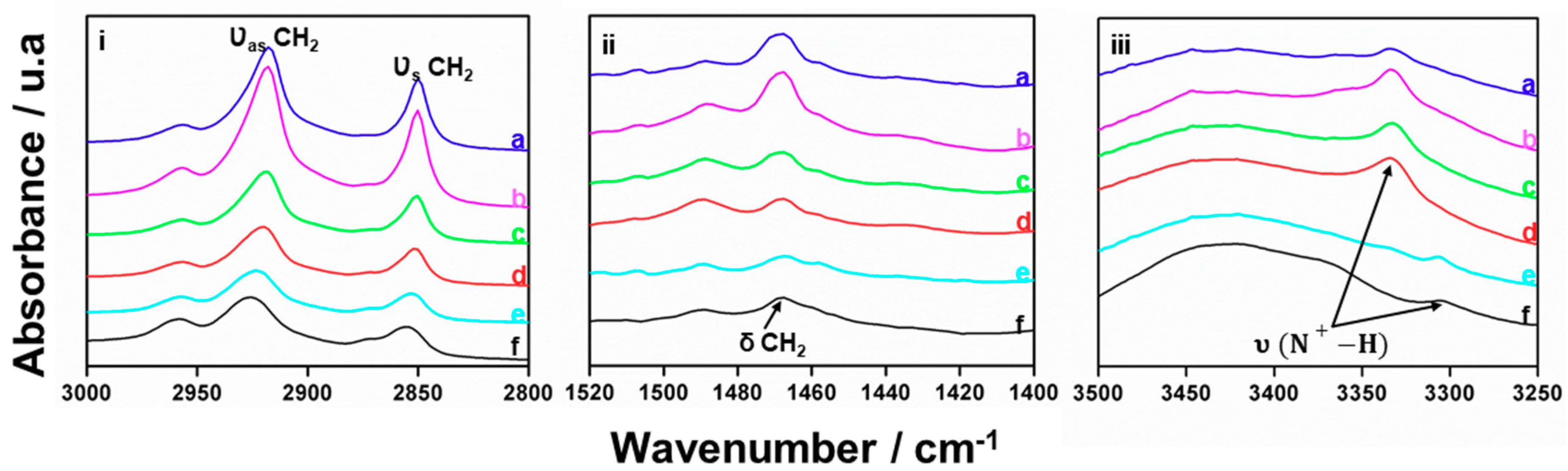
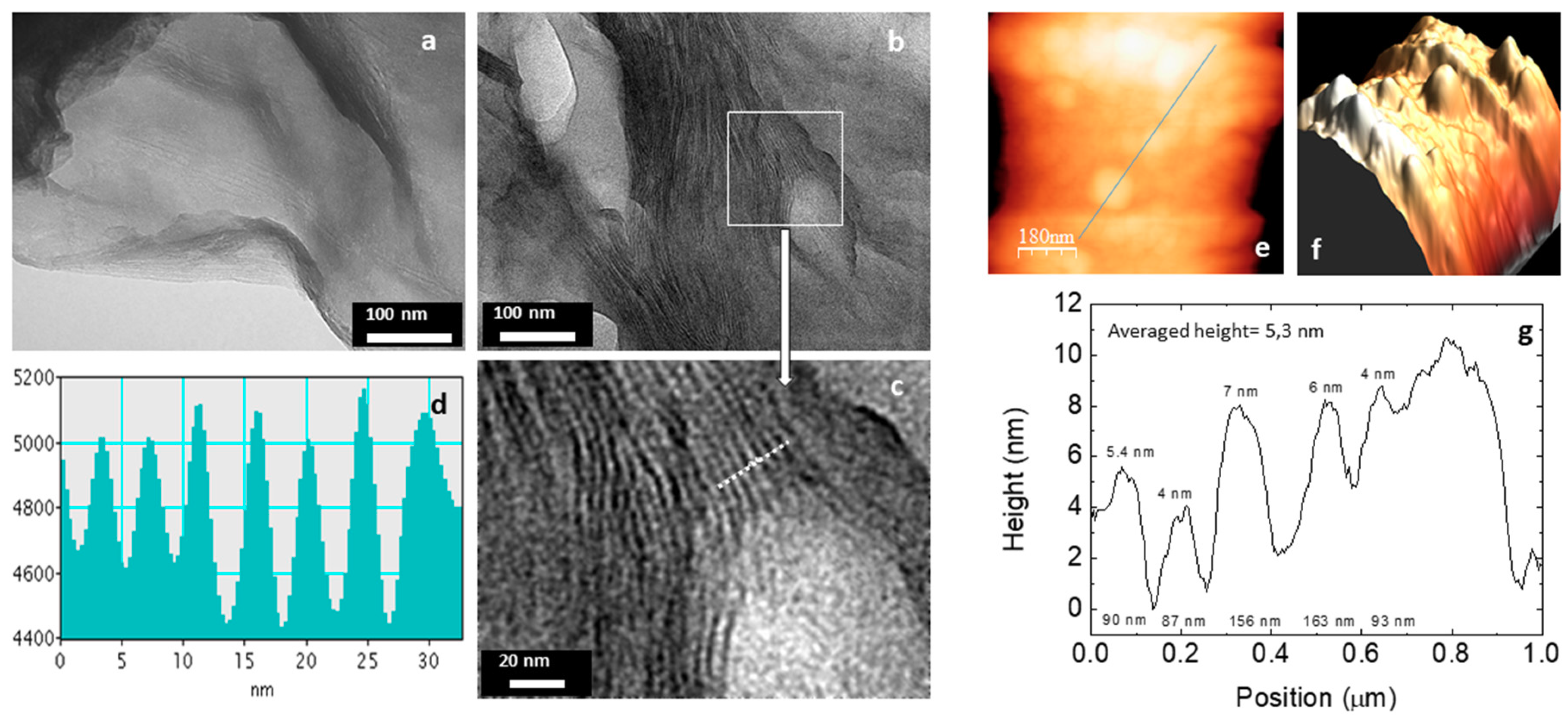
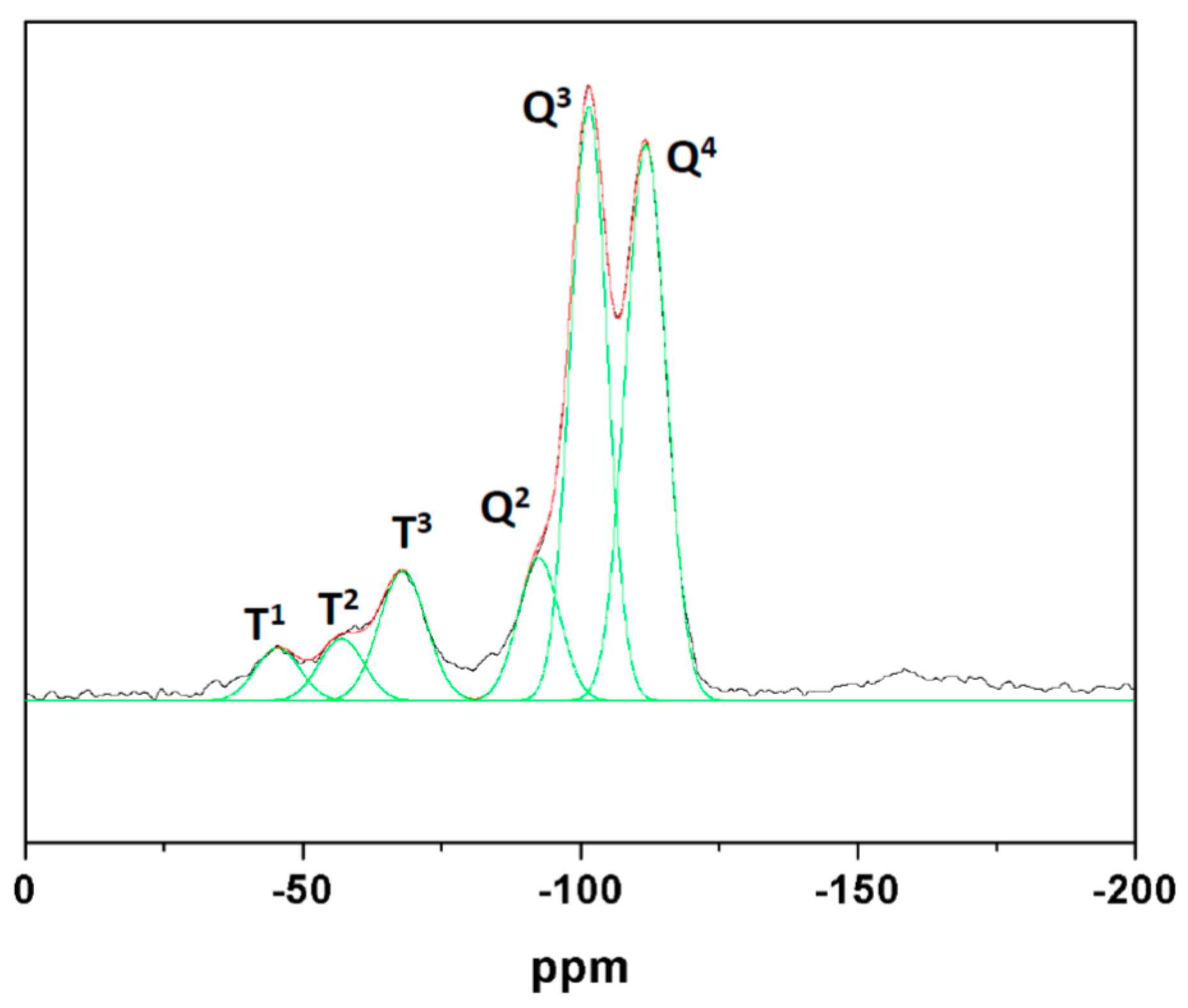
3.2. Characterization
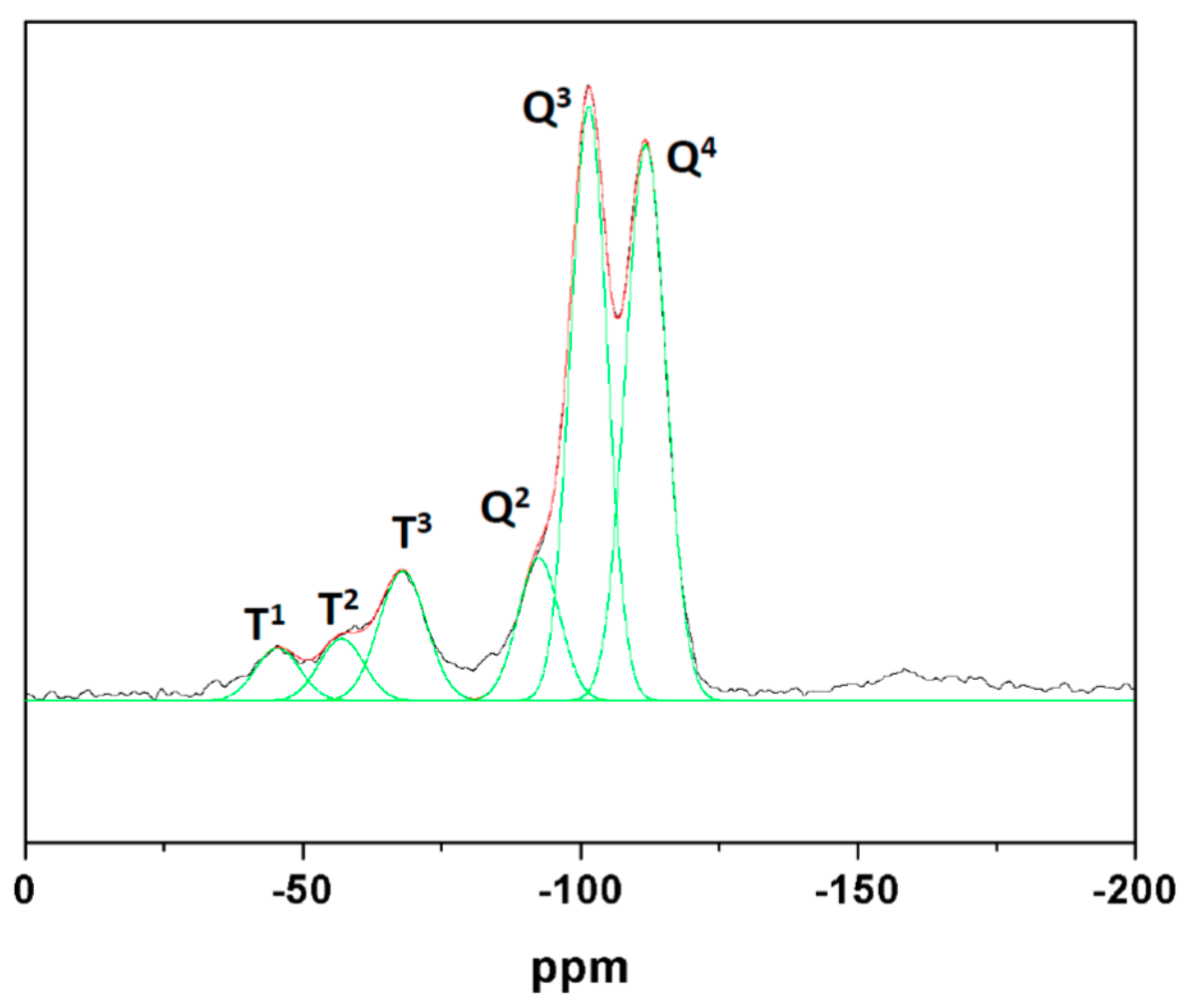
3.3. Functionalized Materials
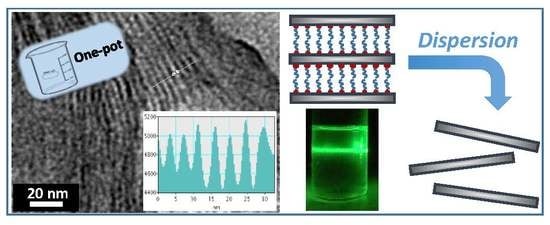
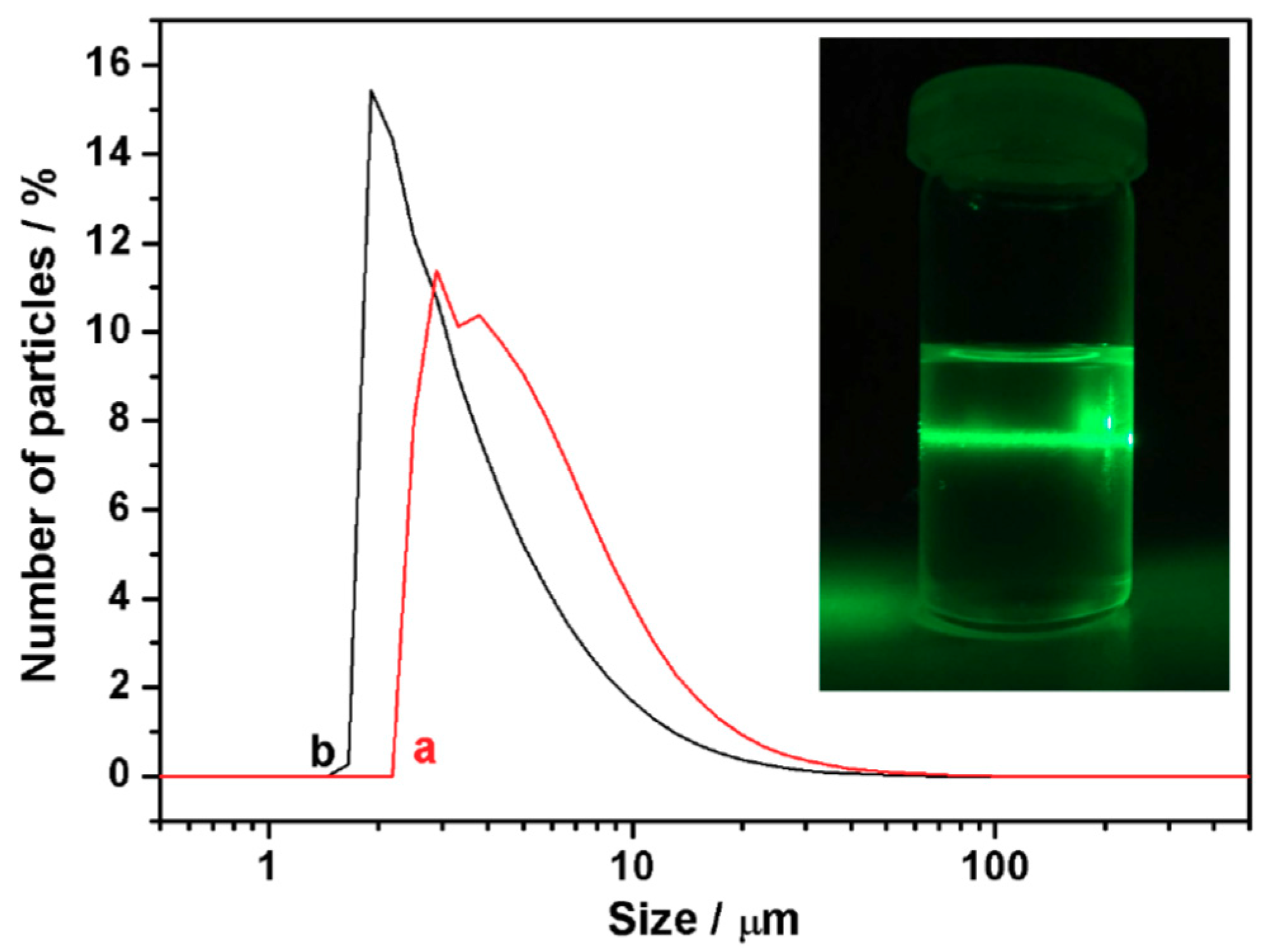
3.4. From Layered Mesostructures to Colloids
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sanchez, C.; Julián, B.; Belleville, P.; Popall, M. Applications of hybrid organic-inorganic nanocomposites. J. Mater. Chem. 2005, 15, 3559–3592. [Google Scholar] [CrossRef]
- Pavlidou, S.; Papaspyrides, C.D. A review on polymer-layered silicate nanocomposites. Prog. Polym. Sci. 2008, 33, 1119–1198. [Google Scholar] [CrossRef]
- Centi, G.; Perathoner, S. Catalysis by layered materials: A review. Micropor. Mesopor. Mater. 2008, 107, 3–15. [Google Scholar] [CrossRef]
- Takahashi, N.; Kuroda, K. Materials design of layered silicates through covalent modification of interlayer surfaces. J. Mater. Chem. 2011, 21, 14336–14353. [Google Scholar] [CrossRef]
- Selvam, T.; Inayat, A.; Schwieger, W. Reactivity and applications of layered silicates and layered double hydroxides. Dalton. Trans. 2014, 43, 10365–10387. [Google Scholar] [CrossRef] [PubMed]
- Shimojima, A.; Kuroda, K. Designed synthesis of nanostructured siloxane-organic hybrids from amphiphilic silicon-based precursors. Chem. Rec. 2006, 6, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Kuroda, K. Ordered mesoporous silica derived from layered silicates. Adv. Funct. Mater. 2009, 19, 511–527. [Google Scholar] [CrossRef]
- Zhou, C.H.; Tong, D.S.; Bao, M.; Du, Z.X.; Ge, Z.H.; Li, X.N. Generation and characterization of catalytic nanocomposite materials of highly isolated iron nanoparticles dispersed in clays. Top. Catal. 2006, 39, 213–219. [Google Scholar] [CrossRef]
- Zhang, D.; Zhou, C.H.; Lin, C.X.; Tong, D.S.; Yu, W.H. Synthesis of clay minerals. Appl. Clay Sci. 2010, 50, 1–11. [Google Scholar] [CrossRef]
- Huo, Q.; Margolese, D.I.; Ciesla, U.; Demuth, D.; Feng, P.; Gier, T.E.; Sieger, P.; Firouzi, A.; Chmelka, B.F.; Schütz, F.; Stucky, G.D. Organization of organic molecules with inorganic molecular species into nanocomposite biphase arrays. Chem. Mater. 1994, 6, 1176–1191. [Google Scholar] [CrossRef]
- Tolbert, S.H.; Landry, C.C.; Stucky, G.D.; Chmelka, B.F.; Noroby, P.; Hanson, J.C.; Monnier, A. Phase transitions in mesostructured silica/surfactant composites: Surfactant packing and the role of charge density matching. Chem. Mater. 2001, 13, 2247–2256. [Google Scholar] [CrossRef]
- Huo, Q.; Margolese, D.I.; Stucky, G.D. Surfactant control of phases in the synthesis of mesoporous silica-based materials. Chem. Mater. 1996, 8, 1147–1160. [Google Scholar] [CrossRef]
- Christiansen, S.C.; Zhao, D.; Janicke, M.T.; Landry, C.C.; Stucky, G.D.; Chmelka, B.F. Molecularly ordered inorganic frameworks in layered silicate surfactant mesophases. J. Am. Chem. Soc. 2002, 123, 4519–4529. [Google Scholar] [CrossRef]
- Wang, L.-Q.; Exarhos, G.L. Study of local molecular ordering in layered surfactant-silicate mesophase composites. J. Phys. Chem. B. 2003, 107, 443–450. [Google Scholar] [CrossRef]
- Díaz, I.; Pérez-Pariente, J.; Terasaki, O. Structural study by transmission and scanning electron microscopy of the time-dependent structural change in M41S mesoporous silica (MCM-41 to MCM-48, and MCM-50). J. Mater. Chem. 2004, 14, 48–53. [Google Scholar] [CrossRef]
- Hedin, N.; Graf, R.; Christiansen, S.C.; Gervais, C.; Hayward, R.C.; Eckert, J.; Chmelka, B.F. Structure of a surfactant-templated silicate framework in the absence of 3D crystallinity. J. Am. Chem. Soc. 2004, 126, 9425–9432. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.S.; Vartuli, J.C.; Roth, W.J.; Leonowicz, M.E.; Kresge, C.T.; Schmitt, K.D.; Chu, C.T.-W.; Olson, D.H.; Sheppard, E.W.; McCullen, S.B.; et al. A new family of mesoporous molecular sieves prepared with liquid crystal templates. J. Am. Chem. Soc. 1992, 114, 10834–10843. [Google Scholar] [CrossRef]
- Vartuli, J.C.; Schmitt, K.D.; Kresge, C.T.; Roth, W.J.; Leonowicz, M.E.; McCullen, S.B.; Hellring, S.D.; Beck, J.S.; Schlenker, J.L.; Olson, D.H.; Sheppard, E.W. Effect of surfactant/silica molar ratios on the formation of mesoporous molecular sieves: Inorganic mimicry of surfactant liquid-crystal phases and mechanistic implications. Chem. Mater. 1994, 6, 2317–2326. [Google Scholar] [CrossRef]
- Karkamkar, A.J.; Kim, S.-S.; Mahanti, S.D.; Pinnavaia, T.J. Lamellar mesostructured silicas with chemically significant hierarchical morphologies. Adv. Funct. Mater. 2004, 14, 507–512. [Google Scholar] [CrossRef]
- Huang, M.H.; Dunn, B.S.; Zink, J.I. In situ luminescence probing of the chemical and structural changes during formation of dip-coated lamellar phase sodium dodecyl sulfate sol-gel thin films. J. Am. Chem. Soc. 2000, 122, 3739–3745. [Google Scholar] [CrossRef]
- Ogura, T.; Sakai, K.; Sakai, H.; Abe, M. Synthesis of highly ordered mesoporous silica with a lamellar structure using assembly of cationic and anionic surfactant mixtures as a template. J. Phys. Chem. C 2008, 112, 12184–12187. [Google Scholar] [CrossRef]
- Ulagappan, N.; Neeraj; Raju, B.V.N.; Rao, C.N.R. Preparation of lamellar and hexagonal forms of mesoporous silica and zircona by the neutral amine route: Lamellar-hexagonal transformation in the solid state. Chem. Commun. 1996, 19, 2243–2244. [Google Scholar] [CrossRef]
- Tanev, P.T.; Liang, Y.; Pinnavaia, T.J. Assembly of mesoporous lamellar silicas with hierarchical particle architectures. J. Am. Chem. Soc. 1997, 119, 8616–8624. [Google Scholar] [CrossRef]
- Fujimoto, Y.; Shimojina, A.; Kuroda, K. Formation of layered silica-alcohol nanostructured materials from alkoxytrichlorosilanes. Chem. Mater. 2003, 15, 4768–4774. [Google Scholar] [CrossRef]
- Bhattacharyva, S.; Lelong, G.; Sabougni, M.-L. Recent progress in the synthesis and selected applications of MCM-41: A short review. J. Exp. Nanosci. 2006, 1, 375–395. [Google Scholar] [CrossRef]
- Rolison, D.R. Catalytic nanoarchitectures-the importance of nothing and the unimportance of periodicity. Science 2003, 299, 1698–1701. [Google Scholar] [CrossRef] [PubMed]
- Danumah, C.; Bousmina, M.; Kaliaguine, S. Novel polymer nanocomposites from templated mesostructured inorganic materials. Macromolecules 2003, 36, 8208–8209. [Google Scholar] [CrossRef]
- Takahashi, N.; Hata, H.; Kuroda, K. Exfoliation of layered silicates trough immobilization of imidazolium groups. Chem. Mater. 2011, 23, 266–273. [Google Scholar] [CrossRef]
- Bi, Y.; Lambert, J.F.; Millot, Y.; Casale, S.; Blanchard, J.; Zeng, S.; Nie, H.; Li, D. Relevant parameters for obtaining high-surface area materials by delamination of magadiite, a layeredsodium silicate. J. Mater. Chem. 2011, 21, 18403–18411. [Google Scholar] [CrossRef]
- Cabrera, S.; El Haskouri, J.; Alamo, J.; Beltrán, A.; Mendioroz, S.; Marcos, M.D.; Amorós, P. Surfactant-assisted synthesis of mesoporous alumina showing continuously adjustable pore sizes. Adv. Mater. 1999, 11, 379–381. [Google Scholar] [CrossRef]
- Cabrera, S.; El Haskouri, J.; Beltrán-Porter, A.; Beltrán-Porter, D.; Amorós, P. Enhanced surface area in thermally stable pure mesoporous TiO2. Solid State Sci. 2000, 2, 513–518. [Google Scholar] [CrossRef]
- El Haskouri, J.; Guillem, C.; Latorre, J.; Beltrán, A.; Beltrán, D.; Amorós, P. S+I− ionic formation mechanism to new mesoporous aluminum phosphonates and diphosphonates. Chem. Mater. 2004, 16, 4359–4372. [Google Scholar] [CrossRef]
- El Haskouri, J.; Cabrera, S.; Gómez-García, C.J.; Guillem, C.; Latorre, J.; Beltrán, A.; Beltrán, D.; Marcos, M.D.; Amorós, P. High cobalt content mesopous silicas. Chem. Mater. 2004, 16, 2805–2813. [Google Scholar] [CrossRef]
- El Haskouri, J.; Moragues, A.; Beltrán, A.; Murcia-Mascarós, S.; Plazaola, F.; Legarra, E.; Mauri-Aucejo, A.; Brotons-Gisbert, M.; Sánchez-Royo, J.F.; Beltrán, D.; Amorós, P. Mesoporous iron phosphate/phosphonate hybrid materials. Micropor. Mesopor. Mat. 2014, 187, 14–22. [Google Scholar] [CrossRef]
- El Haskouri, J.; Cabrera, S.; Guillem, C.; Latorre, J.; Beltrán, A.; Beltrán, D.; Marcos, M.D.; Amorós, P. Atrane precursors in the one-pot surfactant assisted synthesis of high zirconium content porous silicas. Chem. Mater. 2002, 14, 5015–5022. [Google Scholar] [CrossRef]
- Huerta, L.; Guillem, C.; Latorre, J.; Beltrán, A.; Beltrán, D.; Amorós, P. Large monolithic silica-based macrocellular foams with trimodal pre system. Chem. Commun. 2003, 1448–1449. [Google Scholar] [CrossRef]
- El Haskouri, J.; Morales, J.M.; Ortiz de Zárate, D.; Fernández, L.; Latorre, J.; Guillem, C.; Beltrán, A.; Beltrán, D.; Amorós, P. Nanoparticulated silicas with bimodal porosity: Chemical control of the pore sizes. Inorg. Chem. 2008, 47, 8267–8277. [Google Scholar] [CrossRef] [PubMed]
- Moragues, A.; Guillem, C.; Mauri-Aucejo, A.; Tortajada, M.; Beltrán, A.; Beltrán, D.; Amorós, P. Enlarged pore size in nanoparticulated bimodal porous silicas: Improving accessibility. Micropor. Mesopor. Mat. 2016, 221, 150–158. [Google Scholar] [CrossRef]
- Fernández, L.; Viruela-Martín, P.; Latorre, J.; Guillem, C.; Beltrán, A.; Amorós, P. Molecular precursors of mesostructured silica materials in the atrane route: A DFT/GIAO/NBO theoretical study. J. Mol. Struct THEOCHEM 2007, 822, 89–102. [Google Scholar] [CrossRef]
- Cabrera, S.; El Haskouri, J.; Guillem, C.; Latorre, J.; Beltrán, A.; Beltrán, D.; Marcos, M.D.; Amorós, P. Generalised syntheses of ordered mesoporous oxides: The atrane route. Solid State Sci. 2000, 2, 405–420. [Google Scholar] [CrossRef]
- El Haskouri, J.; Cabrera, S.; Caldés, M.; Alamo, J.; Beltrán-Porter, A.; Marcos, M.D.; Amorós, P.; Beltrán-Porter, D. Ordered mesoporous materials: Composition and topology control through chemistry. Int. J. Inorg. Chem. 2001, 3, 1157–1163. [Google Scholar] [CrossRef]
- Lin, H.-P.; Mou, C.-Y. Structural and morphological control of cationic surfactant-templated mesoporous silica. Acc. Chem. Res. 2002, 35, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Firouzi, A.; Atef, F.; Oertli, A.G.; Stucky, G.D.; Chmelka, B.F. Alkaline lyotropic silicate-surfactant liquid crystals. J. Am. Chem. Soc. 1997, 119, 3596–3610. [Google Scholar] [CrossRef]
- He, X.; Zhu, B.; Huang, J.; Zhao, G. A study on organized assemblies in the aqueous systems of alkylammonium chlorides. J. Colloid Interface Sci. 1999, 220, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Tanev, P.; Pinnavaia, T.J. Mesoporous silica molecular sieves prepared by ionic and neutral surfactant templating: A comparison of physical properties. Chem. Mater. 1996, 8, 2068–2079. [Google Scholar] [CrossRef]
- Zhang, W.; Pauly, T.R.; Pinnavaia, T.J. Tailoring the framework and textural mesopores of HMS molecular sieves through an electrically neutral (S0I0) assembly pathway. Chem. Mater. 1997, 9, 2491–2498. [Google Scholar] [CrossRef]
- Matulis, D.; Bloomfielfd, A. Thermodynamics of the hydrophobic effect. I. Coupling of aggregation and pK(a) shifts in solutions of aliphatic amines. Biophys. Chem. 2001, 93, 37–51. [Google Scholar] [CrossRef]
- Laughlin, R.G. The Aqueous Phase Behaviour of Surfactants, 1st ed.; Academic Press: New York, NY, USA, 1994; pp. 181–236. ISBN 0-12-437745-9. [Google Scholar]
- Vaia, R.A.; Teukolsky, R.K.; Giannelis, E.P. Interlayer structure and molecular environment of alkylammonium layered silicates. Chem. Mater. 1994, 6, 1017–1022. [Google Scholar] [CrossRef]
- Wang, X.; Liu, J.; Du, H.; Miller, J.D. States of adsorbed dodecyl amine and water at a silica surface as revealed by vibrational spectroscopy. Langmuir 2010, 26, 3407–3414. [Google Scholar] [CrossRef] [PubMed]
- Weers, J.G.; Scheuing, D.R. Fourier Transform Infrared Spectroscopy in Colloid and Interface Science; Scheuing, D.R., Ed.; ACS Symposium Ser. 447; American Chemical Society: Washington, DC, USA, 1990; pp. 87–122. [Google Scholar]
- Chernyshova, I.V.; Rao, K.H.; Vidyadhar, A. Mechanisms of adsorption of log-chain alkylamines on silicates. A spectroscopic study. 1. Quartz. Langmuir 2000, 16, 8071–8084. [Google Scholar] [CrossRef]
- Fyfe, C.A. Solid State NMR for Chemists, 1st ed.; CFC Press: Guelph, ON, Canada, 1983; pp. 337–361. ISBN 10: 0889550387. [Google Scholar]
- Alam, N.; Mokaya, R. Crystalline mesoporous silicates from layered precursors. J. Mater. Chem. 2008, 18, 1383–1391. [Google Scholar] [CrossRef]
- de A. A. Soler-Illia, G.J.; Sanchez, C.; Lebeau, B.; Patarin, J. Chemical strategies to design textured materials: From microporous and mesoporous oxides to nanonetworks and hierarchical structures. Chem. Rev. 2002, 102, 4093–4138. [Google Scholar] [CrossRef]
- El Haskouri, J.; Ortiz de Zárate, D.; Guillem, C.; Beltrán-Porter, A.; Caldés, M.; Marcos, M.D.; Beltrán-Porter, D.; Latorre, J.; Amorós, P. Hierarchical porous nanosized organosilicas. Chem. Mater. 2002, 14, 4502–4504. [Google Scholar] [CrossRef]
- Nicolosi, V.; Chhowalla, M.; Kanatzidis, M.G.; Strano, M.S.; Coleman, J.N. Liquid exfoliation of layered materials. Science 2013, 340, 1226419. [Google Scholar] [CrossRef]
- Lee, D.W.; Yoo, B.R. Advanced silica/polymer composites: Materials and applications. J. Ind. Eng. Chem. 2016, 36, 1–12. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | n | d (001) a/nm | Water b/% | S0 Amine b/% | S+ Amine b/% | S0/S+ Ratio |
|---|---|---|---|---|---|---|
| UVM-Ln | 8 | 2.74 | 3.5 | 26 | 13 | 2.0 |
| UVM-Ln | 10 | 3.35 | 3.4 | 28 | 19 | 1.5 |
| UVM-Ln | 12 | 4.16 | 4.9 | 33 | 21 | 1.6 |
| UVM-Ln | 14 | 4.92 | 2.2 | 33 | 24 | 1.4 |
| UVM-Ln | 16 | 5.48 | 1.4 | 41 | 24 | 1.7 |
| UVM-Ln | 18 | 5.91 | 1.7 | 44 | 26 | 1.7 |
| Sample | n | d (001) a/nm | Si/Al b/Nominal | Si/Al c/Real | Si(Q/Si(T) b/Nominal | Si(Q)/Si(T) d/Real |
|---|---|---|---|---|---|---|
| Al-UVM-Ln | 16 | 5.36 | 19 | 22 | - | - |
| Al-UVM-Ln | 16 | 5.33 | 9 | 7 | - | - |
| Al-UVM-Ln | 16 | 5.29 | 4 | 5 | - | - |
| Al-UVM-Ln | 16 | 4.03 | 2 | 3 | - | - |
| Epoxy-UVM-Ln | 12 | 4.13 | - | - | 12.5 | 4.5 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burguete, P.; Morales, J.M.; Fernández, L.; El Haskouri, J.; Latorre, J.; Guillem, C.; Pérez-Pla, F.; Cros, A.; Beltrán, D.; Beltrán, A.; et al. Layered-Expanded Mesostructured Silicas: Generalized Synthesis and Functionalization. Nanomaterials 2018, 8, 817. https://doi.org/10.3390/nano8100817
Burguete P, Morales JM, Fernández L, El Haskouri J, Latorre J, Guillem C, Pérez-Pla F, Cros A, Beltrán D, Beltrán A, et al. Layered-Expanded Mesostructured Silicas: Generalized Synthesis and Functionalization. Nanomaterials. 2018; 8(10):817. https://doi.org/10.3390/nano8100817
Chicago/Turabian StyleBurguete, Pedro, José Manuel Morales, Lorenzo Fernández, Jamal El Haskouri, Julio Latorre, Carmen Guillem, Francisco Pérez-Pla, Ana Cros, Daniel Beltrán, Aurelio Beltrán, and et al. 2018. "Layered-Expanded Mesostructured Silicas: Generalized Synthesis and Functionalization" Nanomaterials 8, no. 10: 817. https://doi.org/10.3390/nano8100817