3.2. Small Irn Clusters on Hydrated γ-Al2O3(110)
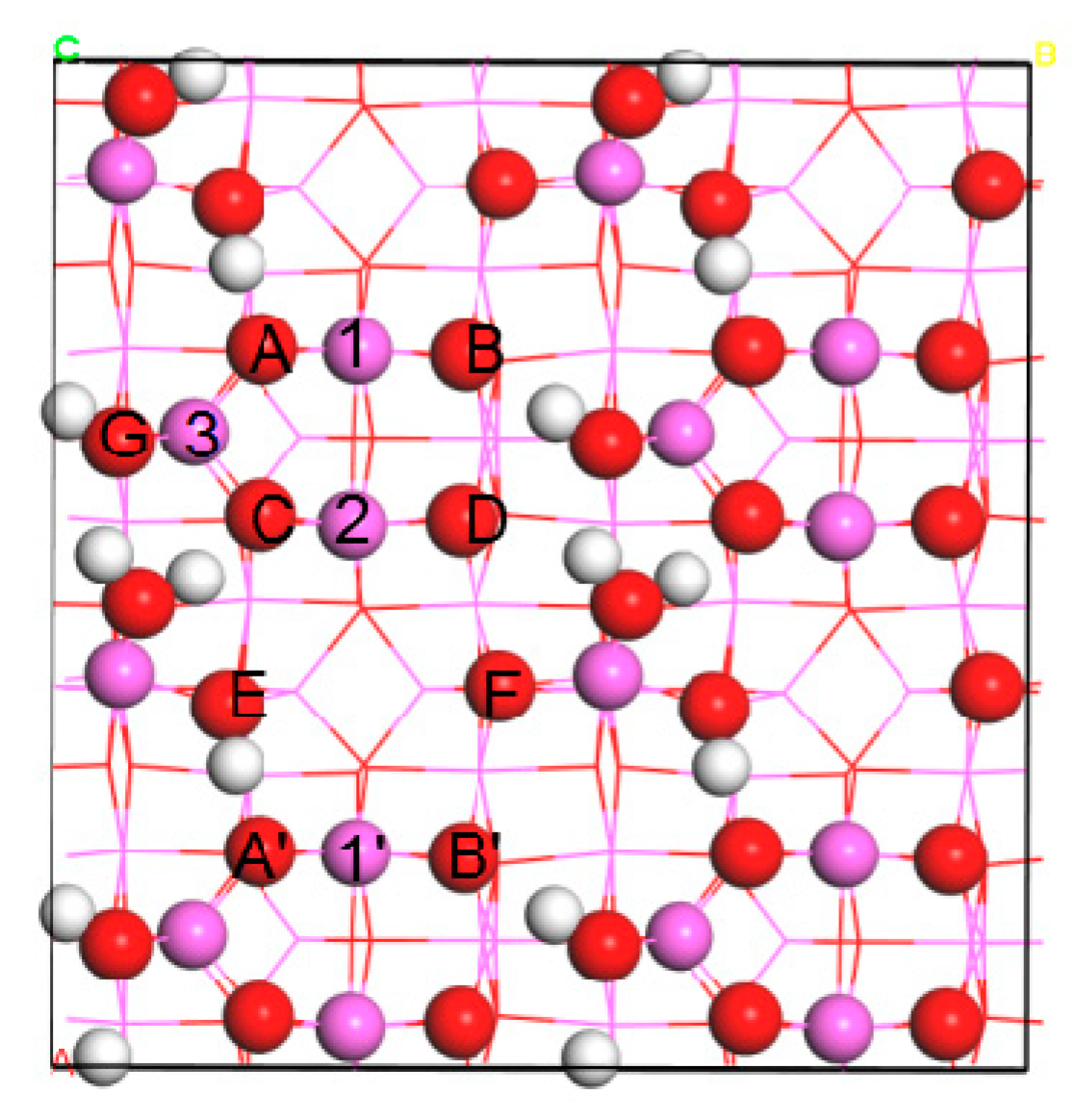
For Ir adsorption, we considered a series of adsorption sites including seven top (O(A), O(B), O(C), O(D), O(F), Al(1), and Al(2)), ten bridge (O(A)-O(C), O(B)-O(D), O(D)-O(F), O(B′)-O(F), Al(1)-Al(2), Al(1′)-Al(2), O(A)-Al(1), O(B)-Al(1), O(C)-Al(2), and O(D)-Al(2)), and three hollow sites (O(A)-O(C)-Al(3), O(D)-O(F)-Al(2), and O(B′)-O(E′)-O(F)) (
Figure 1). The most energetically favorable structures and corresponding adsorption energies E
ads are summarized in
Figure 2 and
Table S2, respectively.
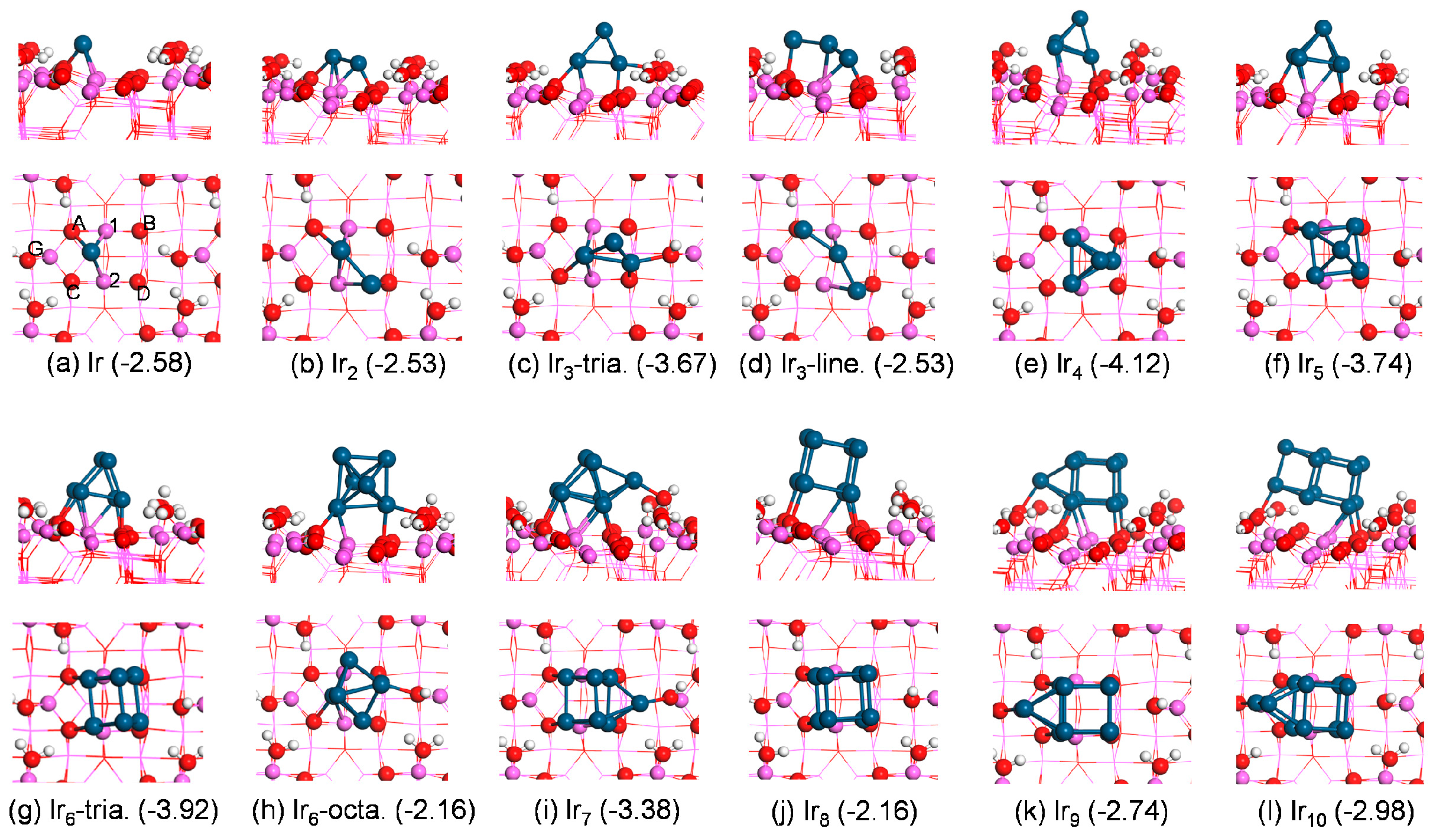
As shown in
Figure 2, a single Ir atom preferred to bond to surface Al(1), Al(2), and O(A) atoms yielding E
ads of −2.58 eV. For Ir
2 adsorption, both Ir atoms bonded to the surface, forming three Ir–Al bonds and two Ir–O bonds with E
ads of −2.53 eV. Two Ir atoms bonded to the same surface Al center.
Unlike the case in the gas phase, the adsorbed Ir3 preferred the triangular configuration over the linear one. The adsorbed triangular structure presented Eads of −3.67 eV with respect to the gas-phase triangular trimer, which was 1.14 eV lower in energy than the linear structure, while in the gas phase the former was 0.26 eV higher than the latter. The adsorbed triangular trimer presented a stand-up configuration with two Ir atoms binding to the surface and one Ir atom pointing to the air. While in the adsorbed linear trimer, each Ir atom bonded to the surface.
For Ir
4 adsorption, all attempts to obtain the square planar Ir
4 cluster (the most stable configuration in gas phase) converted to the geometry with a bent rhombus on the surface. The reconstruction of small deposited metal clusters on the support was also observed for Pd
n clusters (n = 1–7) adsorption on α–Al
2O
3 (0001). Nigam and Majumder [
26] found that the Pd
4 deformed from the tetrahedral configuration in the gas phase into the bent rhombus in the adsorbed state on the Al
2O
3 surface. The most stable structure was a tetrahedron with three Ir atoms bonding to the surface and resulting in E
ads of −4.12 eV. Our results agree with the earlier experimental and theoretical reports. Using EXAFS spectroscopy, Argo et al. [
9] found an Ir–Ir first-shell coordination number in γ-Al
2O
3-supported Ir
4 nanoparticles of about 3. Our previous study showed that Rh
4 also prefers the tetrahedral frame on γ-Al
2O
3 surfaces [
25].
The adsorbed Ir5 retained the square pyramid geometry upon adsorption on the surface, with four Ir atoms bonding to the substrate and yielding Eads of −3.74 eV.
Analogous to the case in the gas phase, the most energetically preferred Ir
6 geometry was a triangular prism with E
ads of −3.92 eV. In the adsorbed state, four Ir atoms made contact with the substrate and two Ir atoms kept away from the surface. The most stable octahedral structure of adsorbed Ir
6 was less stable by 1.76 eV (higher in energy) than the triangular prism on the γ-Al
2O
3 support, and in the gas phase the energy difference between each was 0.70 eV. The coordinates RMSD (root-mean-square deviation) between the triangular prism configuration and the octahedral structure for the Ir
6 cluster were ~0.94 in the gas phase and ~0.98 for the supported case, respectively, which is consistent with the energy difference between the two. The octahedral adsorbed Ir
6 cluster bonded to the substrate via two Ir atoms (
Figure 2h). This was inconsistent with the experimental observation [
9,
26,
27], but in agreement with the previous calculations [
13,
28,
29]. Experimentally, the octahedral Ir
6 commonly exists in the form of Ir
6 complexes [
9]. Argo et al. [
9] used the octahedral frame of [Ir
6(CO)
15]
2− upon its decarbonylation to obtain an Ir
6 cluster on the γ-Al
2O
3 support and found that Ir
6 maintained the octahedral frame after decarbonylation using EXAFS measurements. Here, the CO ligands helped it to keep the octahedral frame, while several theoretical studies [
13,
28,
29] found that the bare gas-phase Ir
6 cluster without any ligand favored a triangular prism with D
3h symmetry over the O
h octahedral structure.
For Ir
7 adsorption, the energetically preferred structure yielded a similar configuration to the gas-phase cluster with E
ads of −3.38 eV. The additional Ir atom to the adsorbed triangular prism Ir
6 cluster bonded to the oxygen atom in a surface hydroxyl. A surface distortion (
Figure 2i) was observed upon the adsorption of the Ir
7 cluster, where the bonded hydroxyl moved upwards to make contact with the Ir atom. The similar adsorbate-induced support rearrangement was found for triangular Ir
3 adsorption on γ-Al
2O
3 (
Figure 2c).
The most stable configuration for Ir
8 adsorption on alumina was a cubic Ir
8 cluster with four Ir atoms bonding to the surface and resulting in E
ads of −2.16 eV (
Figure 2j). The most favorable structures for Ir
9 and Ir
10 adsorption exhibited E
ads of −2.74 and −2.98 eV, respectively. As shown in
Figure 2k,l, one Ir atom in the adsorbed Ir
9 and Ir
10 cluster bonded to the oxygen center in a surface hydroxyl group.
Similar to our previous finding for Rh adsorption on the hydrated γ-Al
2O
3(110) surface [
25], Ir
n clusters preferred to adsorb in the valley of the surface O and Al sites instead of on the covering hydroxyls layer, indicating the nanosized metal cluster can retard the transformation of γ-Al
2O
3 to AlOOH by pre-adsorption on the transformation site. This is consistent with the previous experimental [
30] and theoretical [
25] observations, confirming that it may be a general effect for a large number of nanosized transition metals.
In summary, Irn preferred to adsorb on the no-hydroxyls-covered area. When cluster diameter (to simplify, the cluster diameter is defined by the longest distance of M–M in the cluster) was smaller than 4.11 Å (e.g., n = 1–6 and 8), the Irn cluster bonded to the surface O and Al atoms only. When cluster diameter was larger than 4.21 Å (n = 7, 9, and 10), besides bonding to the surface O and Al atoms, the Irn cluster bonded to the oxygen of surface hydroxyl as well. The γ–Al2O3 (110) support changed the morphological features and the cluster stability of Ir3 and Ir4 upon their adsorption on the support. The Ir–Ir distance of adsorbed Irn (n = 2–10) in the basal plane underwent an elongation compared with the gas-phase Irn clusters.
3.3. Adsorption of Ethylene on Bare Irn
For ethylene adsorption on the bare Irn cluster, three adsorption modes were considered: di-σ mode, with two carbon ends of ethylene binding to two substrate atoms; π mode, with two carbon ends of ethylene binding to one substrate atom; and a bridge-top (B-T) mode, with one carbon bridging two substrate atoms and the other carbon binding to one substrate atom.
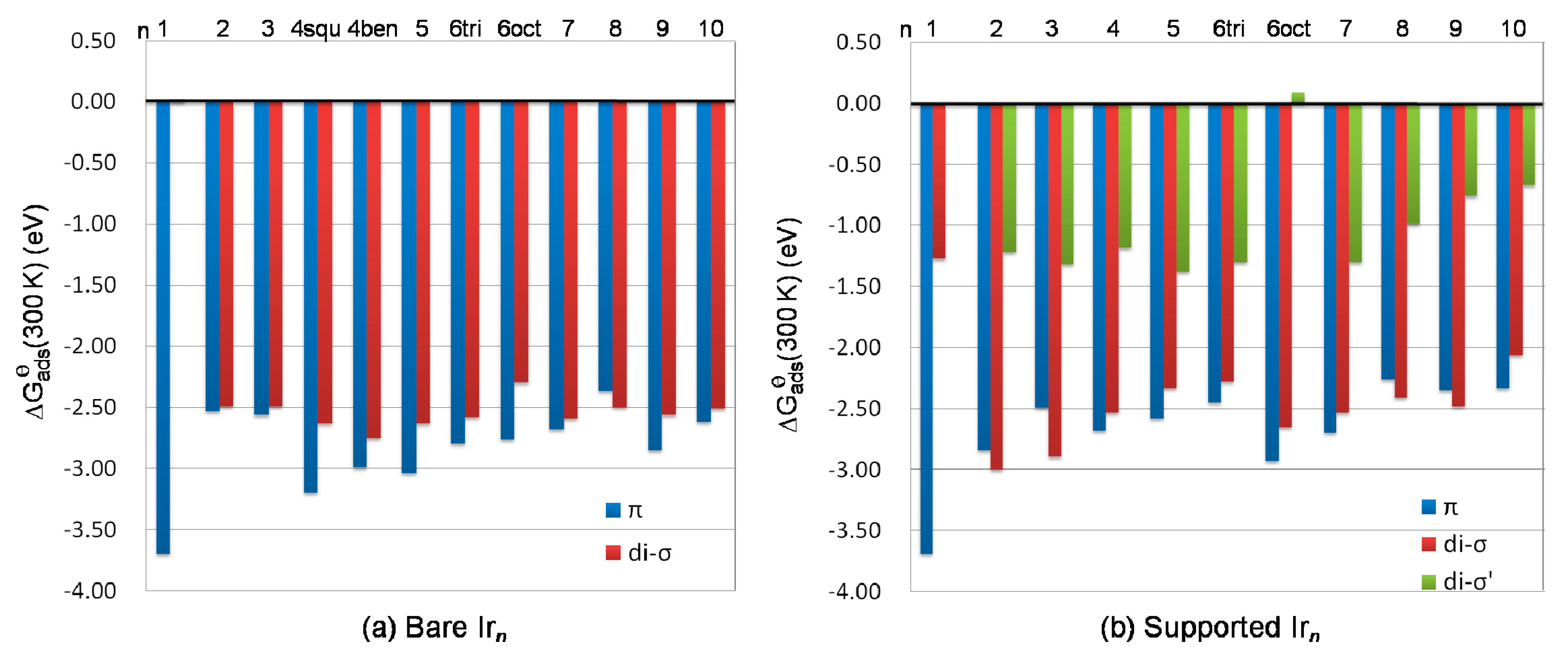
Ethylene adsorption on atomic Ir yielded the largest E
ads of −2.96 eV, suggesting the strongest ethylene binding of all considered Ir
n (n = 1–10) clusters (
Table 1). Ethylene adsorption on the Ir
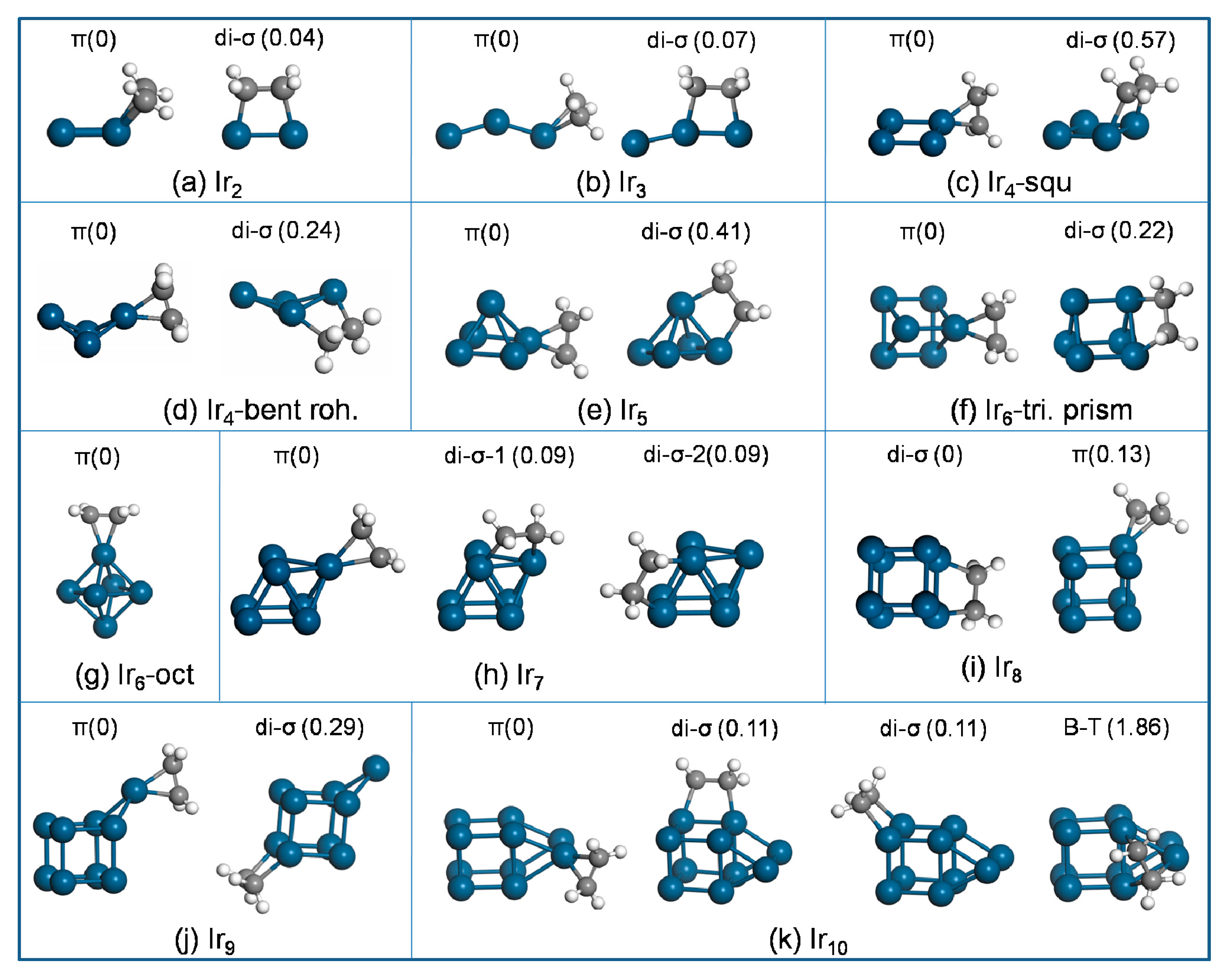
2 cluster (
Figure 3a) via π-bound mode yielded an adsorption energy E
ads of −1.79 eV, which was very close to the di-σ-bound mode that featured E
ads of −1.75 eV. This suggests very similar adsorbate binding for both modes at 0 K.
Geometry optimization of ethylene adsorption on the bare Ir
3 cluster yielded three different local-energy minima, and those with the lowest total energy for each mode are shown in
Figure 3b. The E
ads, −1.82 eV (π-bound mode) and −1.75 eV (di-σ-bound mode), differ only slightly. The adsorption of ethylene induced a strong deformation of the Ir
3 cluster, where Ir
3 changed from the linear structure in the free phase to the bent configuration with the adsorbate. The deformation energy E
def(Ir
3) reached as high as 0.23 (0.15) eV for the π (di-σ)-bound mode.
For ethylene adsorption on the square Ir
4 cluster (
Figure 3c), the calculations found two local equilibrium geometries for each mode. The π-bound state was more favorable than the di-σ-bound mode, where E
ads of the π-bound mode was −2.46 eV and the di-σ-bound state yielded E
ads of −1.89 eV. To compare with the supported Ir
4 cluster, ethylene adsorption on the bent-rhombus Ir
4 cluster was studied as well. Ethylene adsorption yielded four di-σ and two π local minima, and the energetically preferred structure for each mode is provided in
Figure 3d. The di-σ mode was less stable than the π mode by 0.24 eV (higher in energy).
Ethylene adsorption on Ir5 (two local equilibrium geometries for each mode are found) preferred the π-bound state to the di-σ-bound mode, which accounts for Eads of −2.30 eV (π) and −1.80 eV (di-σ).
Ethylene stabilizes on the triangular-prism Ir
6 cluster, and the most favorable adsorption geometries are shown in
Figure 3f. Ethylene adsorption on the triangular-prism Ir
6 preferred the π state to the di-σ mode. The corresponding adsorption energies were −2.06 and −1.84 eV for the π-bound state and di-σ-bound mode, respectively. Similarly, ethylene adsorption on the octahedral Ir
6 cluster (
Figure 3g) via the π-bound state was more stable than it was via the di-σ-bound mode, with an energy difference of 0.46 eV.
Geometry optimization of ethylene adsorption on the Ir
7 cluster yielded three π and four di-σ local-energy minima. The most stable adsorption geometries of ethylene on the Ir
7 cluster for each mode are sketched in
Figure 3h. Our results show that the π structure yields E
ads of −1.94 eV, which is more favorable than the di-σ structure by 0.09 eV (lower in energy).
The energetically preferred adsorption geometries of ethylene on the Ir
8 cluster are sketched in
Figure 3i. The calculations yield E
ads of −1.63 eV (π) and −1.76 eV (di-σ), suggesting stronger binding for the di-σ structure compared with the π state.
For ethylene adsorption on Ir
9, the calculations found three π and five di-σ local-energy minima structures, and the most stable one is shown in
Figure 3j. The most stable π and di-σ structures yielded E
ads of −2.11 and −1.82 eV, respectively, indicating a stronger stability of the π mode.
The most stable adsorption geometry of ethylene on the bare Ir
10 cluster via π-bound mode (of three local equilibrium geometries found) is sketched in
Figure 3k. The stability of ethylene adsorption decreased in the sequence of π > di-σ > B-T. Among the obtained three local equilibrium geometries, the most stable π structure yielded E
ads of −1.88 eV, which was 0.11 eV lower than the most stable di-σ structure (of seven local equilibrium geometries). Two di-σ structures yielded identical E
ads of −1.77 eV. The B-T configuration (
Figure 3k) was the least stable, with much smaller E
ads of −0.02 eV. Note that the B-T structure was only available on Ir
10. All attempts to obtain the B-T configuration on the other Ir
n clusters resulted in either the π structure or the di-σ mode.
In summary, the stability of ethylene adsorption on the bare Irn clusters decreased in the sequence of π > di-σ > B-T with one exception of Ir8 where the di-σ structure was energetically preferred over the π structure.
To analyze the adsorption energy in more details, we divided it into three contributions according to E
ads = E
def(C
2H
4) + E
def(Ir
n) + E
int. From the data summarized in
Table 1, we can see that the ethylene deformation energies for the di-σ and π structures were within the energy range of 1.53–1.85 and 0.45–0.53 eV, respectively. The deformation energy of ethylene accompanied the adsorption of ethylene along with the C−C bond elongation. The di-σ structure always induced a greater elongation regarding the gas phase than the π mode in our studies, which is in agreement with previous theoretical studies [
31,
32]. The C−C bond distance in the di-σ mode enlarged to ~1.51Å from 1.33 Å in the gas phase, while the π mode caused a smaller C−C bond extension (~1.43 Å).
Further analysis shows that the deformation of the adsorbate was much stronger than that of the substrate. As the deformation energies of the adsorbed ethylene were larger than 0.45 eV, the deformation energies of Ir
n clusters were rather small, below 0.23 eV. The energy cost for the deformation could have been compensated by the interaction energy between the adsorbates and the substrate. Despite the cluster–ethylene interaction energy in the di-σ mode always being larger than that in the π mode, it could not compensate for the energy cost difference of the deformation between two modes on most of the Ir
n clusters (excluding Ir
8). As a result, ethylene preferred to adsorb on the bare clusters via the π mode, except for the Ir
8 cluster. The reverse preference of adsorption mode on Ir
8 was the same with Ir(111) [
33], where the di-σ mode was more favorable than the π mode.
3.4. Adsorption of Ethylene on Al2O3(110)-Supported Irn
Next, we studied the adsorption of ethylene on hydrated γ-Al
2O
3(110)-supported Ir
n clusters. For ethylene adsorption on Ir
n/γ-Al
2O
3, besides the three scenarios described on the bare Ir
n cluster, one more scenario was considered: the di-σ′ mode at the interface with one carbon atom on the Ir
n cluster and one carbon atom on the γ-Al
2O
3 support. Therefore, for ethylene adsorption on Ir
n/γ-Al
2O
3, we considered four possible adsorption geometries, including three modes on the supported Ir
n cluster (π, B-T, and di-σ) and one mode at the interface (di-σ′). The most stable configuration for each mode and their corresponding energies are summarized in
Figure 4 and
Figure 5 and
Table 2.
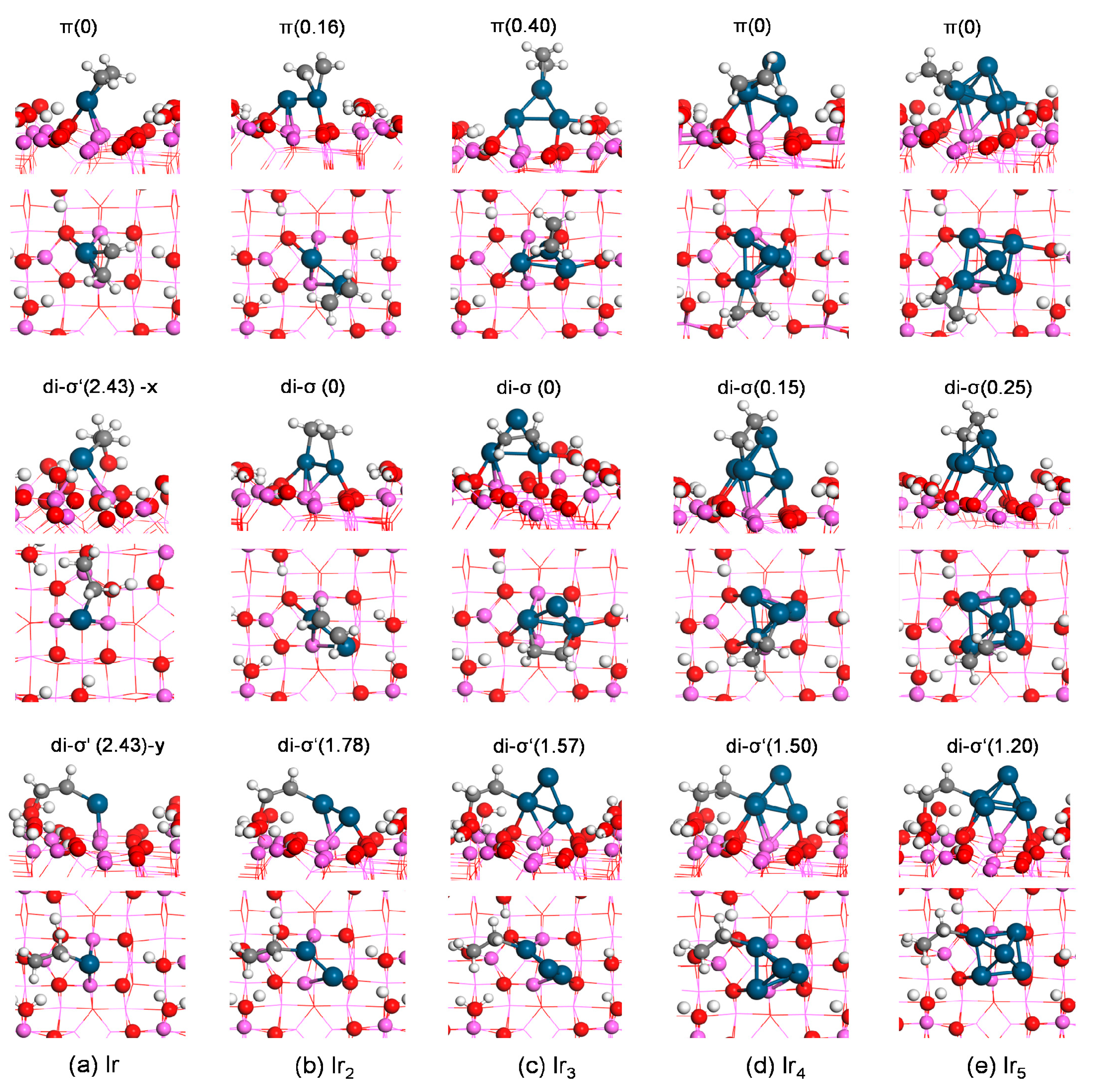
Ethylene adsorption yielded almost the identical adsorption energy of −2.95 eV both on Ir1/γ-Al2O3(110) and on atomic Ir. The di-σ′ mode at the interface, where one carbon atom of ethylene bonds to the Ir atom and the other carbon end bonds to the oxygen site of the surface hydroxyl group, was less stable by 2.42 eV in energy.
For ethylene adsorption on Ir
2/γ-Al
2O
3(110), the di-σ structure was energetically more favorable than the π state (
Figure 4b). The adsorption energy of the di-σ mode was −2.26 eV, while the π mode gave E
ads of −2.10 eV. As was the case for Ir
1/γ-Al
2O
3(110), the di-σ′ structure at the interface resulted in much higher E
ads of −0.48 eV, indicating this structure was less favorable than the di-σ and π structures on the supported Ir
2 cluster thermodynamically.
Three π and three di-σ structures were obtained for ethylene adsorption on Ir
3/γ-Al
2O
3(110) through geometry optimization. In the most stable π mode (
Figure 4c), ethylene preferred to adsorb on the upper Ir site while in the most favorable di-σ mode, two carbon ends bonded to the bottom Ir centers. The adsorption energies of the di-σ structure and the π mode were −1.99 and −1.75 eV, respectively, indicating the preference of the di-σ structure. The most stable di-σ′ structure at the interface (of three obtained structures) yielded E
ads of −0.58 eV.
Our calculations obtained five di-σ, six π, three monodentate (M) with one carbon end adsorbing on a metal site (
Figure S1a), and two di-σ′ structures at the interface for ethylene adsorption on Ir
4/γ-Al
2O
3. The most stable π mode yielded E
ads of −1.94 eV. While the most stable di-σ structure yielded E
ads of −1.79 eV (
Figure 4d). The di-σ′ structure at the interface with one carbon end at a bottom Ir site and one carbon end at the oxygen O(G) site in the hydroxyl group of the γ-Al
2O
3(110) surface yielded E
ads of −0.44 eV. The most stable M mode yielded E
ads of –0.66 eV. The stability of the ethylene adsorption mode decreased in the order of π > di-σ > M > di-σ′ (at interface).
For ethylene adsorption on Ir
5/γ-Al
2O
3, ethylene binded to one bottom Ir atom resulting in E
ads of −1.84 eV in the most stable π mode (of four local equilibrium geometries found). Ethylene adsorbed on the substrate through its two C atoms bonding to one bottom Ir and one upper Ir (di-σ) yielding E
ads of −1.59 eV. Two di-σ′ structures at the interface yielded very close E
ads, with an energy difference of 0.08 eV (the more stable one is shown in
Figure 4e).
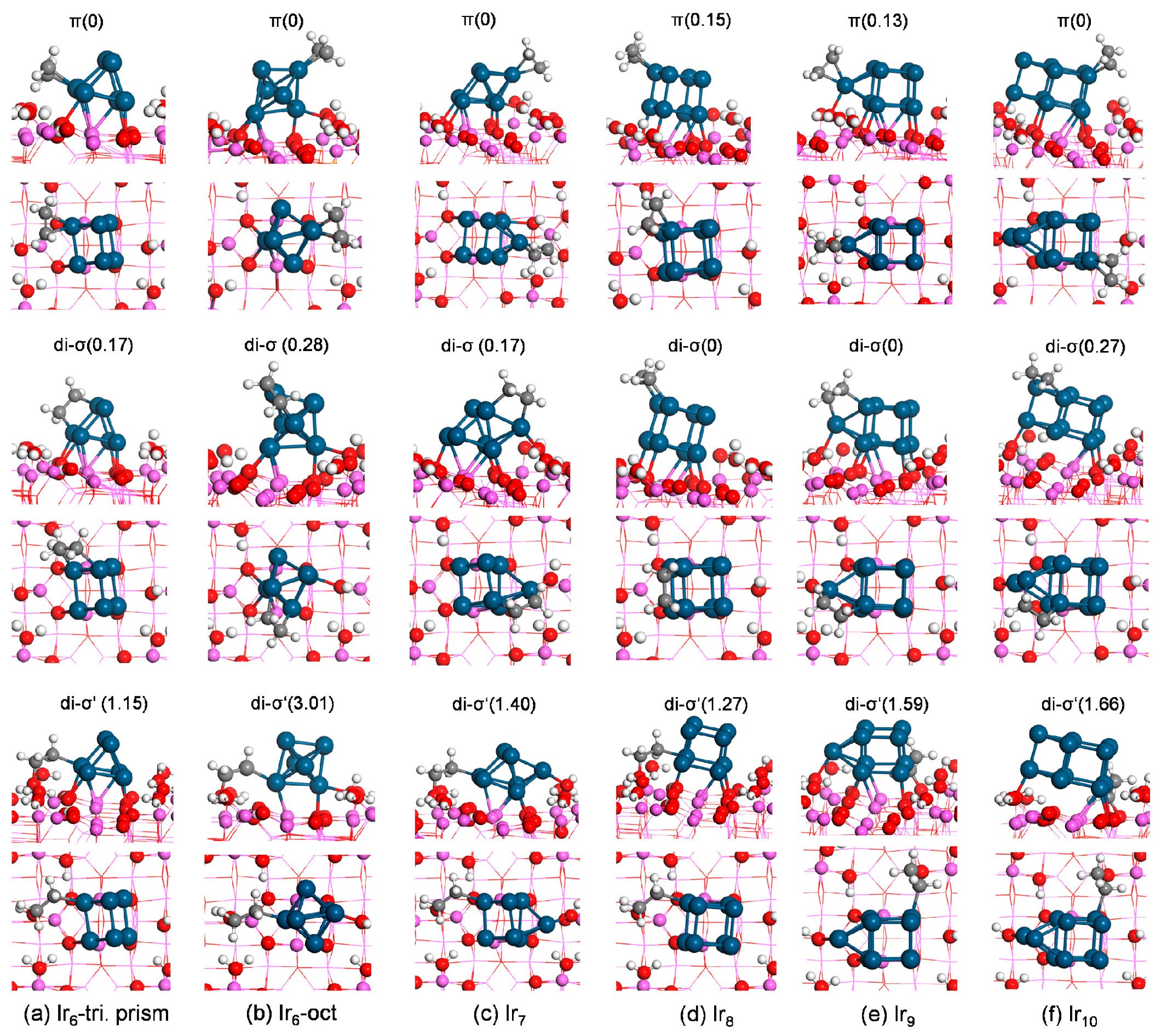
On Ir
6/γ-Al
2O
3, the most stable π-bound ethylene yielded E
ads of −1.71 eV (
Figure 5a). It adsorbed on a bottom Ir atom. The most stable di-σ-bound structure yielded an adsorption energy of −1.54 eV and bridged one top and one bottom Ir atoms. The most stable di-σ′ structure at the interface (of two local equilibrium geometries) on Ir
6/γ-Al
2O
3(110), with one carbon atom on a bottom Ir atom and one carbon atom on the oxygen site of surface hydroxyl, yielded E
ads of −0.56 eV.
Although the most stable supported Ir
6 exhibited a triangular-prism configuration and the octahedral structure was less stable by 1.76 eV (higher in energy), as discussed in
Section 3.2, the supported octahedral Ir
6 cluster was observed by the experiments. Therefore, the adsorption of ethylene on the most stable supported octahedral Ir
6 cluster was studied for comparison. On Ir
6oct/γ-Al
2O
3, the most stable π-bound ethylene yielded E
ads of −2.19 eV (
Figure 5b). It adsorbed on a top Ir atom with two Ir–C bond lengths both of 2.11 Å. The most stable di-σ-bound structure yielded an adsorption energy of −1.91 eV. It bridged one top Ir atom and one middle Ir atom. The present results agree with the previous experimental observation [
9] and theoretical results [
34,
35]. Qi et al. [
34] and Valero et al. [
35] discovered that the π adsorption mode was more stable than the di-σ mode for ethylene adsorption on Ir
4-C/γ-Al
2O
3(110) and Pd
4/γ-Al
2O
3(110) catalyst. Argo et al. [
9] observed that the π adsorption mode was directly relevant to the hydrogenation reaction on Ir
n/γ-Al
2O
3 (n = 4 and 6). The most stable M structure (of three local-energy minimum structures,
Figure S1b) yielded E
ads of −0.62 eV. The di-σ′ (at interface) structure yielded E
ads of +0.82 eV suggesting the adsorption was meta-stable and strongly endothermic.
For ethylene adsorption on Ir
7/γ-Al
2O
3, five local equilibrium geometries for both the π and di-σ modes were found. The most stable π state resulted in E
ads of −1.96 eV (
Figure 5c). The most stable di-σ structure was less stable than the π state, with E
ads of −1.79 eV. The di-σ′ structure at the interface yielded E
ads of −0.56 eV. Therefore, the stability of these structures decreases in the order π > di-σ > di-σ′ (at interface).
We found three π and four di-σ-bound local-energy minimum structures for ethylene adsorption on Ir
8/γ-Al
2O
3. Analogous to the case on the bare Ir
8 cluster, the di-σ configuration was energetically preferred over the π state by 0.18 eV (lower in energy). For the most stable π and di-σ structures, ethylene preferred to adsorb on the upper Ir atoms away from the interface (
Figure 5d). The most stable di-σ′ structure at the interface among two obtained local minima yielded an adsorption energy of −0.25 eV, suggesting less stability than the other two modes.
For ethylene adsorption on Ir
9/γ-Al
2O
3, geometry optimization obtained four π and three di-σ structures. In the most stable π and di-σ structures, ethylene preferred to adsorb on the Ir atom, capping on the face of the cube (
Figure 5e). Unlike the case on the bare Ir
9 cluster, the π structure was less stable than the di-σ state. The corresponding adsorption energies E
ads were −1.61 and −1.74 eV for the π and di-σ structures, respectively. For ethylene adsorption at the interface, the adsorption was nearly neutral, with E
ads of −0.02 eV.
For ethylene adsorption on Ir
10/γ-Al
2O
3, the most stable π (of five local minima) and di-σ (of four local equilibrium geometries) structures are provided in
Figure 5f. Ethylene adsorbed on the upper Ir atom of the cube in the most stable π mode. In the most stable di-σ mode, ethylene adsorbed on the top surface away from the interface where it bridged one Ir atom of the cube and one caped Ir atom. The most stable π structure yielded an adsorption energy of −1.59 eV, 0.27 eV lower in energy than the most stable di-σ state and suggesting the preference of π structure. Similar to the case on supported Ir
9, ethylene adsorption at the interface of Ir
10/γ-Al
2O
3, with one carbon bonding to one surface oxygen atom and the other carbon bonding to one bottom Ir site, was slightly endothermic, with E
ads of +0.07 eV.
As shown in
Table 1 and
Table 2, the di-σ mode (either at the interface or on the Ir
n cluster) caused a stronger distortion of the adsorbed ethylene associated with the larger deformation energy E
def(C
2H
4) than the π state. This was due to the fundamental nature of the bonds. The bonding in these modes involved a rehybridization of the carbon centers. In
Table 1 and
Table 2, we provide the mean hybridization value according to the work of [
35]. According to this work, the mean hybridization value of the gas-phase ethylene—whose carbon center exhibits
sp2 hybridization—was 2, and the carbon center with
sp3 hybridization (e.g., C in ethane) yielded the mean hybridization value of 3. We found the mean hybridization value of the di-σ′ mode at the interface was about 3, indicating a complete rehybridization of the carbon centers from
sp2 in the gas phase to
sp3 in the adsorbed state. The di-σ structure at the interface exhibited the same structure with the gas-phase ethane where two H atoms of ethane were substituted by one Ir atom and one surface O (see
Figure 3a). The mean hybridization values of the di-σ and π states on the Ir
n cluster were ~2.45 and ~2.85, indicating a weaker rehybridization of the carbon centers in the π state than the di-σ structures regarding the gas-phase ethylene (the mean hybridization value was 2). These results reveal an electron transfer from the support to π* orbitals of ethylene. A linear relationship between the deformation energy of ethylene and the mean hybridization value of the carbon centers can be observed in
Figure S2.
It is noted that Ir6oct/γ-Al2O3 exhibited the largest deformation energy upon ethylene adsorption, suggesting the strongest reconstruction among all considered Irn/γ-Al2O3. Because of the steric hindrance effect, which limits space at the interface, the supported octahedral Ir6 rearranged itself to accommodate the adsorbed ethylene molecule. Meanwhile the ethylene-support interaction energy was not large enough to balance the deformation energies, resulting in the largest positive Eads of +0.82 eV and indicating the adsorption at the interface was the weakest among all the considered configurations and strongly meta-stable.
3.6. Analysis of Electronic Properties
To know more about the charge redistribution upon the adsorption, we examined the local charge flow for adsorbed monomer, Ir
4, and Ir
10 systems using electron density difference maps. The electron density difference (∆
ρ) was calculated by
where
ρ(Ads/Sub) is the total electron density of the adsorbates/substrate complex,
ρ(Ads)
fix, and
ρ(Sub)
fix are the electron densities of the isolated adsorbates and substrate in the same geometry as the adsorbed state, respectively.
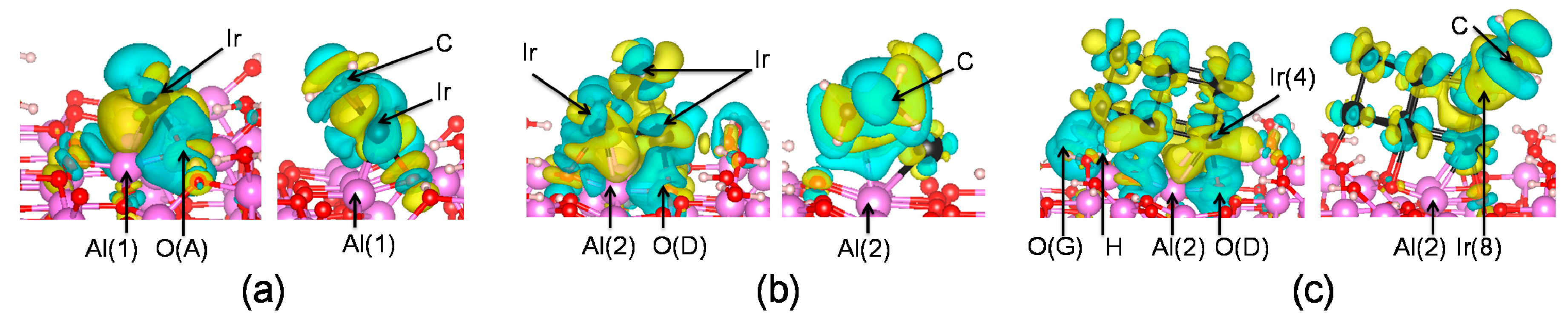
In the electron density difference maps (
Figure 7), some
d orbitals of Ir were depleted upon Ir
n cluster adsorption on the surface, which was associated with the charge redistribution of the formed Ir–O and Ir–Al bonds. Oxygen atoms, which bond to Ir atoms, lose electrons during the adsorption process, causing decreased electron density along the Ir–O bond, while in the regions of the Ir–Al bond, electron density increases. Similar phenomena have been observed for Pd [
40] and Rh [
25] cluster adsorption on
γ-Al
2O
3. The depletion of Rh (Pd)
d orbitals during its adsorption on
γ-Al
2O
3 was balanced by increased electron density along the Rh(Pd)–Al bond.
As shown in
Figure 7, electrons previously accumulated along Ir–Al bonds in Ir
n/γ-Al
2O
3 were transferred to the Ir
n/C
2H
4 part upon C
2H
4 adsorption on γ-Al
2O
3-supported Ir
n. This suggests that the adsorption of ethylene on γ-Al
2O
3-supported Ir
n influences the charge distribution at the metal–alumina interface. The following projected density of states (PDOS) analysis further confirms this statement.
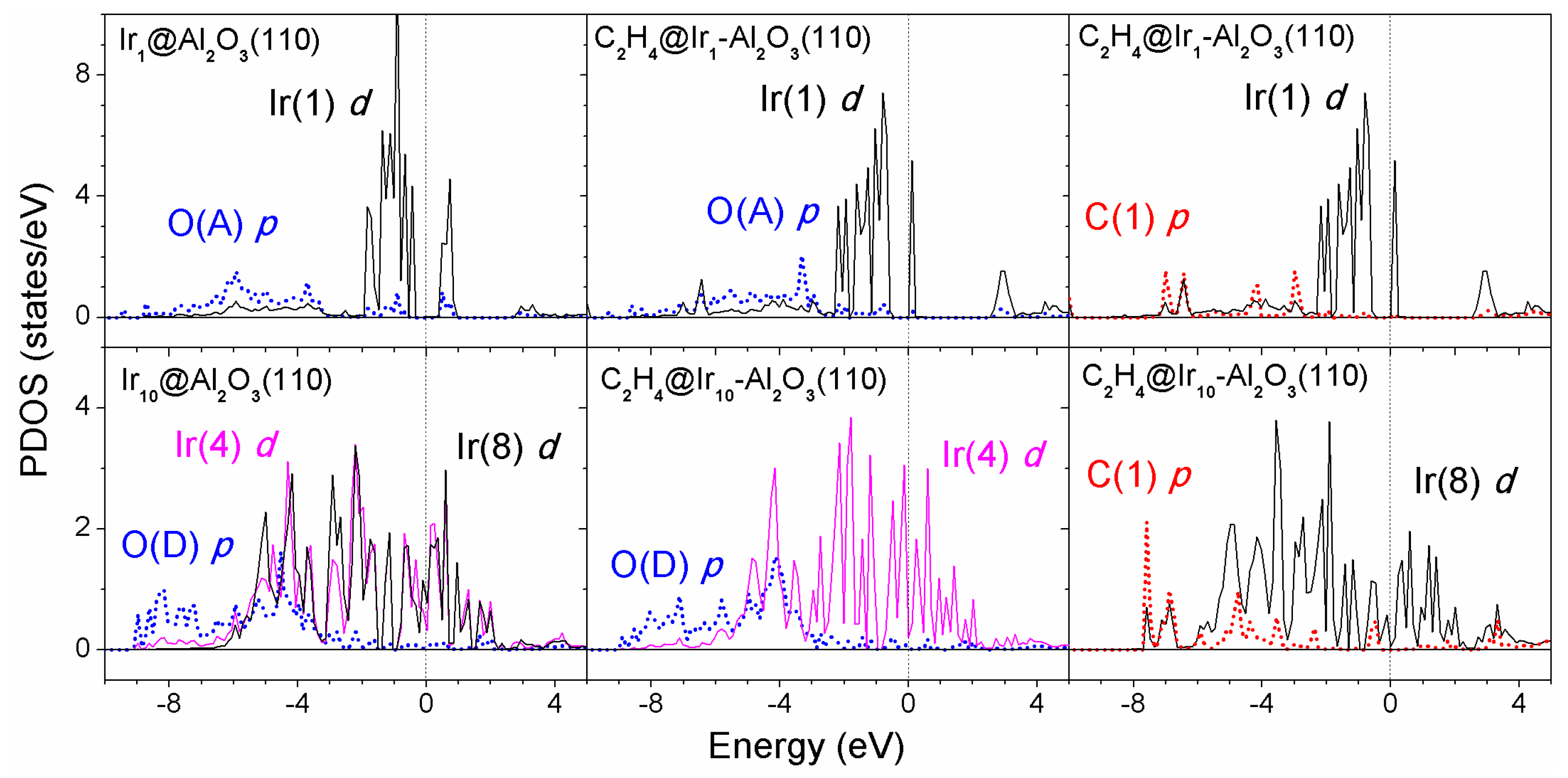
The PDOS are summarized in
Figure 8 for the Ir
1/
γ-Al
2O
3 and Ir
10/
γ-Al
2O
3 systems before and after ethylene adsorption. O(A)-Ir(1) and O(D)-Ir(4) were chosen to analyze the metal–support interaction for Ir
1/
γ-Al
2O
3 and Ir
10/
γ-Al
2O
3, respectively.
Before ethylene adsorption, the d band states of Ir monomer on the alumina support were quite localized and showed an energy gap of ~0.8 eV, while for the supported Ir10 cluster, broad and delocalized d states (either Ir(4) or Ir(8) atoms) showed up and exhibited a finite density of states at the Fermi level. After ethylene adsorption, the d band states became a little smoother for the supported Ir monomer but still localized at the metal cluster, while for the supported Ir10 cluster the d band states became sharper upon ethylene adsorption. We note that the Ir atom in contact with the support but away from the adsorbed ethylene—Ir(4) as labeled—yielded sharper d band states after ethylene adsorption compared with the case before ethylene adsorption. This means the adsorbed ethylene induced charge redistribution at the iridium–alumina interface. The p orbitals of oxygen (carbon) atoms strongly mixed with the low-energy (typically below −3 eV) d states of Ir atoms.
3.7. Effect of Adsorbed Ethylene on Nucleation of Irn Clusters on γ-Al2O3
We notice that the di-σ′ adsorption mode at the interface of Irn(n = 1–8)/γ-Al2O3 occurred at the same place. This raises the question of whether the pre-adsorbed ethylene at the interface would have affected the growth of Irn clusters on the support. To answer this question, we calculated the nucleation energy according to the following equations.
The nucleation or growth energy of Ir
n clusters from a combination of an adsorbed monomer and an Ir
n−1 was defined by
For pre-adsorbed ethylene at the interface (di-σ′ mode) of Ir
n/
γ-Al
2O
3, the nucleation energy E
nuc was obtained by
For the nucleation of the gas-phase Ir
n clusters, the nucleation energy E
nuc was calculated using
For pre-adsorbed ethylene on the gas-phase Ir
n clusters, the nucleation energy was defined by
Only the most favorable structure is considered for each cluster size.
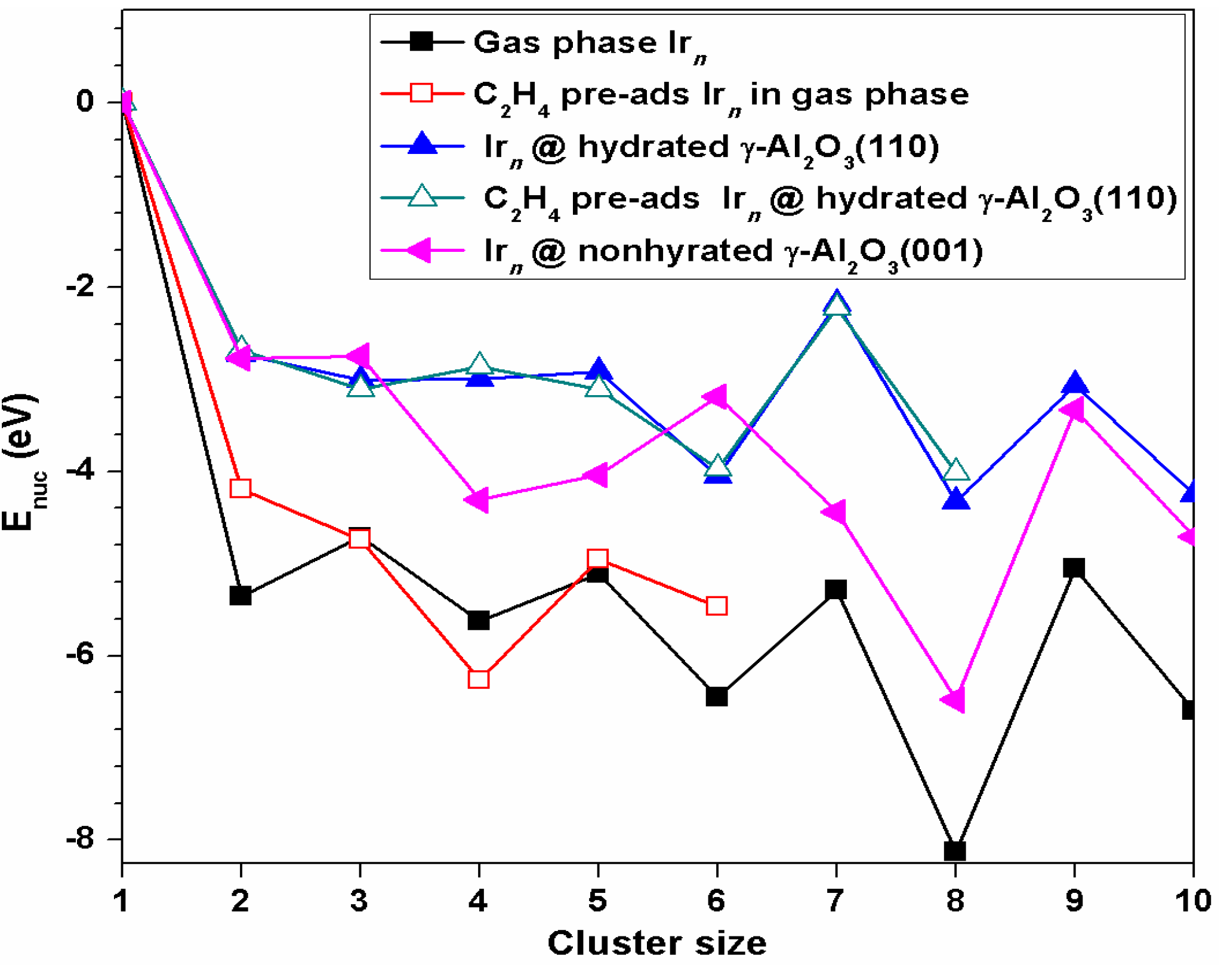
As shown in
Figure 9, the nucleation energies for all Ir cluster sizes we considered were negative, indicating the critical cluster size for Ir growth is 2. Each nucleation step was exothermic, indicating the nucleation process is thermodynamically favorable. In other words, Ir atoms prefer to grow into nanosize Ir
n clusters atom by atom both in the gas phase and on the
γ-Al
2O
3 surface, which is consistent with the experimental observation. Yentekakis et al. [
41] observed the Ir particle agglomeration on
γ-Al
2O
3 in the methane dry reformation reaction using transmission electron microscopy.
An E
nuc comparison between hydrated (110) and non-hydrated (001)
γ-Al
2O
3 surfaces revealed that except for Ir
3 and Ir
6 clusters, the nucleation of Ir
n clusters were less favorable on the hydrated
γ-Al
2O
3 (110) than the non-hydrated
γ-Al
2O
3 (001). For Ir
3 and Ir
6 clusters, the trend was reversed. The corresponding E
nuc for Ir
3 and Ir
6 on the hydrated (110) surface was 0.26 and 0.86 eV lower than those on the non-hydrated (001) surface, respectively. It should be pointed out that Ir
6 exhibited different adsorption configurations on two surfaces. Chen et al. [
13] found the most stable Ir
6 adsorption configuration was an octahedron structure on the non-hydrated
γ-Al
2O
3(001) surface, while on the hydrated
γ-Al
2O
3(110) we found the octahedron structure of Ir
6 was less stable by 1.76 eV (higher in energy) than the triangular prism (which was the most stable in the gas phase also).
A comparison of the nucleation energies of Irn on the hydrated γ-Al2O3(110) and pre-adsorbed ethylene at the interface (di-σ′ mode) of Irn/γ-Al2O3(110) suggests that the pre-adsorbed ethylene facilitated the nucleation from the even-sized supported Irn to the odd-sized Irn clusters, but hindered the nucleation from the odd-sized Irn to the even-sized Irn clusters. For ethylene pre-adsorbed Irn in the gas phase via the π mode, the pre-adsorbed ethylene hindered the nucleation of Irn (n = 2, 5, and 6), facilitated the nucleation of Ir4 from the Ir3 cluster, and had no effect on the nucleation of Ir3 from Ir2.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}