Reactivity of Atomically Functionalized C-Doped Boron Nitride Nanoribbons and Their Interaction with Organosulfur Compounds

 , , , and
, , , and
Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
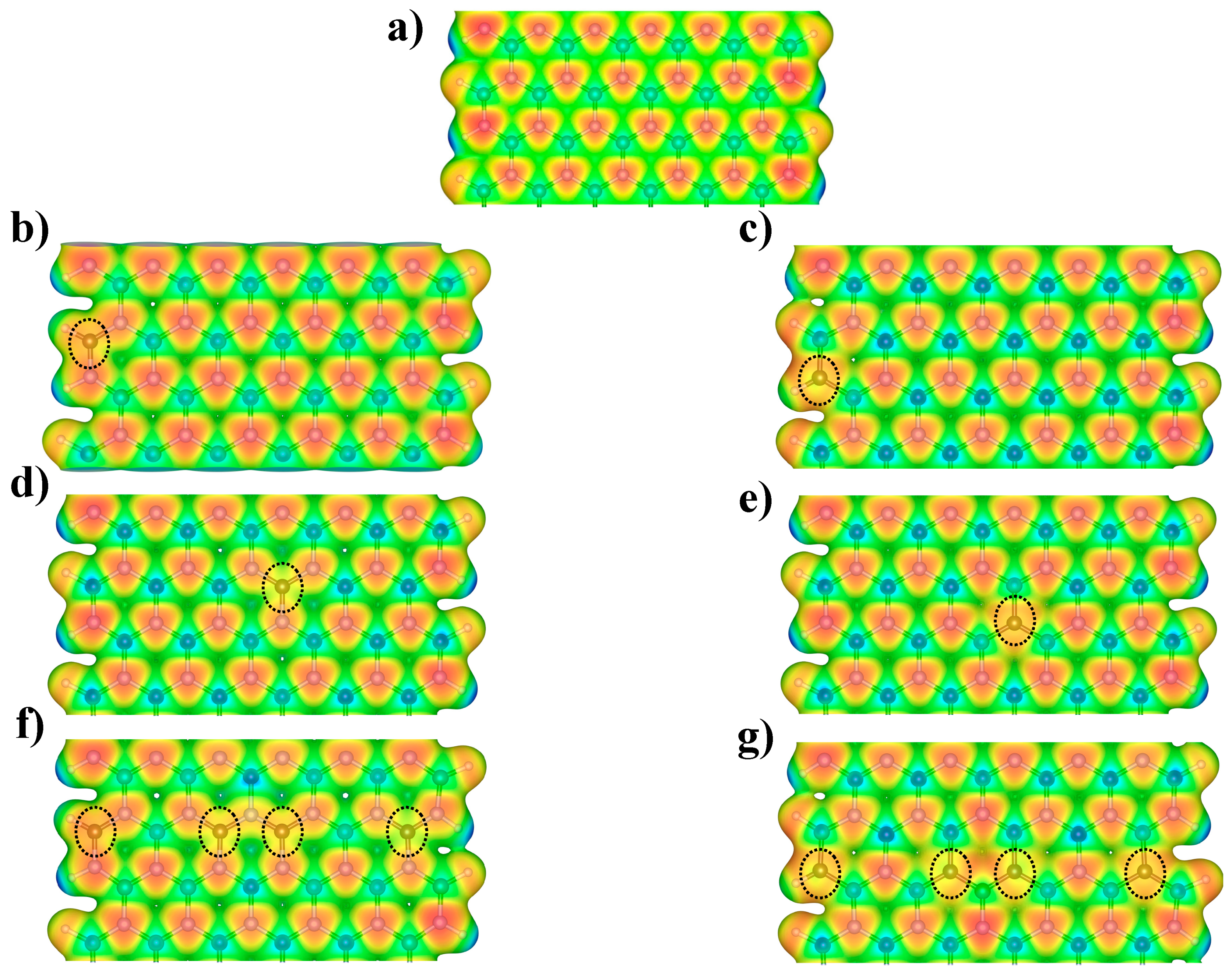
3.1. Energetic Stability and Electronic Properties of C-Doped BNNRs
3.2. Reactivity of C-Doped BNNRs
3.3. Interaction of Organosulfur Compounds on C-Doped BNNRs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Telg, H.; Maultzsch, J.; Reich, S.; Hennrich, F.; Thomsen, C. Chirality Distribution and Transition Energies of Carbon Nanotubes. Phys. Rev. Lett. 2004, 93, 177401. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Valencia, J.R.; Dienel, T.; Gröning, O.; Shorubalko, I.; Mueller, A.; Jansen, M.; Amsharov, K.; Ruffieux, P.; Fasel, R. Controlled synthesis of single-chirality carbon nanotubes. Nature 2014, 512, 61. [Google Scholar] [CrossRef] [PubMed]
- Wilder, J.W.G.; Venema, L.C.; Rinzler, A.G.; Smalley, R.E.; Dekker, C. Electronic structure of atomically resolved carbon nanotubes. Nature 1998, 391, 59. [Google Scholar] [CrossRef]
- Castro Neto, A.H.; Guinea, F.; Peres, N.M.R.; Novoselov, K.S.; Geim, A.K. The electronic properties of graphene. Rev. Mod. Phys. 2009, 81, 109–162. [Google Scholar] [CrossRef] [Green Version]
- Son, Y.-W.; Cohen, M.L.; Louie, S.G. Energy Gaps in Graphene Nanoribbons. Phys. Rev. Lett. 2006, 97, 216803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paine, R.T.; Narula, C.K. Synthetic routes to boron nitride. Chem. Rev. 1990, 90, 73–91. [Google Scholar] [CrossRef]
- Wentorf, R.H., Jr. Cubic Form of Boron Nitride. J. Chem. Phys. 1957, 26, 956. [Google Scholar] [CrossRef]
- Sōma, T.; Sawaoka, A.; Saito, S. Characterization of wurtzite type boron nitride synthesized by shock compression. Mater. Res. Bull. 1974, 9, 755–762. [Google Scholar] [CrossRef]
- Ooi, N.; Rajan, V.; Gottlieb, J.; Catherine, Y.; Adams, J.B. Structural properties of hexagonal boron nitride. Model. Simul. Mater. Sci. Eng. 2006, 14, 515. [Google Scholar] [CrossRef]
- Liu, L.; Feng, Y.P.; Shen, Z.X. Structural and electronic properties of h-BN. Phys. Rev. B 2003, 68, 104102. [Google Scholar] [CrossRef]
- Hod, O. Graphite and Hexagonal Boron-Nitride have the Same Interlayer Distance. Why? J. Chem. Theory Comput. 2012, 8, 1360–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Tang, T.-T.; Girit, C.; Hao, Z.; Martin, M.C.; Zettl, A.; Crommie, M.F.; Shen, Y.R.; Wang, F. Direct observation of a widely tunable bandgap in bilayer graphene. Nature 2009, 459, 820. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Taniguchi, T.; Kanda, H. Direct-bandgap properties and evidence for ultraviolet lasing of hexagonal boron nitride single crystal. Nat. Mater. 2004, 3, 404. [Google Scholar] [CrossRef] [PubMed]
- Chopra, N.G.; Luyken, R.J.; Cherrey, K.; Crespi, V.H.; Cohen, M.L.; Louie, S.G.; Zettl, A. Boron Nitride Nanotubes. Science 1995, 269, 966–967. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; Zhou, G.; Duan, W.; Wu, J.; Gu, B.-L. Tremendous Spin-Splitting Effects in Open Boron Nitride Nanotubes: Application to Nanoscale Spintronic Devices. J. Am. Chem. Soc. 2006, 128, 8453–8458. [Google Scholar] [CrossRef] [PubMed]
- Dhungana, K.; Pati, R. Boron Nitride Nanotubes for Spintronics. Sensors 2014, 14, 17655. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Peralta, J.E. Magnetic Boron Nitride Nanoribbons with Tunable Electronic Properties. Nano Lett. 2008, 8, 2210–2214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, H.; Zhi, C.; Zhang, Z.; Wei, X.; Wang, X.; Guo, W.; Bando, Y.; Golberg, D. “White Graphenes”: Boron Nitride Nanoribbons via Boron Nitride Nanotube Unwrapping. Nano Lett. 2010, 10, 5049–5055. [Google Scholar] [CrossRef]
- Lin, Z.; McNamara, A.; Liu, Y.; Moon, K.-s.; Wong, C.-P. Exfoliated hexagonal boron nitride-based polymer nanocomposite with enhanced thermal conductivity for electronic encapsulation. Compos. Sci. Technol. 2014, 90, 123–128. [Google Scholar] [CrossRef]
- Sakurai, H.; Tobias, H.J.; Park, K.; Zarling, D.; Docherty, K.S.; Kittelson, D.B.; McMurry, P.H.; Ziemann, P.J. On-line measurements of diesel nanoparticle composition and volatility. Atmos. Environ. 2003, 37, 1199–1210. [Google Scholar] [CrossRef]
- Song, C. An overview of new approaches to deep desulfurization for ultra-clean gasoline, diesel fuel and jet fuel. Catal. Today 2003, 86, 211–263. [Google Scholar] [CrossRef]
- Valencia, D.; García-Cruz, I.; Uc, V.H.; Ramírez-Verduzco, L.F.; Aburto, J. Refractory Character of 4,6-Dialkyldibenzothiophenes: Structural and Electronic Instabilities Reign Deep Hydrodesulfurization. Chem. Select 2018, 3, 8849–8856. [Google Scholar] [CrossRef]
- Srivastav, A.; Srivastava, V.C. Adsorptive desulfurization by activated alumina. J. Hazard. Mater. 2009, 170, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Xu, W.; Zhang, X.; Yan, Y. A surface-imprinted polymer for removing dibenzothiophene from gasoline. Microchim. Acta 2010, 171, 441–449. [Google Scholar] [CrossRef]
- Babich, I.V.; Moulijn, J.A. Science and technology of novel processes for deep desulfurization of oil refinery streams: A review. Fuel 2003, 82, 607–631. [Google Scholar] [CrossRef]
- Xiao, J.; Bian, G.; Zhang, W.; Li, Z. Adsorption of Dibenzothiophene on Ag/Cu/Fe-Supported Activated Carbons Prepared by Ultrasonic-Assisted Impregnation. J. Chem. Eng. Data 2010, 55, 5818–5823. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Du, A.J.; Smith, S.C.; Lu, G.Q. First-principle studies of electronic structure and C-doping effect in boron nitride nanoribbon. Chem. Phys. Lett. 2007, 447, 181–186. [Google Scholar] [CrossRef]
- Ivanov, A.S.; Morris, A.J.; Bozhenko, K.V.; Pickard, C.J.; Boldyrev, A.I. Inorganic Double-Helix Structures of Unusually Simple Lithium–Phosphorus Species. Angew. Chem. Int. Ed. 2012, 51, 8330–8333. [Google Scholar] [CrossRef] [PubMed]
- Jana, G.; Pan, S.; Rodríguez-Kessler, P.L.; Merino, G.; Chattaraj, P.K. Adsorption of Molecular Hydrogen on Lithium–Phosphorus Double-Helices. J. Phys. Chem. C 2018, 122, 27941–27946. [Google Scholar] [CrossRef]
- Kan, E.-J.; Li, Z.; Yang, J.; Hou, J.G. Half-Metallicity in Edge-Modified Zigzag Graphene Nanoribbons. J. Am. Chem. Soc. 2008, 130, 4224–4225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayers, P.W.; Yang, W.; Bartolotti, L.J. Chemical Reactivity Theory: A Density Functional View; Chattaraj, P.K., Ed.; CRC Press: Boca Raton, FL, USA, 2009; p. 610. [Google Scholar]
- Yang, W.; Mortier, W.J. The use of global and local molecular parameters for the analysis of the gas-phase basicity of amines. J. Am. Chem. Soc. 1986, 108, 5708–5711. [Google Scholar] [CrossRef] [PubMed]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Park, C.-H.; Louie, S.G. Energy Gaps and Stark Effect in Boron Nitride Nanoribbons. Nano Lett. 2008, 8, 2200–2203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galvan, M.; Vela, A.; Gazquez, J.L. Chemical reactivity in spin-polarized density functional theory. J. Phys. Chem. 1988, 92, 6470–6474. [Google Scholar] [CrossRef]
- Pérez, P.; Andrés, J.; Safont, V.S.; Tapia, O.; Contreras, R. Spin-Philicity and Spin-Donicity as Auxiliary Concepts To Quantify Spin-Catalysis Phenomena. J. Phys. Chem. A 2002, 106, 5353–5357. [Google Scholar] [CrossRef]
- Navarro-Santos, P.; Ricardo-Chávez, J.L.; Reyes-Reyes, M.; Rivera, J.L.; López-Sandoval, R. Tuning the electronic properties of armchair carbon nanoribbons by a selective boron doping. J. Phys. Condens. Matter 2010, 22, 505302. [Google Scholar] [CrossRef]
- Dumitrică, T.; Hua, M.; Yakobson, B.I. Endohedral silicon nanotubes as thinnest silicide wires. Phys. Rev. B 2004, 70, 241303. [Google Scholar] [CrossRef]
- Barone, V.; Hod, O.; Scuseria, G.E. Electronic Structure and Stability of Semiconducting Graphene Nanoribbons. Nano Lett. 2006, 6, 2748–2754. [Google Scholar] [CrossRef]
- Hod, O.; Barone, V.; Peralta, J.E.; Scuseria, G.E. Enhanced Half-Metallicity in Edge-Oxidized Zigzag Graphene Nanoribbons. Nano Lett. 2007, 7, 2295–2299. [Google Scholar] [CrossRef] [Green Version]
- Morell, C.; Grand, A.; Toro-Labbé, A. New Dual Descriptor for Chemical Reactivity. J. Phys. Chem. A 2005, 109, 205–212. [Google Scholar] [CrossRef]
- Saha, S.; Dinadayalane, T.C.; Leszczynska, D.; Leszczynski, J. DFT-based reactivity study of (5,5) armchair boron nitride nanotube (BNNT). Chem. Phys. Lett. 2013, 565, 69–73. [Google Scholar] [CrossRef]
- Martínez, J.I.; Moncada, J.L.; Larenas, J.M. The dual descriptor to measure local reactivity on Buckminster fullerenes: An analysis within the framework of conceptual DFT. J. Mol. Model. 2010, 16, 1825–1832. [Google Scholar] [CrossRef]
- Cerón, M.L.; Calatayud, M. Application of dual descriptor to understand the activity of Cu/ZrO2 catalysts in the water gas shift reaction. J. Mol. Model. 2017, 23, 34. [Google Scholar] [CrossRef]
- Cárdenas, C.; Rabi, N.; Ayers, P.W.; Morell, C.; Jaramillo, P.; Fuentealba, P. Chemical Reactivity Descriptors for Ambiphilic Reagents: Dual Descriptor, Local Hypersoftness, and Electrostatic Potential. J. Phys. Chem. A 2009, 113, 8660–8667. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, R. Nanocarbon materials for oxidative-adsorptive desulfurization using air oxygen under mild conditions. Diam. Relat. Mater. 2017, 73, 161–168. [Google Scholar] [CrossRef]
- Rivera, J.L.; Navarro-Santos, P.; Hernandez-Gonzalez, L.; Guerra-Gonzalez, R. Reactivity of Alkyldibenzothiophenes Using Theoretical Descriptors. J. Chem. 2014, 2014, 8. [Google Scholar] [CrossRef]
- López-Albarrán, P.; Navarro-Santos, P.; Garcia-Ramirez, M.A.; Ricardo-Chávez, J.L. Dibenzothiophene adsorption at boron doped carbon nanoribbons studied within density functional theory. J. Appl. Phys. 2015, 117, 234301. [Google Scholar] [CrossRef]
- Rivera, J.L.; Navarro-Santos, P.; Guerra-Gonzalez, R.; Lima, E. Interaction of Refractory Dibenzothiophenes and Polymerizable Structures. Int. J. Polym. Sci. 2014, 2014, 11. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Gómez, B.; Martínez-Magadán, J.M. A Theoretical Study of Dibenzothiophene Absorbed on Open-Ended Carbon Nanotubes. J. Phys. Chem. B 2005, 109, 14868–14875. [Google Scholar] [CrossRef]
- Goering, J.; Burghaus, U. Adsorption kinetics of thiophene on single-walled carbon nanotubes (CNTs). Chem. Phys. Lett. 2007, 447, 121–126. [Google Scholar] [CrossRef]
- Denis, P.A.; Iribarne, F. Thiophene adsorption on Single Wall Carbon Nanotubes and graphene. J. Mol. Struct. Theochem. 2010, 957, 114–119. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M × N | Pristine | BNNR_BE | BNNR_NE | BNNR_BC | BNNR_NC |
|---|---|---|---|---|---|
| 12 × 2 | 6.32 (−5.55) | 6.29 (−5.41) | 6.30 (−5.39) | 6.27 (−5.39) | 6.29 (−5.38) |
| 16 × 2 | 6.48 (−5.68) | 6.45 (−5.57) | 6.46 (−5.55) | 6.44 (−5.56) | 6.45 (−5.55) |
| 24 × 2 | 6.65 (−5.82) | 6.63 (−5.74) | 6.63 (−5.73) | 6.62 (−5.73) | 6.63 (−5.73) |
| 30 × 2 | 6.72 (−5.88) | 6.71 (−5.82) | 6.71 (−5.81) | 6.70 (−5.81) | 6.71 (−5.80) |
| 16 × 4 | 6.53 (−5.74) | 6.46 (−5.62) | 6.47 (−5.61) | 6.45 (−5.61) | 6.46 (−5.61) |
| Complex | Thio | B-Thio | DBT | 4-MDBT | 4,6-DMDBT | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| <<1>> | <<2>> | <<1>> | <<2>> | <<1>> | <<2>> | <<1>> | <<2>> | <<1>> | <<2>> | ||
| BNNR_BE | −16.6 | −17.1 | −11.8 | −9.9 | −13.1 | −14.1 | −15.6 | −16.2 | −18.4 | −18.5 | |
| BNNR_BC | −15.1 | −16.3 | −13.3 | −10.2 | −18.6 | −15.1 | −16.4 | −17.0 | −18.2 | −19.0 | |
| BNNR_NE | −12.6 | −12.6 | −6.9 | −5.5 | −18.0 | −8.8 | −11.0 | −10.3 | −13.6 | −12.7 | |
| BNNR_NC | −12.1 | −13.8 | −12.3 | −6.2 | −23.2 | −11.9 | −15.2 | −13.3 | −14.9 | −15.6 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villanueva-Mejia, F.; Navarro-Santos, P.; Rodríguez-Kessler, P.L.; Herrera-Bucio, R.; Rivera, J.L. Reactivity of Atomically Functionalized C-Doped Boron Nitride Nanoribbons and Their Interaction with Organosulfur Compounds. Nanomaterials 2019, 9, 452. https://doi.org/10.3390/nano9030452
Villanueva-Mejia F, Navarro-Santos P, Rodríguez-Kessler PL, Herrera-Bucio R, Rivera JL. Reactivity of Atomically Functionalized C-Doped Boron Nitride Nanoribbons and Their Interaction with Organosulfur Compounds. Nanomaterials. 2019; 9(3):452. https://doi.org/10.3390/nano9030452
Chicago/Turabian StyleVillanueva-Mejia, Francisco, Pedro Navarro-Santos, Peter Ludwig Rodríguez-Kessler, Rafael Herrera-Bucio, and José Luis Rivera. 2019. "Reactivity of Atomically Functionalized C-Doped Boron Nitride Nanoribbons and Their Interaction with Organosulfur Compounds" Nanomaterials 9, no. 3: 452. https://doi.org/10.3390/nano9030452