Designing a Novel Monolayer β-CSe for High Performance Photovoltaic Device: An Isoelectronic Counterpart of Blue Phosphorene

Abstract
:1. Introduction
2. Methods
3. Results and Discussions
3.1. Geometric and Electronic Structures
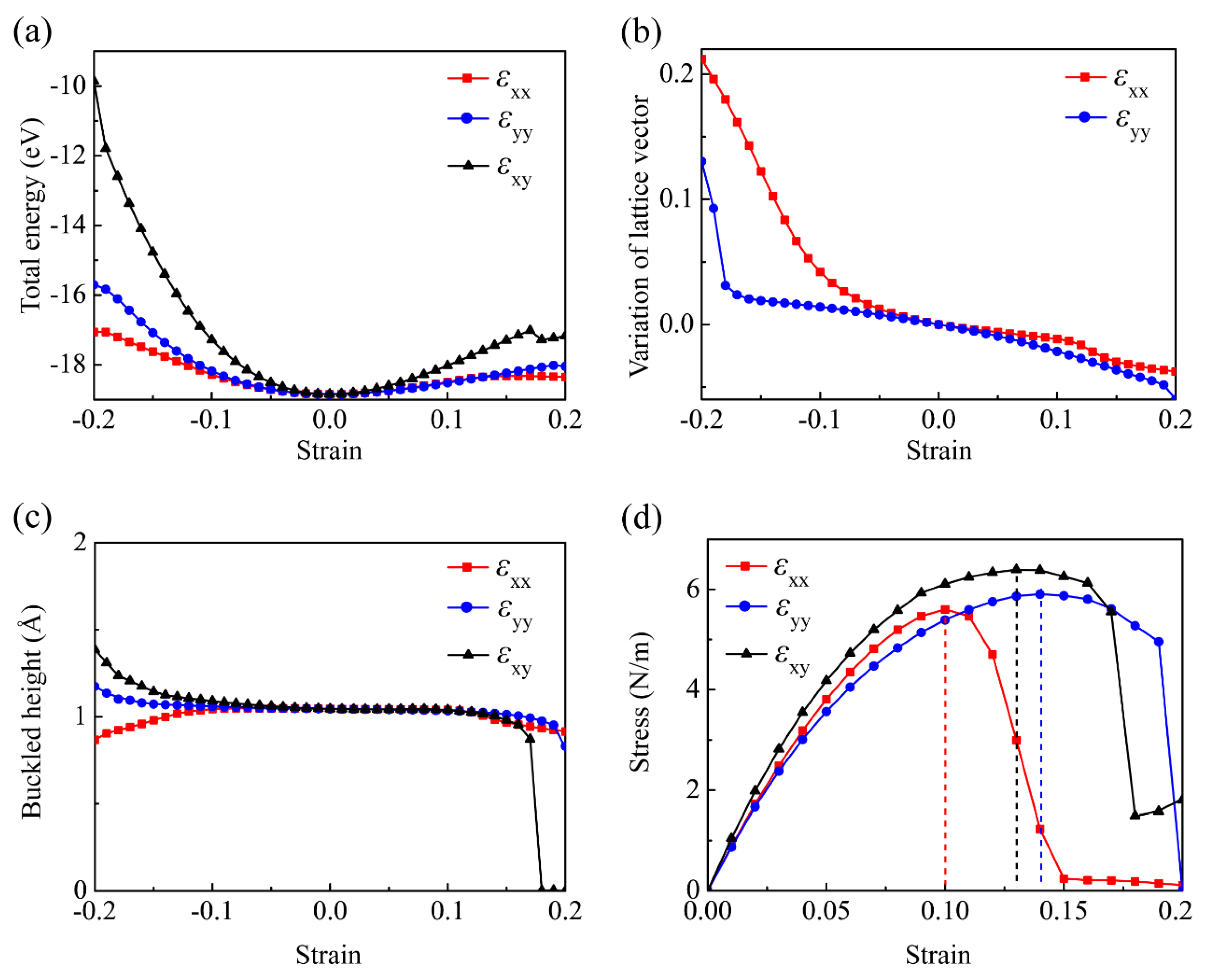
3.2. Mechanical Properties
3.3. Strain Dependence of the Electronic Structures
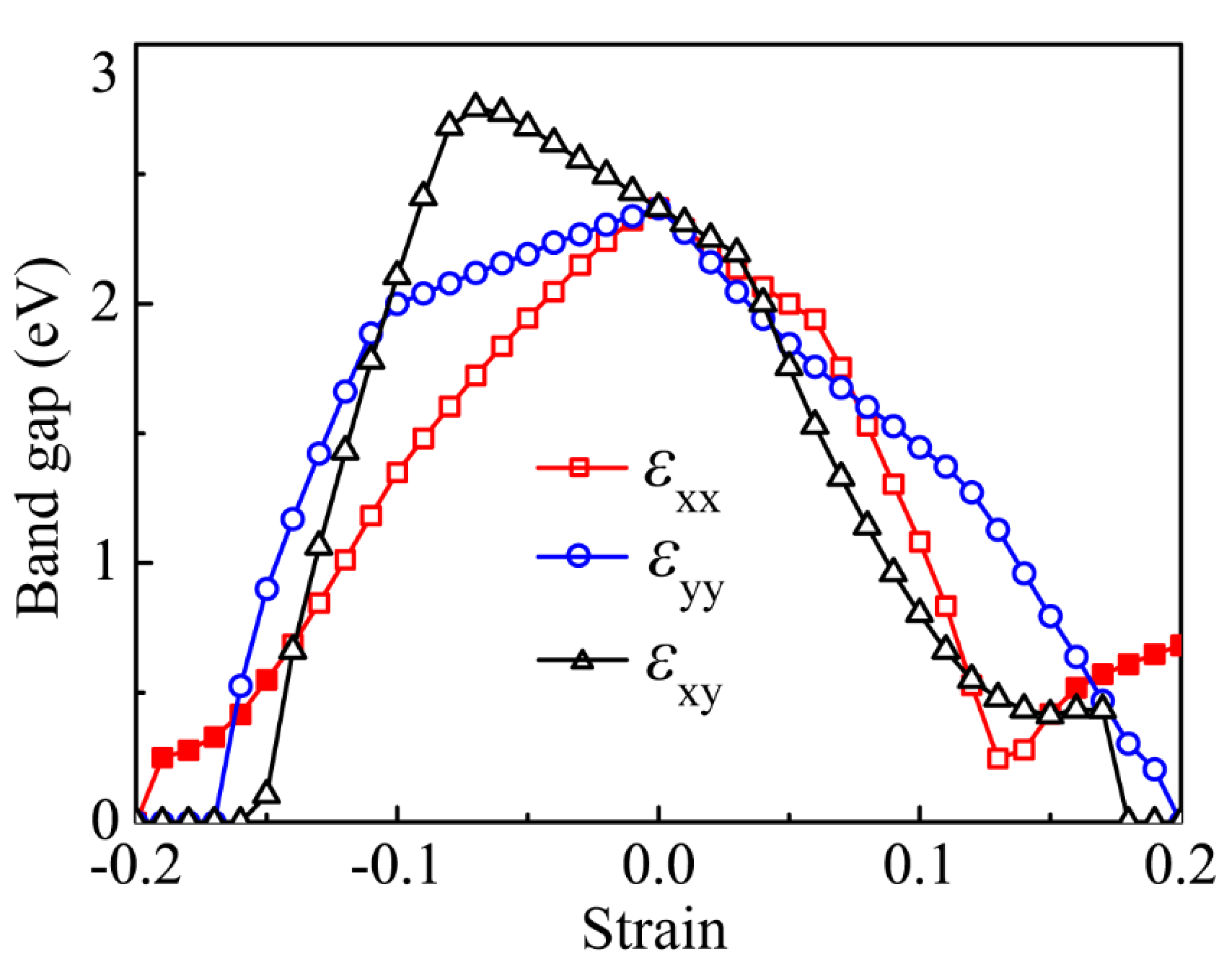
3.4. Strain Dependence of the Bandgap
3.5. The Dependence of Carrier Effective Masse on Strain
3.6. The Type-II vdW p-n α-CSe/β-CSe Hetrostructure as a Metal-Free Photocatalyst
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chhowalla, M.; Shin, H.S.; Eda, G.; Li, L.-J.; Loh, K.P.; Zhang, H. The chemistry of two-dimensional layered transition metal dichalcogenide nanosheets. Nat. Chem. 2013, 5, 263–275. [Google Scholar] [CrossRef]
- Li, M.-Y.; Shi, Y.; Cheng, C.-C.; Lu, L.-S.; Lin, Y.-C.; Tang, H.-L.; Tsai, M.-L.; Chu, C.-W.; Wei, K.-H.; He, J.-H.; et al. Epitaxial growth of a monolayer WSe2-MoS2 lateral p-n junction with an atomically sharp interface. Science 2015, 349, 524–528. [Google Scholar] [CrossRef]
- Kim, K.S.; Zhao, Y.; Jang, H.; Lee, S.Y.; Kim, J.M.; Kim, K.S.; Ahn, J.-H.; Kim, P.; Choi, J.-Y.; Hong, B.H. Large-scale pattern growth of graphene films for stretchable transparent electrodes. Nature 2009, 457, 706–710. [Google Scholar] [CrossRef]
- Wang, Q.H.; Kalantar-Zadeh, K.; Kis, A.; Coleman, J.N.; Strano, M.S. Electronics and optoelectronics of two-dimensional transition metal dichalcogenides. Nat. Nanotechnol. 2012, 7, 699–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Murali, S.; Stoller, M.D.; Ganesh, K.J.; Cai, W.; Ferreira, P.J.; Pirkle, A.; Wallace, R.M.; Cychosz, K.A.; Thommes, M.; et al. Carbon-Based Supercapacitors Produced by Activation of Graphene. Science 2011, 332, 1537. [Google Scholar] [CrossRef]
- Liu, C.; Yu, Z.; Neff, D.; Zhamu, A.; Jang, B.Z. Graphene-Based Supercapacitor with an Ultrahigh Energy Density. Nano Lett. 2010, 10, 4863–4868. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, Y.; Huang, Y.; Zhang, F.; Yang, X.; Ma, Y.; Chen, Y. Preventing Graphene Sheets from Restacking for High-Capacitance Performance. J. Phys. Chem. C 2011, 115, 23192–23197. [Google Scholar] [CrossRef]
- Ji, L.; Rao, M.; Zheng, H.; Zhang, L.; Li, Y.; Duan, W.; Guo, J.; Cairns, E.J.; Zhang, Y. Graphene Oxide as a Sulfur Immobilizer in High Performance Lithium/Sulfur Cells. J. Am. Chem. Soc. 2011, 133, 18522–18525. [Google Scholar] [CrossRef]
- Hu, L.-H.; Wu, F.-Y.; Lin, C.-T.; Khlobystov, A.N.; Li, L.-J. Graphene-modified LiFePO4 cathode for lithium ion battery beyond theoretical capacity. Nat. Commun. 2013, 4, 1687. [Google Scholar] [CrossRef]
- Xiao, J.; Mei, D.; Li, X.; Xu, W.; Wang, D.; Graff, G.L.; Bennett, W.D.; Nie, Z.; Saraf, L.V.; Aksay, I.A.; et al. Hierarchically Porous Graphene as a Lithium–Air Battery Electrode. Nano Lett. 2011, 11, 5071–5078. [Google Scholar] [CrossRef]
- Huang, L.; Li, Y.; Wei, Z.; Li, J. Strain induced piezoelectric effect in black phosphorus and MoS2 van der Waals heterostructure. Sci. Rep. 2015, 5, 16448. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, Y.; Liu, N.; Han, Y.; Zhang, X.; Huang, H.; Lifshitz, Y.; Lee, S.-T.; Zhong, J.; Kang, Z. Metal-free efficient photocatalyst for stable visible water splitting via a two-electron pathway. Science 2015, 347, 970–974. [Google Scholar] [CrossRef]
- Shalom, M.; Gimenez, S.; Schipper, F.; Herraiz-Cardona, I.; Bisquert, J.; Antonietti, M. Controlled Carbon Nitride Growth on Surfaces for Hydrogen Evolution Electrodes. Angew. Chem. 2014, 126, 3728–3732. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Sun, H.; Peng, W. 2D Transition Metal Dichalcogenides and Graphene-Based Ternary Composites for Photocatalytic Hydrogen Evolution and Pollutants Degradation. Nanomaterials 2017, 7, 62. [Google Scholar] [CrossRef]
- Li, L.; Yu, Y.; Ye, G.J.; Ge, Q.; Ou, X.; Wu, H.; Feng, D.; Chen, X.H.; Zhang, Y. Black phosphorus field-effect transistors. Nat. Nanotechnol. 2014, 9, 372–377. [Google Scholar] [CrossRef] [Green Version]
- Nazir, G.; Khan, F.M.; Aftab, S.; Afzal, M.A.; Dastgeer, G.; Rehman, A.M.; Seo, Y.; Eom, J. Gate Tunable Transport in Graphene/MoS2/(Cr/Au) Vertical Field-Effect Transistors. Nanomaterials 2018, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Kou, L.; Frauenheim, T.; Chen, C. Phosphorene as a Superior Gas Sensor: Selective Adsorption and Distinct I-V Response. J. Phys. Chem. Lett. 2014, 5, 2675–2681. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Wei, X.; Kysar, J.W.; Hone, J. Measurement of the Elastic Properties and Intrinsic Strength of Monolayer Graphene. Science 2008, 321, 385–388. [Google Scholar] [CrossRef]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef]
- Castro Neto, A.H.; Guinea, F.; Peres, N.M.R.; Novoselov, K.S.; Geim, A.K. The electronic properties of graphene. Rev. Mod. Phys. 2009, 81, 109–162. [Google Scholar] [CrossRef] [Green Version]
- Mak, K.F.; Lee, C.; Hone, J.; Shan, J.; Heinz, T.F. Atomically Thin MoS2: A New Direct-Gap Semiconductor. Phys. Rev. Lett. 2010, 105, 136805. [Google Scholar] [CrossRef]
- Radisavljevic, B.; Radenovic, A.; Brivio, J.; Giacometti, V.; Kis, A. Single-layer MoS2 transistors. Nat. Nanotechnol. 2011, 6, 147–150. [Google Scholar] [CrossRef]
- Lu, J.; Carvalho, A.; Chan, X.K.; Liu, H.; Liu, B.; Tok, E.S.; Loh, K.P.; Castro Neto, A.H.; Sow, C.H. Atomic Healing of Defects in Transition Metal Dichalcogenides. Nano Lett. 2015, 15, 3524–3532. [Google Scholar] [CrossRef]
- Sa, B.; Li, Y.-L.; Qi, J.; Ahuja, R.; Sun, Z. Strain Engineering for Phosphorene: The Potential Application as a Photocatalyst. J. Phys. Chem. C 2014, 118, 26560–26568. [Google Scholar] [CrossRef] [Green Version]
- Qiao, J.; Kong, X.; Hu, Z.-X.; Yang, F.; Ji, W. High-mobility transport anisotropy and linear dichroism in few-layer black phosphorus. Nat. Commun. 2014, 5, 4475. [Google Scholar] [CrossRef]
- Yang, W.; Chen, G.; Shi, Z.; Liu, C.-C.; Zhang, L.; Xie, G.; Cheng, M.; Wang, D.; Yang, R.; Shi, D.; et al. Epitaxial growth of single-domain graphene on hexagonal boron nitride. Nat. Mater. 2013, 12, 792–797. [Google Scholar] [CrossRef]
- Cai, Y.; Zhang, G.; Zhang, Y.-W. Electronic Properties of Phosphorene/Graphene and Phosphorene/Hexagonal Boron Nitride Heterostructures. J. Phys. Chem. C 2015, 119, 13929–13936. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Wu, F.; Li, J. Structural anisotropy results in strain-tunable electronic and optical properties in monolayer GeX and SnX (X = S, Se, Te). J. Chem. Phys. 2016, 144, 114708. [Google Scholar] [CrossRef]
- Kamal, C.; Chakrabarti, A.; Ezawa, M. Direct band gaps in group IV-VI monolayer materials: Binary counterparts of phosphorene. Phys. Rev. B 2016, 93, 125428. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, X.; Liu, W.-C.; Wang, Y. Stacking effect on electronic, photocatalytic and optical properties: A comparison between bilayer and monolayer SnS. Comput. Mater. Sci. 2019, 158, 272–281. [Google Scholar] [CrossRef]
- Zhang, Q.; Xin, T.; Lu, X.; Wang, Y. Optoelectronic Properties of X-Doped (X = O, S, Te) Photovoltaic CSe with Puckered Structure. Materials 2018, 11, 431. [Google Scholar] [CrossRef]
- Brent, J.R.; Lewis, D.J.; Lorenz, T.; Lewis, E.A.; Savjani, N.; Haigh, S.J.; Seifert, G.; Derby, B.; O’Brien, P. Tin(II) Sulfide (SnS) Nanosheets by Liquid-Phase Exfoliation of Herzenbergite: IV-VI Main Group Two-Dimensional Atomic Crystals. J. Am. Chem. Soc. 2015, 137, 12689–12696. [Google Scholar] [CrossRef]
- Jiang, J.; Wong, C.P.Y.; Zou, J.; Li, S.; Wang, Q.; Chen, J.; Qi, D.; Wang, H.; Eda, G.; Chua, D.H.C.; et al. Two-step fabrication of single-layer rectangular SnSe flakes. 2D Mater. 2017, 4, 021026. [Google Scholar] [CrossRef]
- Sirikumara, H.I.; Jayasekera, T. Tunable indirect-direct transition of few-layer SnSe via interface engineering. J. Phys. Condens. Matter. 2017, 29, 425501. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Tomanek, D. Semiconducting layered blue phosphorus: A computational study. Phys. Rev. Lett. 2014, 112, 176802. [Google Scholar] [CrossRef]
- Zeng, J.; Cui, P.; Zhang, Z. Half Layer By Half Layer Growth of a Blue Phosphorene Monolayer on a GaN(001) Substrate. Phys. Rev. Lett. 2017, 118, 046101. [Google Scholar] [CrossRef]
- Zhang, J.L.; Zhao, S.; Han, C.; Wang, Z.; Zhong, S.; Sun, S.; Guo, R.; Zhou, X.; Gu, C.D.; Yuan, K.D.; et al. Epitaxial Growth of Single Layer Blue Phosphorus: A New Phase of Two-Dimensional Phosphorus. Nano Lett. 2016, 16, 4903–4908. [Google Scholar] [CrossRef]
- Zhu, Z.; Guan, J.; Liu, D.; Tománek, D. Designing Isoelectronic Counterparts to Layered Group V Semiconductors. ACS Nano 2015, 9, 8284–8290. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.Y.; Li, K.Y.; Lu, Y.; Huang, Y.; Shao, X.H. High thermoelectric performances of monolayer SnSe allotropes. Nanoscale 2017, 9, 16093–16100. [Google Scholar] [CrossRef]
- Huang, L.; Li, J. Tunable electronic structure of black phosphorus/blue phosphorus van der Waals p-n heterostructure. Appl. Phys. Lett. 2016, 108, 083101. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Hafner, J. Ab-initio simulations of materials using VASP: Density-functional theory and beyond. J. Comput. Chem. 2008, 29, 2044–2078. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple (vol 77, pg 3865, 1996). Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef]
- Mi, K.; Xie, J.; Si, M.S.; Gao, C.X. Layer-stacking effect on electronic structures of bilayer arsenene. EPL 2017, 117, 27002. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef]
- Togo, A.; Oba, F.; Tanaka, I. First-principles calculations of the ferroelastic transition between rutile-type andCaCl2-typeSiO2 at high pressures. Phys. Rev. B 2008, 78, 134106. [Google Scholar] [CrossRef]
- Shi, L.-B.; Zhang, Y.-Y.; Xiu, X.-M.; Dong, H.-K. Structural characteristics and strain behavior of two-dimensional C3N: First principles calculations. Carbon 2018, 134, 103–111. [Google Scholar] [CrossRef]
- Wang, H.; Li, Q.; Gao, Y.; Miao, F.; Zhou, X.-F.; Wan, X.G. Strain effects on borophene: Ideal strength, negative Possion’s ratio and phonon instability. New J. Phys. 2016, 18, 073016. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, L.; Yao, G.; Wang, H. Unique mechanical responses of layered phosphorus-like group-IV monochalcogenides. J. Appl. Phys. 2019, 125, 082519. [Google Scholar] [CrossRef]
- Wang, H.; Liu, E.; Wang, Y.; Wan, B.; Ho, C.-H.; Miao, F.; Wan, X.G. Cleavage tendency of anisotropic two- dimensional materials: ReX2 (X = S, Se) and WTe2. Phys. Rev. B 2017, 96, 165418. [Google Scholar] [CrossRef]
- Jiang, J.W.; Park, H.S. Negative poisson’s ratio in single-layer black phosphorus. Nat. Commun. 2014, 5, 4727. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Ming, P.; Li, J. Ab initio calculation of ideal strength and phonon instability of graphene under tension. Phys. Rev. B 2007, 76, 064120. [Google Scholar] [CrossRef]
- Yang, C.; Yu, Z.; Lu, P.; Liu, Y.; Ye, H.; Gao, T. Phonon instability and ideal strength of silicene under tension. Comput. Mater. Sci. 2014, 95, 420–428. [Google Scholar] [CrossRef]
- Huang, M.; Yan, H.; Heinz, T.F.; Hone, J. Probing Strain-Induced Electronic Structure Change in Graphene by Raman Spectroscopy. Nano Lett. 2010, 10, 4074–4079. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Wei, Q.; Copple, A. Strain-engineered direct-indirect band gap transition and its mechanism in two-dimensional phosphorene. Phys. Rev. B 2014, 90, 085402. [Google Scholar] [CrossRef]
- Ghosh, B.; Nahas, S.; Bhowmick, S.; Agarwal, A. Electric field induced gap modification in ultrathin blue phosphorus. Phys. Rev. B 2015, 91, 115433. [Google Scholar] [CrossRef]
- Wang, J.; Li, X.; You, Y.; Yang, X.; Wang, Y.; Li, Q. Interfacial coupling induced direct Z-scheme water splitting in metal-free photocatalyst: C3N/g-C3N4 heterojunctions. Nanotechnology 2018, 29, 365401. [Google Scholar] [CrossRef]
- Martín-Sánchez, J.; Trotta, R.; Piredda, G.; Schimpf, C.; Trevisi, G.; Seravalli, L.; Frigeri, P.; Stroj, S.; Lettner, T.; Reindl, M.; et al. Reversible Control of In-Plane Elastic Stress Tensor in Nanomembranes. Adv. Opt. Mater. 2016, 4, 682–687. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Space Group | Cohesive Energy (eV/atom) | Lattice Constants (Å) | Bond Length (Å) | Bond Angle (deg) | |
|---|---|---|---|---|---|---|
| a | b | |||||
| β-CSe | P3m1 | −3.79 | 3.065 | 5.22 | 2.055 | θ = 96.45 |
| β-P | P3m1 | −5.23 | 3.28/ | 2.261 | θ = 92.907 | |
| Material | PBE Gap (type) eV | HSE Gap (type) eV | m*h/me Zigzag Direction | m*h/me Armchair Direction | m*e/me Zigzag Direction | m*e/me Armchair Direction |
|---|---|---|---|---|---|---|
| β-CSe | 1.54 (In) | 2.37 (In) | 0.718 | 0.795 | 0.23 | 1.027 |
| β-P | 1.94 (In) | 2.7 (In) | 0.588 | 0.486 | 0.353 | 0.794 |
| Direction | f (N/m) | Young’s Modulus (N/m) | Poisson’s Ratio | |
|---|---|---|---|---|
| Armchair | 5.6 | 0.10 | 86.14 | 0.14 |
| Zigzag | 5.90 | 0.14 | 83.47 | 0.16 |
| Biaxial | 6.4 | 0.13 | 99.15 | 0.09 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Q.; Feng, Y.; Chen, X.; Zhang, W.; Wu, L.; Wang, Y. Designing a Novel Monolayer β-CSe for High Performance Photovoltaic Device: An Isoelectronic Counterpart of Blue Phosphorene. Nanomaterials 2019, 9, 598. https://doi.org/10.3390/nano9040598
Zhang Q, Feng Y, Chen X, Zhang W, Wu L, Wang Y. Designing a Novel Monolayer β-CSe for High Performance Photovoltaic Device: An Isoelectronic Counterpart of Blue Phosphorene. Nanomaterials. 2019; 9(4):598. https://doi.org/10.3390/nano9040598
Chicago/Turabian StyleZhang, Qiang, Yajuan Feng, Xuanyu Chen, Weiwei Zhang, Lu Wu, and Yuexia Wang. 2019. "Designing a Novel Monolayer β-CSe for High Performance Photovoltaic Device: An Isoelectronic Counterpart of Blue Phosphorene" Nanomaterials 9, no. 4: 598. https://doi.org/10.3390/nano9040598