Alkali Hydrolysis of Sulfated Cellulose Nanocrystals: Optimization of Reaction Conditions and Tailored Surface Charge



Abstract
:1. Introduction
2. Materials and Methods
2.1. Raw Materials
2.2. Nanocrystal Preparation
2.3. Surface Desulfation of H2SO4-Hydrolyzed CNCs
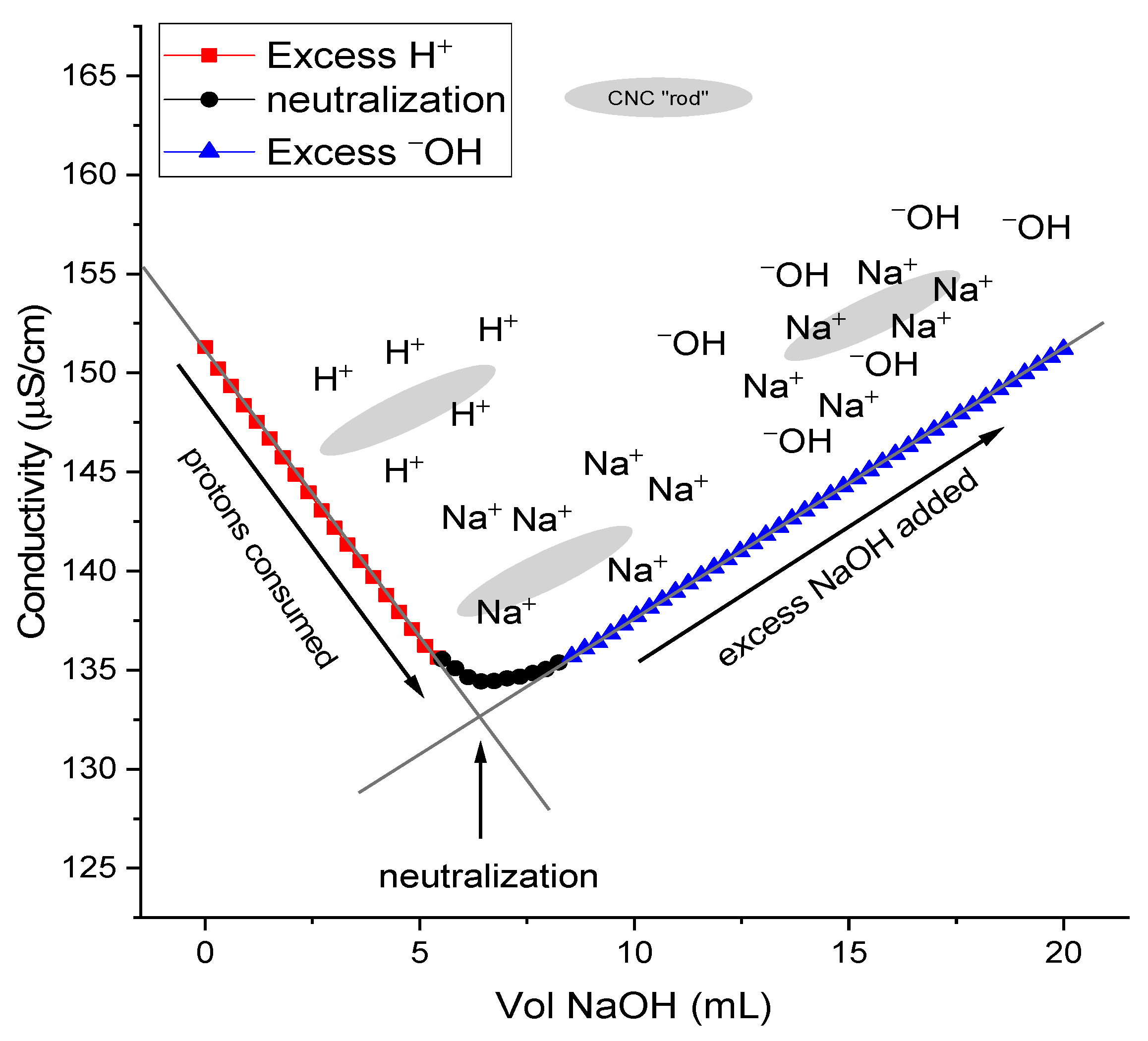
2.4. Conductometric Titration
2.5. Atomic Force Microscopy
2.6. ζ-Potential
2.7. Design of Experiments (DOE)
3. Results and Discussion
4. Conclusions
- Hydrolysis of sulfate half-esters on the CNC surface occurred over a broad range of different conditions: [NaOH] (<0.1 M to >2.0 M), CNC wt% (~0.5 to ≥2.0 wt%) and time (>0 to ≤6 h).
- Above 0.1–0.2 M NaOH there is only a minor observed difference in overall efficacy of –OSO3– removal.
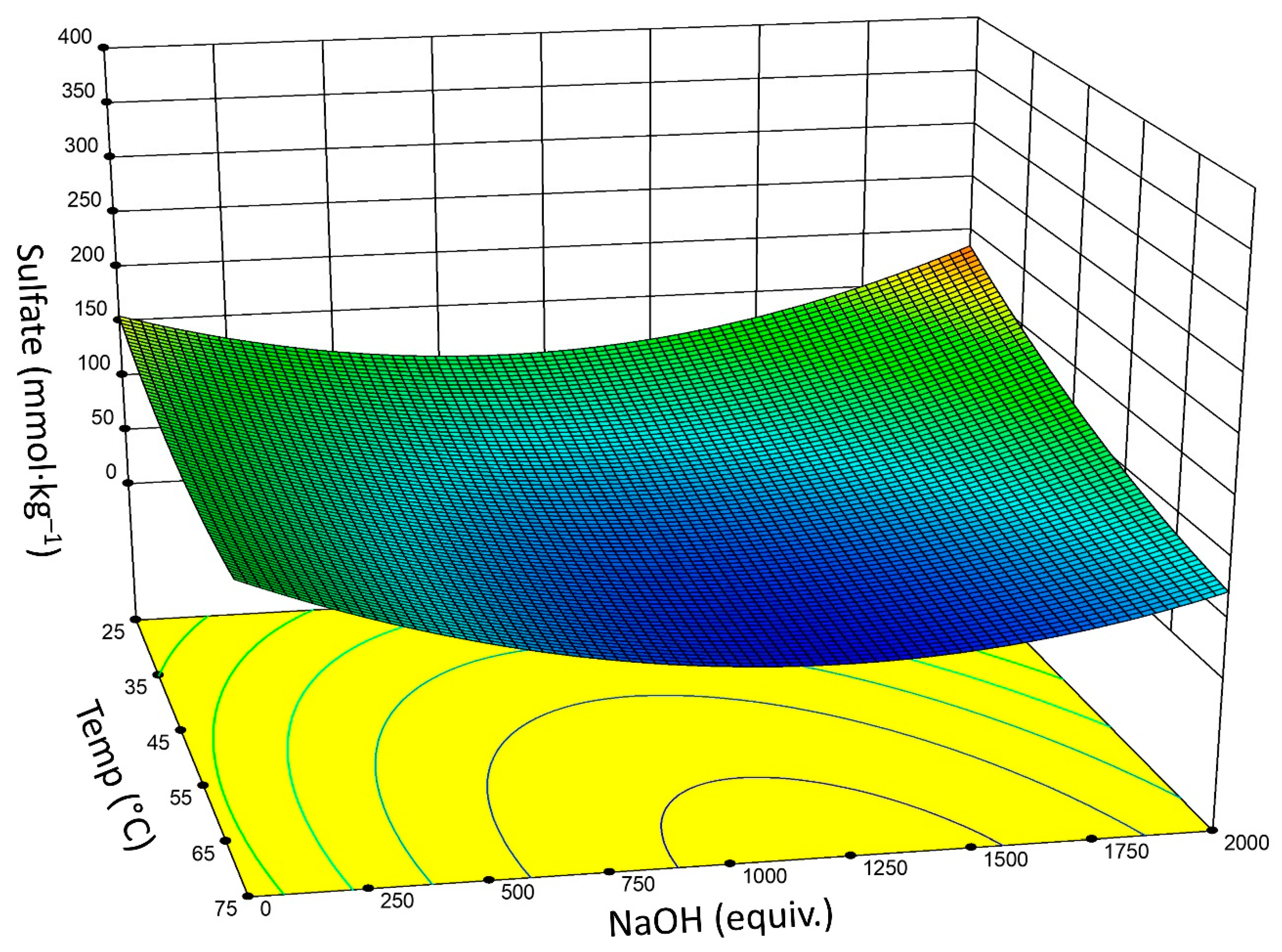
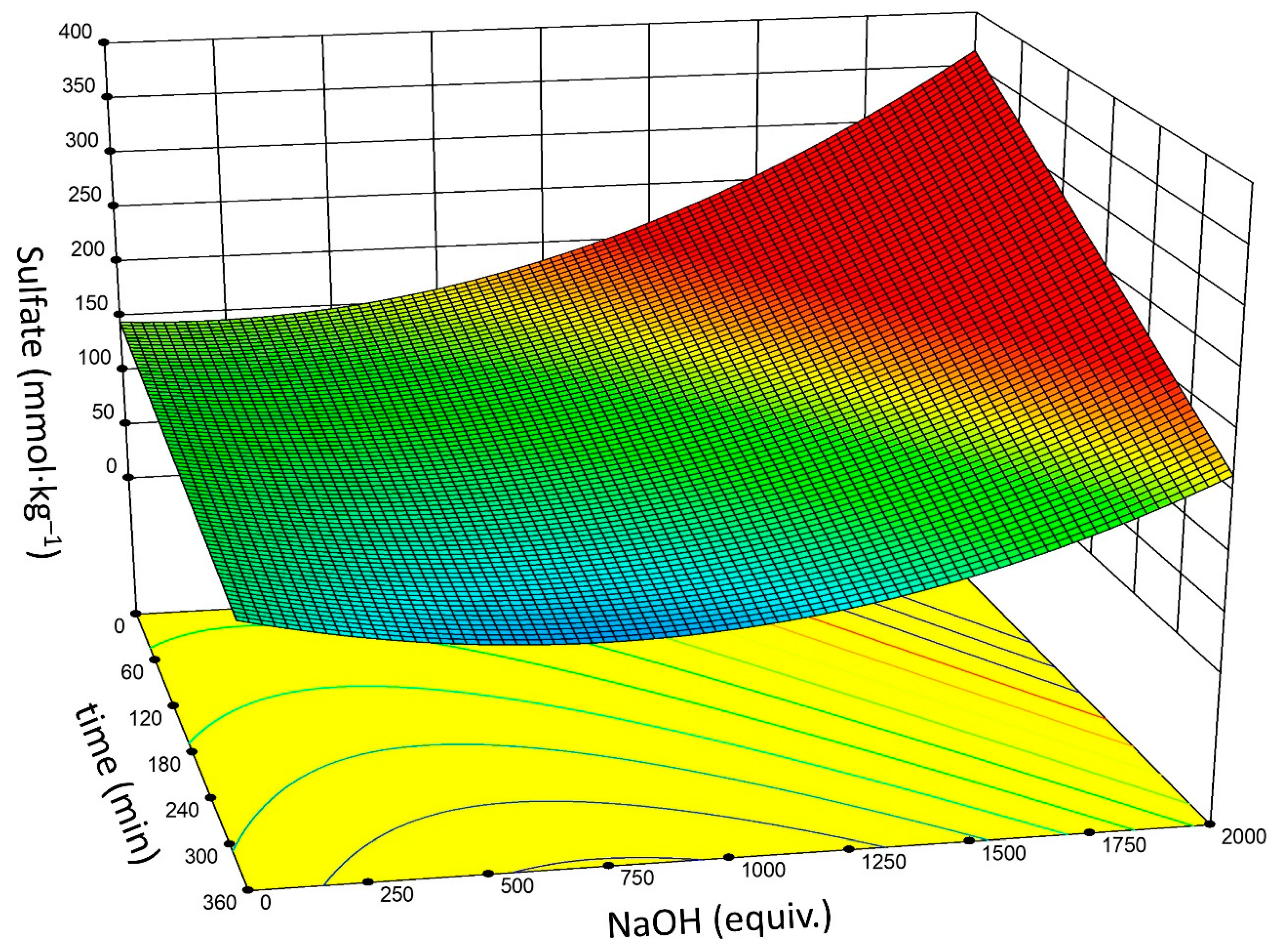
- Based upon DOE analysis, reaction time, temperature, and NaOH concentration are significant factors for effective sulfate half-ester removal. There is a two-factor interaction between reaction time and NaOH concentration. The significance of the NaOH concentration is non-linear.
- Optimal conditions may vary depending on the initial and targeted sulfate concentration: 60–120 equivalents NaOH and a reaction time of 3–6 h gives Δ(–OSO3–) of ~60%.
- The traditional conditions (1.5 M NaOH, 60 °C, 5 h) typically remove about one-third to one-half of the available –OSO3– groups, dependent upon the selected concentration of CNCs (wt%).
- More desirable conditions still yield colloidally stable CNCs at significantly lower use of NaOH, thus improving work-up.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dong, X.M.; Kimura, T.; Revol, J.-F.; Gray, D.G. Effects of Ionic Strength on the Isotropic−Chiral Nematic Phase Transition of Suspensions of Cellulose Crystallites. Langmuir 1996, 12, 2076–2082. [Google Scholar] [CrossRef]
- Dong, X.M.; Gray, D.G. Induced Circular Dichroism of Isotropic and Magnetically-Oriented Chiral Nematic Suspensions of Cellulose Crystallites. Langmuir 1997, 13, 3029–3034. [Google Scholar] [CrossRef]
- Dong, X.M.; Gray, D.G. Effect of Counterions on Ordered Phase Formation in Suspensions of Charged Rodlike Cellulose Crystallites. Langmuir 1997, 13, 2404–2409. [Google Scholar] [CrossRef]
- Kargarzadeh, H.; Mariano, M.; Gopakumar, D.; Ahmad, I.; Thomas, S.; Dufresne, A.; Huang, J.; Lin, N. Advances in cellulose nanomaterials. Cellulose 2018, 25, 2151–2189. [Google Scholar] [CrossRef]
- Kontturi, E.; Laaksonen, P.; Linder, M.B.; Nonappa; Rojas, O.J.; Ikkala, O. Advanced Materials through Assembly of Nanocelluloses. Adv. Mater. 2018, 30, 1703779. [Google Scholar] [CrossRef] [PubMed]
- Klemm, D.; Cranston, E.D.; Fischer, D.; Gama, M.; Kedzior, S.A.; Kralisch, D.; Kramer, F.; Kondo, T.; Lindstroem, T.; Nietzsche, S.; et al. Nanocellulose as a natural source for groundbreaking applications in materials science: Today’s state. Mater. Today 2018, 21, 720–748. [Google Scholar] [CrossRef]
- Dufresne, A. Cellulose nanomaterial reinforced polymer nanocomposites. Curr. Opin. Colloid Interface Sci. 2017, 29, 1–8. [Google Scholar] [CrossRef]
- Flauzino Neto, W.P.; Mariano, M.; Vieira da Silva, I.S.; Silverio, H.A.; Putaux, J.-L.; Otaguro, H.; Pasquini, D.; Dufresne, A. Mechanical properties of natural rubber nanocomposites reinforced with high aspect ratio cellulose nanocrystals isolated from soy hulls. Carbohydr. Polym. 2016, 153, 143–152. [Google Scholar] [CrossRef]
- Nascimento, D.M.; Nunes, Y.L.; Figueiredo, M.C.B.; de Azeredo, H.M.C.; Aouada, F.A.; Feitosa, J.P.A.; Rosa, M.F.; Dufresne, A. Nanocellulose nanocomposite hydrogels: Technological and environmental issues. Green Chem. 2018, 20, 2428–2448. [Google Scholar] [CrossRef]
- Zhou, S.; You, T.; Zhang, X.; Xu, F. Superhydrophobic Cellulose Nanofiber-Assembled Aerogels for Highly Efficient Water-in-Oil Emulsions Separation. ACS Appl. Nano Mater. 2018, 1, 2095–2103. [Google Scholar] [CrossRef]
- Ben Ayed, E.; Cochereau, R.; Dechance, C.; Capron, I.; Nicolai, T.; Benyahia, L. Water-In-Water Emulsion Gels Stabilized by Cellulose Nanocrystals. Langmuir 2018, 34, 6887–6893. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Spinney, S.; Shi, Z.; Tang, J.; Peng, B.; Luo, J.; Tam, K.C. Amphiphilic Cellulose Nanocrystals for Enhanced Pickering Emulsion Stabilization. Langmuir 2018, 34, 12897–12905. [Google Scholar] [CrossRef] [PubMed]
- Jordan, J.H.; Easson, M.W.; Dien, B.; Thompson, S.; Condon, B.D. Extraction and characterization of nanocellulose crystals from cotton gin motes and cotton gin waste. Cellulose 2019, 26, 5959–5979. [Google Scholar] [CrossRef]
- Siqueira, G.; Bras, J.; Dufresne, A. Cellulosic bionanocomposites: A review of preparation, properties and applications. Polymer 2010, 2, 728–765. [Google Scholar] [CrossRef]
- Vanderfleet, O.M.; Cranston, E.D.; Osorio, D.A. Optimization of cellulose nanocrystal length and surface charge density through phosphoric acid hydrolysis. Philos Trans. A Math. Phys. Eng. Sci. 2018, 376. [Google Scholar] [CrossRef] [PubMed]
- Vanderfleet, O.M.; Reid, M.S.; Bras, J.; Heux, L.; Godoy-Vargas, J.; Panga, M.K.R.; Cranston, E.D. Insight into thermal stability of cellulose nanocrystals from new hydrolysis methods with acid blends. Cellulose 2018, 26, 507–528. [Google Scholar] [CrossRef]
- Camarero Espinosa, S.; Kuhnt, T.; Foster, E.J.; Weder, C. Isolation of thermally stable cellulose nanocrystals by phosphoric acid hydrolysis. Biomacromolecules 2013, 14, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Dhar, P.; Bhasney, S.M.; Kumar, A.; Katiyar, V. Acid functionalized cellulose nanocrystals and its effect on mechanical, thermal, crystallization and surfaces properties of poly (lactic acid) bionanocomposites films: A comprehensive study. Polymer 2016, 101, 75–92. [Google Scholar] [CrossRef]
- Reid, M.S.; Villalobos, M.; Cranston, E.D. Benchmarking Cellulose Nanocrystals: From the Laboratory to Industrial Production. Langmuir 2017, 33, 1583–1598. [Google Scholar] [CrossRef]
- Cheng, M.; Qin, Z.; Chen, Y.; Hu, S.; Ren, Z.; Zhu, M. Efficient Extraction of Cellulose Nanocrystals through Hydrochloric Acid Hydrolysis Catalyzed by Inorganic Chlorides under Hydrothermal Conditions. ACS Sustain. Chem. Eng. 2017, 5, 4656–4664. [Google Scholar] [CrossRef]
- Lin, N.; Dufresne, A. Surface chemistry, morphological analysis and properties of cellulose nanocrystals with gradiented sulfation degrees. Nanoscale 2014, 6, 5384–5393. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhao, X.; Zhu, J.Y. Kinetics of Strong Acid Hydrolysis of a Bleached Kraft Pulp for Producing Cellulose Nanocrystals (CNCs). Ind. Eng. Chem. Res. 2014, 53, 11007–11014. [Google Scholar] [CrossRef]
- Chen, L.; Wang, Q.; Hirth, K.; Baez, C.; Agarwal, U.P.; Zhu, J.Y. Tailoring the yield and characteristics of wood cellulose nanocrystals (CNC) using concentrated acid hydrolysis. Cellulose 2015, 22, 1753–1762. [Google Scholar] [CrossRef]
- Sasaki, M.; Kabyemela, B.; Malaluan, R.; Hirose, S.; Takeda, N.; Adschiri, T.; Arai, K. Cellulose hydrolysis in subcritical and supercritical water. J. Supercrit. Fluids 1998, 13, 261–268. [Google Scholar] [CrossRef]
- Kumar, S.; Gupta, R.B. Hydrolysis of Microcrystalline Cellulose in Subcritical and Supercritical Water in a Continuous Flow Reactor. Ind. Eng. Chem. Res. 2008, 47, 9321–9329. [Google Scholar] [CrossRef]
- Luo, J.; Semenikhin, N.; Chang, H.; Moon, R.J.; Kumar, S. Post-sulfonation of cellulose nanofibrils with a one-step reaction to improve dispersibility. Carbohydr. Polym. 2018, 181, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Bian, H.; Wang, Q.; Zhang, N.; Liang, Y.; Dong, D. Direct functionalization of cellulose nanocrystals with polymer brushes via UV-induced polymerization: Access to novel heterogeneous visible-light photocatalysts. RSC Adv. 2016, 6, 53062–53068. [Google Scholar] [CrossRef]
- Abitbol, T.; Kam, D.; Levi-Kalisman, Y.; Gray, D.G.; Shoseyov, O. Surface Charge Influence on the Phase Separation and Viscosity of Cellulose Nanocrystals. Langmuir 2018, 34, 3925–3933. [Google Scholar] [CrossRef] [PubMed]
- Wolfenden, R.; Yuan, Y. Monoalkyl sulfates as alkylating agents in water, alkylsulfatase rate enhancements, and the “energy-rich” nature of sulfate half-esters. Proc. Natl. Acad. Sci. USA 2007, 104, 83–86. [Google Scholar] [CrossRef] [PubMed]
- March, J.; Smith, M.B. March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 6th ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007. [Google Scholar]
- Hasani, M.; Cranston, E.D.; Westman, G.; Gray, D.G. Cationic surface functionalization of cellulose nanocrystals. Soft Matter 2008, 4, 2238–2244. [Google Scholar] [CrossRef]
- Jiang, F.; Esker, A.R.; Roman, M. Acid-Catalyzed and Solvolytic Desulfation of H2SO4-Hydrolyzed Cellulose Nanocrystals. Langmuir 2010, 26, 17919–17925. [Google Scholar] [CrossRef]
- Lewis, L.; Derakhshandeh, M.; Hatzikiriakos, S.G.; Hamad, W.Y.; MacLachlan, M.J. Hydrothermal Gelation of Aqueous Cellulose Nanocrystal Suspensions. Biomacromolecules 2016, 17, 2747–2754. [Google Scholar] [CrossRef]
- Lin, N.; Geze, A.; Wouessidjewe, D.; Huang, J.; Dufresne, A. Biocompatible Double-Membrane Hydrogels from Cationic Cellulose Nanocrystals and Anionic Alginate as Complexing Drugs Codelivery. ACS Appl. Mater. Interfaces 2016, 8, 6880–6889. [Google Scholar] [CrossRef]
- Dorris, A.; Gray, D.G. Gelation of cellulose nanocrystal suspensions in glycerol. Cellulose 2012, 19, 687–694. [Google Scholar] [CrossRef]
- Abitbol, T.; Palermo, A.; Moran-Mirabal, J.M.; Cranston, E.D. Fluorescent Labeling and Characterization of Cellulose Nanocrystals with Varying Charge Contents. Biomacromolecules 2013, 14, 3278–3284. [Google Scholar] [CrossRef]
- Kalashnikova, I.; Bizot, H.; Cathala, B.; Capron, I. Modulation of Cellulose Nanocrystals Amphiphilic Properties to Stabilize Oil/Water Interface. Biomacromolecules 2012, 13, 267–275. [Google Scholar] [CrossRef]
- Ellebracht, N.C.; Jones, C.W. Amine functionalization of cellulose nanocrystals for acid-base organocatalysis: Surface chemistry, cross-linking, and solvent effects. Cellulose 2018. [Google Scholar] [CrossRef]
- Pandey, A.; Derakhshandeh, M.; Kedzior, S.A.; Pilapil, B.; Shomrat, N.; Segal-Peretz, T.; Bryant, S.L.; Trifkovic, M. Role of interparticle interactions on microstructural and rheological properties of cellulose nanocrystal stabilized emulsions. J. Colloid Interface Sci. 2018, 532, 808–818. [Google Scholar] [CrossRef]
- Zoppe, J.O.; Johansson, L.-S.; Seppala, J. Manipulation of cellulose nanocrystal surface sulfate groups toward biomimetic nanostructures in aqueous media. Carbohydr. Polym. 2015, 126, 23–31. [Google Scholar] [CrossRef]
- Zoppe, J.O.; Ruottinen, V.; Ruotsalainen, J.; Rönkkö, S.; Johansson, L.-S.; Hinkkanen, A.; Järvinen, K.; Seppälä, J. Synthesis of Cellulose Nanocrystals Carrying Tyrosine Sulfate Mimetic Ligands and Inhibition of Alphavirus Infection. Biomacromolecules 2014, 15, 1534–1542. [Google Scholar] [CrossRef]
- Lokanathan, A.R.; Uddin, K.M.A.; Rojas, O.J.; Laine, J. Cellulose Nanocrystal-Mediated Synthesis of Silver Nanoparticles: Role of Sulfate Groups in Nucleation Phenomena. Biomacromolecules 2014, 15, 373–379. [Google Scholar] [CrossRef]
- Cherhal, F.; Cousin, F.; Capron, I. Influence of Charge Density and Ionic Strength on the Aggregation Process of Cellulose Nanocrystals in Aqueous Suspension, as Revealed by Small-Angle Neutron Scattering. Langmuir 2015, 31, 5596–5602. [Google Scholar] [CrossRef]
- Hasani, M.; Cranston, E.D.; Westman, G.; Gray, D.G. Cationic surface functionalization of cellulose nanocrystals [Erratum to document cited in CA151:339450]. Soft Matter 2015, 11, 7440. [Google Scholar] [CrossRef]
- Kloser, E.; Gray, D.G. Surface Grafting of Cellulose Nanocrystals with Poly(ethylene oxide) in Aqueous Media. Langmuir 2010, 26, 13450–13456. [Google Scholar] [CrossRef]
- Beck, S.; Bouchard, J. Auto-catalyzed acidic desulfation of cellulose nanocrystals. Nord. Pulp Pap. Res. J. 2014, 29, 6–14. [Google Scholar] [CrossRef]
- Zoppe, J.O.; Xu, X.; Kanel, C.; Orsolini, P.; Siqueira, G.; Tingaut, P.; Zimmermann, T.; Klok, H.-A. Effect of Surface Charge on Surface-Initiated Atom Transfer Radical Polymerization from Cellulose Nanocrystals in Aqueous Media. Biomacromolecules 2016, 17, 1404–1413. [Google Scholar] [CrossRef]
- Reid, M.S.; Kedzior, S.A.; Villalobos, M.; Cranston, E.D. Effect of Ionic Strength and Surface Charge Density on the Kinetics of Cellulose Nanocrystal Thin Film Swelling. Langmuir 2017, 33, 7403–7411. [Google Scholar] [CrossRef]
- Wohlhauser, S.; Delepierre, G.; Labet, M.; Morandi, G.; Thielemans, W.; Weder, C.; Zoppe, J.O. Grafting Polymers from Cellulose Nanocrystals: Synthesis, Properties, and Applications. Macromolecules 2018, 51, 6157–6189. [Google Scholar] [CrossRef]
- Beck, S.; Methot, M.; Bouchard, J. General procedure for determining cellulose nanocrystal sulfate half-ester content by conductometric titration. Cellulose 2015, 22, 101–116. [Google Scholar] [CrossRef]
- Abitbol, T.; Kloser, E.; Gray, D.G. Estimation of the surface sulfur content of cellulose nanocrystals prepared by sulfuric acid hydrolysis. Cellulose 2013, 20, 785–794. [Google Scholar] [CrossRef]
- Johnston, L.J.; Jakubek, Z.J.; Beck, S.; Araki, J.; Cranston, E.D.; Danumah, C.; Fox, D.; Li, H.; Wang, J.; Mester, Z.; et al. Determination of sulfur and sulfate half-ester content in cellulose nanocrystals: An interlaboratory comparison. Metrologia 2018, 55, 872–882. [Google Scholar] [CrossRef]
- Foster, E.J.; Moon, R.J.; Agarwal, U.P.; Bortner, M.J.; Bras, J.; Camarero-Espinosa, S.; Chan, K.J.; Clift, M.J.D.; Cranston, E.D.; Eichhorn, S.J.; et al. Current characterization methods for cellulose nanomaterials. Chem. Soc. Rev. 2018, 47, 2609–2679. [Google Scholar] [CrossRef] [Green Version]
- Cherhal, F.; Cousin, F.; Capron, I. Structural Description of the Interface of Pickering Emulsions Stabilized by Cellulose Nanocrystals. Biomacromolecules 2016, 17, 496–502. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Conditions (Final) 1 | –OSO3– (mmol·kg−1) | Reference | |
|---|---|---|---|---|
| Initial | Final | |||
| Acidic | 0.97 wt%, 0.024 M HCl, 80 °C, 2.5 h | 293 | 191 | [32] 1 |
| ×2 | 153 | |||
| ×3 | 103 | |||
| ×4 | 58 | |||
| ×7 | 55 | |||
| 4.39 wt%, 0.025 M HCl, 80 °C, 20 h | 148 ± 12 | 64 ± 8 | [36] | |
| 4.5 wt%, 0.025 M HCl, 80 °C, 20 h | 125 | 47 | [27] 1 | |
| 0.4 wt%, 2.5 N HCl, | 44 | n/a 3 | [43] 1 | |
| 0.5 wt%, 0.05 M HCl, 80 °C, 24 h | 280 | 120 | [38] | |
| 2.0 wt%, 2.5 M HCl, 100 °C, 5 h | ~430 | ~50 | [39] 4,5 | |
| Alkaline | 9 wt%, 2.0 M NaOH, 65 °C, 5 h | 130 ± 95 | n/a 3 | [31,44] 2 |
| 2.78 wt%, 1.0 M NaOH, 60 °C, 5 h | 240 | 80 | [45] | |
| 2.78 wt%, 1.7 M NaOH, 85 °C, 72 h | 240 | 40 | ||
| 2 wt%, 0.1 M NaOH, 23 °C, 20 min | 234 | 222 | [46] 5 | |
| 0.56 wt%, 0.1 M NaOH, 50 °C, 20 min | 244 | 240 | ||
| 0.56 wt%, 0.1 M NaOH, 50 °C, 160 min | 244 | 225 | ||
| 0.55 wt%, 0.85 M NaOH, 50 °C, 20 min | 240 | 228 | ||
| 0.56 wt%, 0.85 M NaOH, 50 °C, 180 min | 240 | 229 | ||
| 1.33 wt%, 0.17 M NaOH, 60 °C, 1 h | 209 | 166 | [24] 1 | |
| 1.33 wt%, 0.33 M NaOH, 60 °C, 1.5 h | 209 | 144 | ||
| 1.33 wt%, 0.50 M NaOH, 60 °C, 2 h | 209 | 90.6 | ||
| 1.33 wt%, 0.67 M NaOH, 60 °C, 3 h | 209 | 56.3 | ||
| 1.0 wt%, 1.0 M NaOH, 60 °C, 5 h | 220 | 40 | [40,41,47]2 | |
| 1.0 wt%, 0.01 M NaOH, 65 °C, 30 min | ~194 | ~165 | [42] 4 | |
| 1.0 wt%, 0.1 M NaOH, 65 °C, 30 min | ~194 | ~152 | ||
| 1.0 wt%, 0.5 M NaOH, 65 °C, 30 min | ~194 | ~142 | ||
| 5.0 wt%, 1.5 M NaOH, 65 °C, 5 h | ~219 | ~125 | [48] 1,4 | |
| 2.0 wt%, 2 M NaOH, 65 °C, 5 h | ~430 | ~190 | [39] 5 | |
| 1.45 wt%, 1.0 M NaOH, 60 °C, 2.5 h | 150 ± 15 | 62 ± 1 | [13] | |
| Batch (#) | H2SO4 (wt%) | Temp (°C) | Time (min) | –OSO3– (mmol·kg−1) | ζ-Potential (mV) | Yield (%) |
|---|---|---|---|---|---|---|
| 1 | 55 | 60 | 150 | 155 | –31.5 ± 1.5 | 43 |
| 2 | 65 | 55 | 60 | 197 | –40.8 ± 0.7 | 21 |
| 3 | 65 | 45 | 90 | 211 | –41.1 ± 1.5 | 32 |
| 4 | 65 | 60 | 90 | 308 | –45.3 ± 1.0 | 14 |
| 5 | 62 | 50 | 30 | 134 | –41.5 ± 1.1 | 39 |
| Sample | CNC (wt%) | Temp (°C) | Time (h) | –OSO3– (mmol·kg−1) | Ref. or exp # | |
|---|---|---|---|---|---|---|
| Initial | Final | |||||
| H-CNC | 3.8 | 70 | 120 | 265 2 | 90 2 | Ref. [46] |
| H-CNC | 2.8 | 85 | 72 | 275 1 | 103 1 | |
| H-CNC | 4.0 | 100 | 2 | 217 2 | 108 2 | |
| Na-CNC | 2.8 | 85 | 72 | 275 1 | 275 1 | |
| Na-CNC | 3.8 | 70 | 120 | 265 2 | 254 2 | |
| H-CNC | 0.50 | 25 | 6 | 211 1 | 204 1 | #1 |
| H-CNC | 0.50 | 75 | 6 | 197 1 | 87 1 | #2 |
| Na-CNC | 0.50 | 25 | 0 | 211 1 | 204 1 | #3 |
| Na-CNC | 0.50 | 75 | 6 | 197 1 | 193 1 | #4 |
| Exp (#) | Time (min) | Temp (°C) | [NaOH] (M) | [CNC] (wt%) | Yield (%) | –OSO3– (mmol·kg−1) | Δ(–OSO3–) (%) | |
|---|---|---|---|---|---|---|---|---|
| Initial | Final | |||||||
| #5 | 150 | 60 | 0.5 | 1.44 | 88 | 155 | 109 | 30% |
| #6 | 150 | 60 | 1.0 | 1.44 | 83 | 155 | 105 | 32% |
| #7 | 300 | 60 | 1.0 | 1.44 | 87 | 155 | 86 | 45% |
| #8 | 300 | 30 | 1.0 | 1.44 | 83 | 155 | 121 | 21% |
| #9 | 900 | 30 | 1.0 | 1.44 | 90 | 155 | 122 | 22% |
| Sample | CNC (wt%) | –OSO3– (mmol·kg−1) | Δ(–OSO3–) | |
|---|---|---|---|---|
| Initial | Final | (%)/net | ||
| #10 | 0.72 | 308 | 213 | 31|31 |
| ×2 | 0.58 | 213 | 162 | 24|47 |
| ×3 | 0.45 | 162 | 103 | 36|67 |
| ×4 | 0.25 | 103 | 69 | 33|78 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jordan, J.H.; Easson, M.W.; Condon, B.D. Alkali Hydrolysis of Sulfated Cellulose Nanocrystals: Optimization of Reaction Conditions and Tailored Surface Charge. Nanomaterials 2019, 9, 1232. https://doi.org/10.3390/nano9091232
Jordan JH, Easson MW, Condon BD. Alkali Hydrolysis of Sulfated Cellulose Nanocrystals: Optimization of Reaction Conditions and Tailored Surface Charge. Nanomaterials. 2019; 9(9):1232. https://doi.org/10.3390/nano9091232
Chicago/Turabian StyleJordan, Jacobs H., Michael W. Easson, and Brian D. Condon. 2019. "Alkali Hydrolysis of Sulfated Cellulose Nanocrystals: Optimization of Reaction Conditions and Tailored Surface Charge" Nanomaterials 9, no. 9: 1232. https://doi.org/10.3390/nano9091232
APA StyleJordan, J. H., Easson, M. W., & Condon, B. D. (2019). Alkali Hydrolysis of Sulfated Cellulose Nanocrystals: Optimization of Reaction Conditions and Tailored Surface Charge. Nanomaterials, 9(9), 1232. https://doi.org/10.3390/nano9091232