1. Introduction
Determining trace quantities of proteins in complex biological mixtures is one of the most important objectives of bioanalytical chemistry. In order to detect the onset of a disease at an early stage for example, highly sensitive assays are desirable that can quantitatively estimate small amounts of antigens/antibodies circulating in bodily fluids. Immunoassays are very important tools in this regard because of their ability to identify and quantitate specific proteins based on the high affinity between an antigen and an antibody. Among the different types of immunoassays developed to date, Enzyme Linked Immunosorbent Assay or ELISA is arguably one of the most practiced ones. The key advantage of using ELISA methods is the inherent signal amplification in this technique which allows one to reliably quantitate very small amounts of proteins present in biological samples [
1]. While the greater sensitivity of the ELISA method is impressive, it is nevertheless desirable to further improve the limit of detection for this technique extending its applicability to more challenging assays [
2].
In an effort to reduce the sample volume requirement in the ELISA method, this immunoassay has been implemented on the microfluidic platform by several researchers in conjunction with different detection/quantitation techniques [
3]. One of the simpler versions among these relies on the use of a non-fluorescent enzyme substrate that gets transformed into a detectable fluorescent product over the course of the assay [
4]. The amount of this fluorescent reporter in most cases, however, has been either detected off-chip or in the analysis channel after a fixed period of enzyme reaction,
i.e., the end-point ELISA format [
5,
6]. In this article, we report a substantial improvement in both the reproducibility and the limit of detection of the fluorescence based microfluidic ELISA method using the kinetic format of the assay. Antibodies to the Blue Tongue Virus (BTV) and Epizootic Hemorrhagic Disease Virus (EHDV) grown using hybridoma cell lines and previously employed to the detection of these viral infections in a variety of ruminants [
7,
8,
9] were used as the analytes for evaluating the performance of these assays. Both BTV and EHDV are arthropod-borne pathogenic species that cause serious illness in livestock such as sheep, goats, cattle and deer [
10,
11]. There is no efficient treatment currently available for these viral infections and therefore their detection at an early stage is often key to preventing their outbreak among artiodactyles in zoos and livestock farms for minimizing economic damages. Interestingly, both of these viruses are known to be transmitted by biting midges (
Culicoides species) and their infections tend to be confounding due to antigenic similarity [
12]. As a result, there is a need for developing assays that would allow the specific detection of each these conditions in a time and cost effective manner. To this end, several ELISA methods have been successfully developed [
7,
8,
9] whose utility in point-of-care diagnostic applications can be significantly improved through their implementation on portable platforms. In this work, we demonstrate a microfluidic ELISA for quantitatively determining the levels of BTV (serotype 11) and EHDV (serotype 2) antibodies with a higher sensitivity than currently possible on commercial microwell plates. Our experiments show that when the concentrations of these analytes in the microfluidic assays are arrived at by comparing the rate of signal generation rather than the signal itself, the noise in the system is substantially reduced. This leads to a more reliable quantitation particularly under conditions when the change in signal over the enzyme reaction period is small compared to the background fluorescence (signal at the start of the enzyme reaction) in the system as is the case here. While this result was observed to be valid even in the case of microwell plates, the benefit of employing the kinetic format of the assay over its end-point version in microwell based ELISAs was found to be not as significant.
3. Results and Discussion
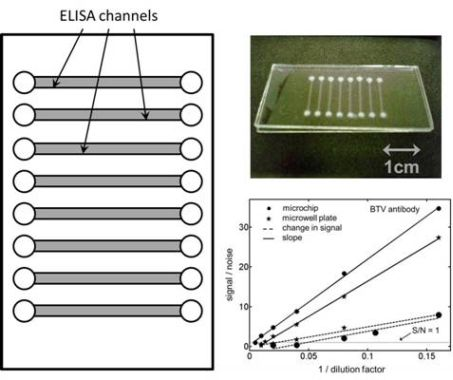
Because a major objective of our research was to compare the sensitivity of the kinetic ELISAs to their end-point versions, it was necessary for us to ensure that the enzyme reaction followed a zeroth order kinetics during the course of the assay. To this end, a series of experiments were performed to establish a suitable enzyme reaction period over which the fluorescence in the microchannels/microwells increased linearly with time. In this regard, we have presented kinetic data from six different ELISA microchannels (see
Figure 1(b)) three of which were incubated with a 50× BTV antibody solution and the other three were blanks for the same analyte. Although all of the concerned microchannels were located on the same microchip in this case, the variability in the observed ELISA signal across them was very similar to those noticed among analysis channels etched on different microchips. In other words, the assay-to-assay variability in our experiments was dominated by the fluctuations in the channel-to-channel characteristics and not by the differences in the microchips. No ELISA channel was used twice for an assay in our study. As may be seen from
Figure 1(b), the fluorescence in these fluidic conduits grew linearly in time with coefficients of determination (R
2 value) greater than 0.97 during the first 30 min of the assay. There onwards the rate of signal generation was observed to slow down deviating from the desirable zeroth order enzyme kinetics [
17]. In this situation, the ELISA reaction period was chosen to be 30 min in all our experiments.
Figure 1(b) also shows that when the same assay is repeated in different microchannels/microchips the variation in the background fluorescence (signal at time = 0) in them was significant compared to the change in signal over the 30 min enzyme reaction period. Interestingly, the rate of signal generation in all these cases was observed to be very reproducible for both the 50× BTV-antibody sample and its corresponding blank. In the case of the 50× BTV-antibody sample for example, the mean value for the background fluorescence across five different ELISA microchannels was measured to be 17.29 ± 0.98 a.u. (5.67% RSD) while the corresponding number for the rate of signal generation in the same conduits was determined to be 0.0511 ± 0.0018 a.u./min (3.52% RSD). In this situation, the average value (using 5 measurements) for the change in fluorescence signal over the 30 min enzyme reaction period in these assays was 1.53 a.u. which was comparable to the standard deviation in the background fluorescence across the different ELISA channels,
i.e., 0.98 a.u. Notice that because the signal and noise level in end-point ELISAs are proportional to these quantities respectively, their similar magnitude implied a lower reliability of the experimental data for this assay format. On the other hand, the small relative standard deviation in the rate of ELISA signal generation,
i.e., 3.52%, suggests that it may be possible to obtain significantly lower limits of detection for the target analyte using the kinetic version of the assay.
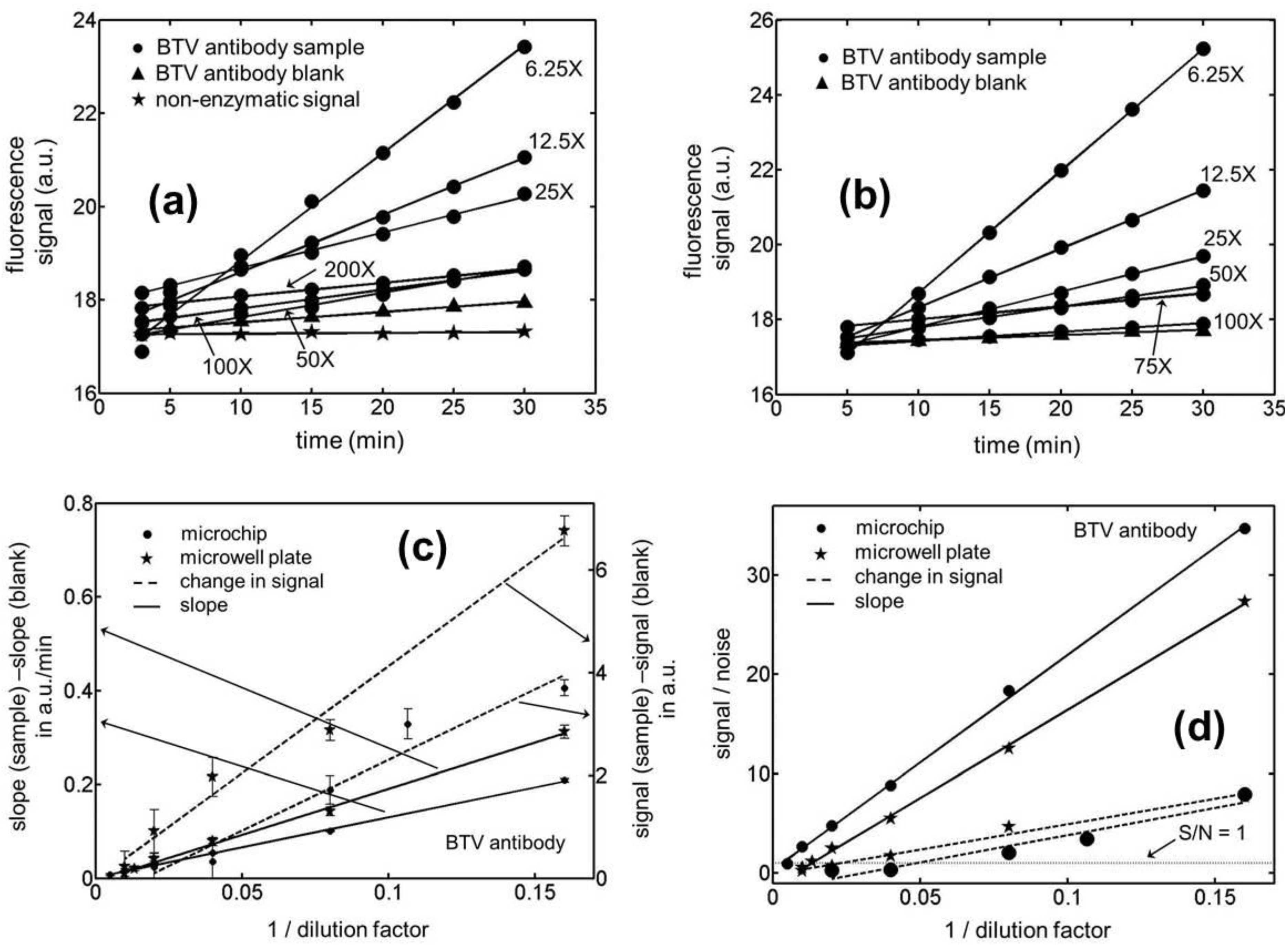
Having established a suitable enzyme reaction period for our assays, fluorescence measurements were made in microchannels/microwells that were incubated with different concentrations of the BTV antibody (see
Figure 2(a,b)). The data shows a fairly linear change in this signal with time for both the microfluidic and microwell based assays. There is however, again a substantial variation in the background fluorescence in going from one assay compartment to the other particularly in the case of the microchannel based ELISAs. As a result, the fluorescence curves for the different sample concentrations are seen to cross each other over the assay period, more frequently for the microchips than for the microwell plates. In
Figure 2(c), we have compared the signal generated by the sample minus the corresponding blank for the end-point and kinetic mode of these assays on both the microchip and microwell platforms. Note that while only one fluorescence measurement was made after a fixed enzyme reaction period in the end-point ELISAs, 6–7 readings were taken over the same duration in the kinetic version of the assay. This enzyme reaction period was chosen to be 30 min in all our experiments which was arrived at based on the results presented in
Figure 1(b). The data presented in this figure suggest that if this reaction time is chosen to be less than 15 min or greater than half-an-hour, the signal to noise ratio in the system is compromised. For the choice of a 30 min enzyme reaction period, the measured signal for both the ELISA formats was observed to vary linearly with the inverse of the dilution factor for the BTV-antibody sample.
Figure 2(c) however shows a significantly larger noise level (measured in terms of the standard deviation for the signal) in the end-point ELISAs which arises due to a substantial variation in the background fluorescence (signal at the start of the enzyme reaction period) going from one assay compartment to the other. Moreover, the disparity in the noise levels for the two ELISA formats was also observed to be substantially greater in the microchannels compared to that in the microwells. While the reasons behind this observation are not completely clear, it is possible that the larger surface roughness in the chemically etched fluidic ducts may have resulted in a larger variability in the quality of the surface coatings within them. In addition, it was also qualitatively observed that the amount of light scattered by the rougher microchannels was greater than that by the microwell plates.
Figure 2.
Temporal variation in the fluorescence signal for Blue Tongue Virus (BTV) antibody samples in (a) microchannels (b) microwells with an active ELISA surface. (c) Calibration curves for the kinetic and end-point ELISAs in microchips and microwell plates for BTV antibody samples. Note that while the rate of signal generation for a sample minus that for its corresponding blank (left axis) has been plotted for quantitating the kinetic ELISAs, the absolute change in signal over a fixed enzyme reaction period minus that for its corresponding blank (right axis) was used as a measure for quantitating the end-point immunoassays. (d) Limit of detection (LOD) curves for the kinetic and end-point ELISAs in microchannels and microwells for the BTV antibody samples. The noise level (N) in these calculations was evaluated as 3-times the standard deviation in the signal (S). The LOD for an assay was estimated as the dilution factor for which S/N = 1.
Figure 2.
Temporal variation in the fluorescence signal for Blue Tongue Virus (BTV) antibody samples in (a) microchannels (b) microwells with an active ELISA surface. (c) Calibration curves for the kinetic and end-point ELISAs in microchips and microwell plates for BTV antibody samples. Note that while the rate of signal generation for a sample minus that for its corresponding blank (left axis) has been plotted for quantitating the kinetic ELISAs, the absolute change in signal over a fixed enzyme reaction period minus that for its corresponding blank (right axis) was used as a measure for quantitating the end-point immunoassays. (d) Limit of detection (LOD) curves for the kinetic and end-point ELISAs in microchannels and microwells for the BTV antibody samples. The noise level (N) in these calculations was evaluated as 3-times the standard deviation in the signal (S). The LOD for an assay was estimated as the dilution factor for which S/N = 1.
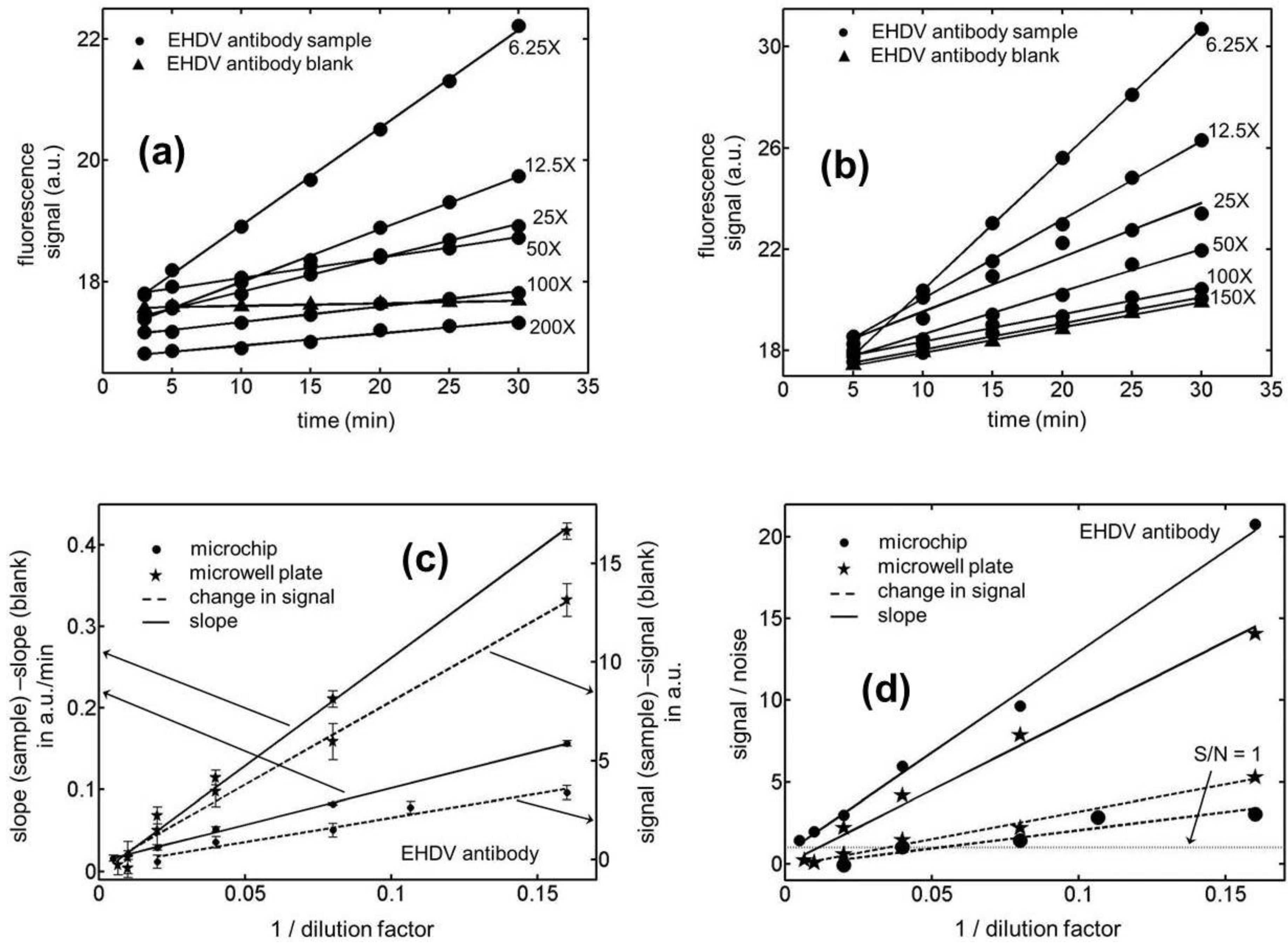
Figure 3.
Temporal variation in the fluorescence signal for Epizootic Hemorrhagic Disease Virus (EHDV) antibody samples in (a) microchannels (b) microwells with an active ELISA surface. (c) Calibration curves for the kinetic and end-point ELISAs in microchips and microwell plates for EHDV antibody samples. (d) Limit of detection (LOD) curves for the kinetic and end-point ELISAs in microchannels and microwells for the EHDV antibody samples. Notice that the x-coordinate in (c) and (d) has been chosen to be the reciprocal of dilution factor rather than the dilution factor itself. Our analyses show that if the latter choice is made, the linear variation between the x and y data points is compromised.
Figure 3.
Temporal variation in the fluorescence signal for Epizootic Hemorrhagic Disease Virus (EHDV) antibody samples in (a) microchannels (b) microwells with an active ELISA surface. (c) Calibration curves for the kinetic and end-point ELISAs in microchips and microwell plates for EHDV antibody samples. (d) Limit of detection (LOD) curves for the kinetic and end-point ELISAs in microchannels and microwells for the EHDV antibody samples. Notice that the x-coordinate in (c) and (d) has been chosen to be the reciprocal of dilution factor rather than the dilution factor itself. Our analyses show that if the latter choice is made, the linear variation between the x and y data points is compromised.
In
Figure 2(d), we have presented the signal to noise ratio (S/N) in our BTV antibody ELISA experiments as a function of the sample dilution factor. The noise level in these evaluations was assumed to be three-times the standard deviation in the magnitude of the signal which establishes the limit of detection for these curves to be the dilution factor when S/N = 1. For all of the measurements presented in this figure, the noise level was estimated based on five independent experiments both for the end-point and kinetic assays. Based on these measurements, the limits of detection (in terms of dilution factors) were established to be 420.1 ± 7.9 and 21.1 ± 4.4 for the kinetic and end-point ELISAs, respectively, in the microfluidic channels. The 95% confidence intervals for these LOD values are estimated to be 420.1 ± 9.8 and 21.1 ± 5.5, respectively, based on the two-sided
t-distribution. This corresponds to a nearly 20-fold reduction in this quantity on adopting the kinetic version of the assay over its end-point format in microchips. Moreover, the data shows a significantly greater reproducibility in the former mode of the immunoassay yielding a relative standard deviation of 1.9% in the limit of detection (LOD) as compared to 20.9% for the latter case. For the microwell based assays, the LODs for the kinetic and end-point ELISAs were estimated to be 142.3 ± 13.4 and 44.2 ± 4.1, respectively. In this situation, the 95% confidence intervals for these quantities turn out to be 142.3 ± 16.7 and 44.2 ± 5.1, respectively, based on the two-sided
t-distribution. This corresponds to about a 3-fold improvement in the smallest concentration that could be reliably quantitated and the relative uncertainly in this figure of merit on adopting the kinetic version of the assay over its end-point format in microwells. On comparing the kinetic ELISAs in microchannels to those in microwells, the former platform has been shown to yield a 3-fold lower LOD with at least 5-fold higher precision. But for the end-point immunoassays the trend reverses and the latter platform was observed to perform twice as well as the microfluidic devices. For the EHDV antibodies, similar trends to those reported above were again noticed for the immunoassays (see
Figure 3). In the case of the microfluidic experiments, the LOD in terms of the dilution factors were estimated to be 307.3 ± 5.9 and 18.8 ± 6.3, respectively, for the kinetic and end-point version of the ELISAs. The corresponding numbers for the microwell plates were 63.1 ± 17.2 and 29.3 ± 8.1, respectively. Based on the two-sided
t-distribution, the 95% confidence intervals for these four LODs can be shown to be 307.3 ± 7.3, 18.8 ± 7.8, 63.1 ± 21.4 and 29.3 ± 10.1, respectively. Therefore, the improvement in LOD yielded by the microchips over the microwell plates was a factor of 4.9 for kinetic EHDV antibody ELISAs while the latter platform performed only about 1.5 fold better than the microchips in the case of the end-point assays. As before, the best reproducibility was obtained for the microchip based kinetic ELISAs with relative standard deviation of 1.9%.

{kind=link}
{kind=link}
{kind=link}
{kind=link}


