
Three Distinct Annotation Platforms Differ in Detection of Antimicrobial Resistance Genes in Long-Read, Short-Read, and Hybrid Sequences Derived from Total Genomic DNA or from Purified Plasmid DNA

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
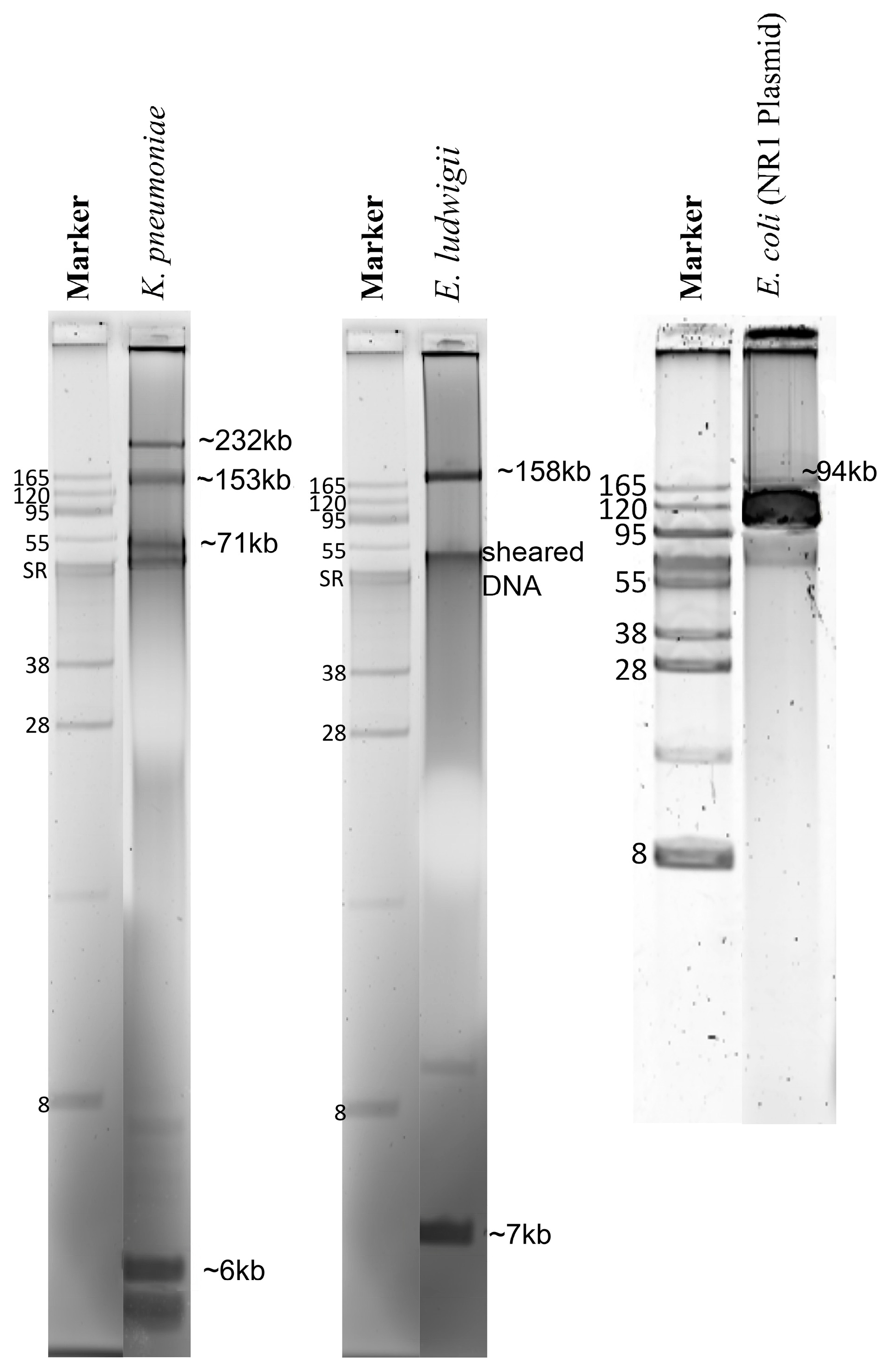
2.1. Concentration and Purity of Total Genomic DNA and Pure Plasmid DNA
2.2. Assembly and Assessment of Total Genomic DNA Preparations
2.3. Plasmid Sequences Assembled from Purified Plasmid DNA vs. Total Genomic DNA
2.4. AMR Genes Detected in Total Genomic DNA vs. Plasmid DNA
2.5. Phenotypic vs. Genotypic Antimicrobial Susceptibility
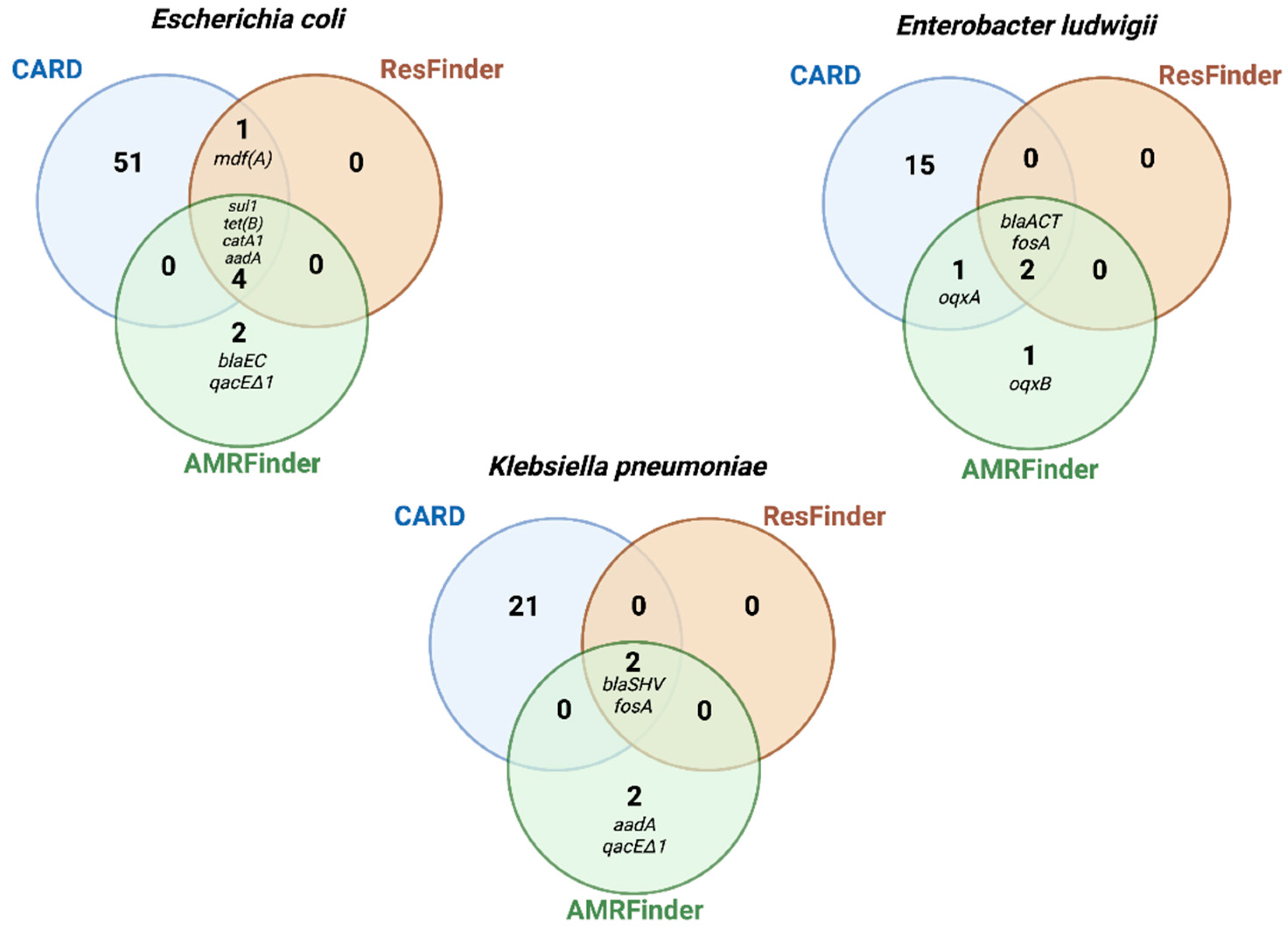
2.6. Comparison of AMR Genes Databases
2.7. Comparision of Sequencing Platforms and Assembly Approaches for Detection of AMR Genes
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains
4.2. Antimicrobial Susceptibility Testing
4.3. Extraction of Total Genomic DNA and Pure Plasmid DNA
4.4. Whole-Genome and Plasmid Sequencing
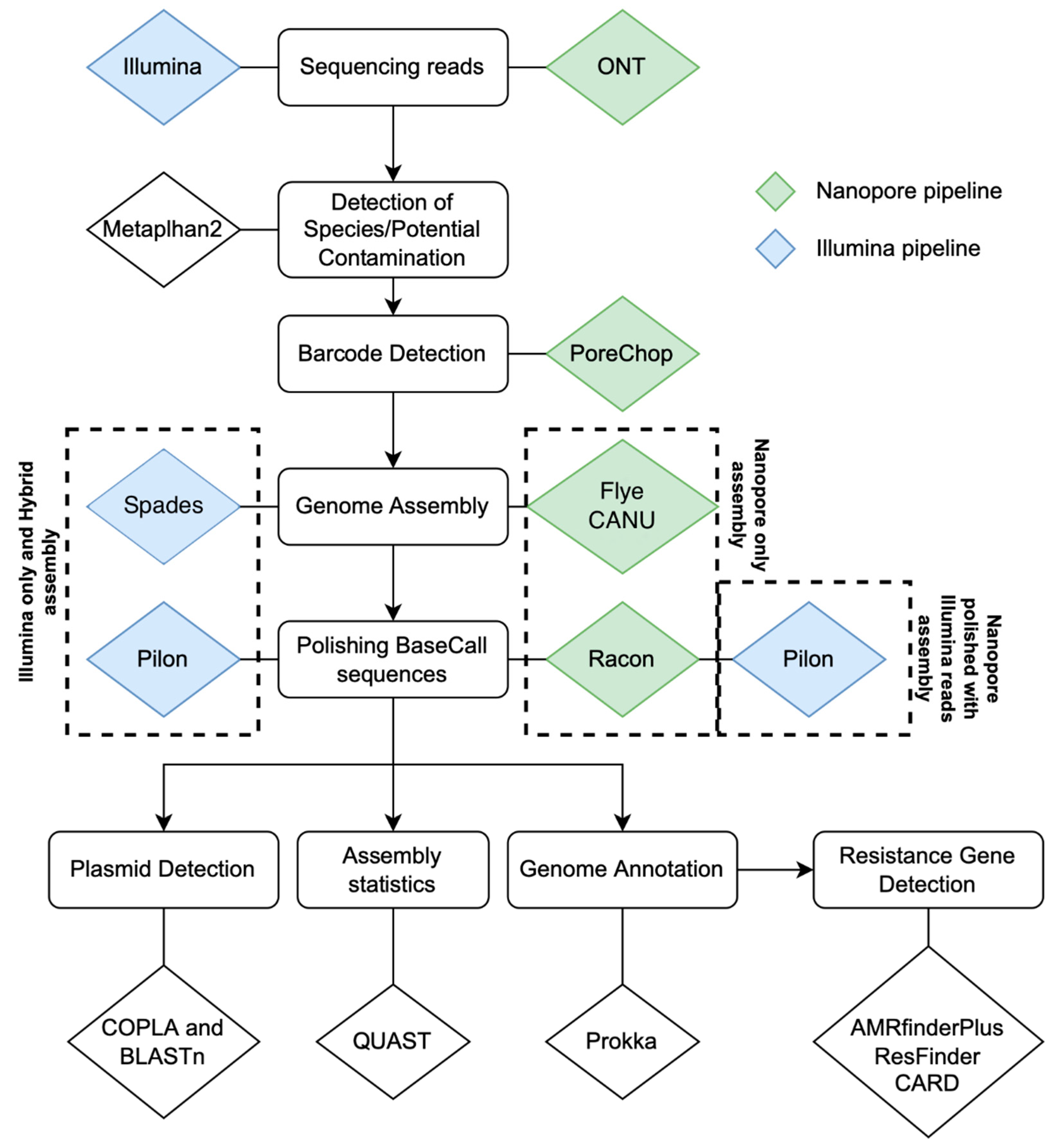
4.5. Contig Assembly and Data Analysis
4.6. Identification of AMR Genes in Whole Genome or Pure Plasmid Sequences
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khan, Z.A.; Siddiqui, M.F.; Park, S. Current and emerging methods of antibiotic susceptibility testing. Diagnostics 2019, 9, 49. [Google Scholar] [CrossRef] [Green Version]
- Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing; Eighteenth informational supplement; CLSI document M100-18; CLS: Wayne, PA, USA, 2018. [Google Scholar]
- The European Committee on Antimicrobial Susceptibility Testing. Breakpoint Tables for Interpretation of MICs and Zone Diameters. Version 9.0. 2019. Available online: http://www.eucast.org (accessed on 1 January 2020).
- Hendriksen, R.S.; Bortolaia, V.; Tate, H.; Tyson, G.H.; Aarestrup, F.; McDermott, P.F. Using genomics to track global antimicrobial resistance. Front. Public Health 2019, 7, 242. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Tyson, G.H.; Chen, Y.; Li, C.; Mukherjee, S.; Young, S.; Lam, C.; Folster, J.P.; Whichard, J.M.; McDermott, P.F. Whole-genome sequencing analysis accurately predicts antimicrobial resistance phenotypes in Campylobacter spp. Appl. Environ. Microbiol. 2016, 82, 459–466. [Google Scholar] [CrossRef] [Green Version]
- Lemon, J.K.; Khil, P.P.; Frank, K.M.; Dekker, J.P. Rapid nanopore sequencing of plasmids and resistance gene detection in clinical isolates. J. Clin. Microbiol. 2017, 55, 3530–3543. [Google Scholar] [CrossRef] [Green Version]
- Golparian, D.; Donà, V.; Sánchez-Busó, L.; Förster, S.; Harris, S.; Endimiani, A.; Low, N.; Unemo, M. Antimicrobial resistance prediction and phylogenetic analysis of Neisseria gonorrhoeae isolates using the Oxford Nanopore MinION sequencer. Sci. Rep. 2018, 8, 17596. [Google Scholar] [CrossRef] [Green Version]
- Tamma, P.D.; Fan, Y.; Bergman, Y.; Pertea, G.; Kazmi, A.Q.; Lewis, S.; Carroll, K.C.; Schatz, M.C.; Timp, W.; Simner, P.J. Applying Rapid Whole-Genome Sequencing to Predict Phenotypic Antimicrobial Susceptibility Testing Results among Carbapenem-Resistant Klebsiella pneumoniae Clinical Isolates. Antimicrob. Agents Chemother. 2019, 63, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, L.; Steglich, M.; Fuchs, S.; Werner, G.; Nübel, U. Comparison of six commercial kits to extract bacterial chromosome and plasmid DNA for MiSeq sequencing. Sci. Rep. 2016, 6, 28063. [Google Scholar] [CrossRef] [Green Version]
- Delaney, S.; Murphy, R.; Walsh, F. A comparison of methods for the extraction of plasmids capable of conferring antibiotic resistance in a human pathogen from complex broiler cecal samples. Front. Microbiol. 2018, 9, 1731. [Google Scholar] [CrossRef]
- Nouws, S.; Bogaerts, B.; Verhaegen, B.; Denayer, S.; Piérard, D.; Marchal, K.; Roosens, N.H.C.; Vanneste, K.; De Keersmaecker, S.C.J. Impact of DNA extraction on Whole Genome Sequencing analysis for characterization and relatedness of Shiga toxin-producing Escherichia coli isolates. Sci. Rep. 2020, 10, 14649. [Google Scholar] [CrossRef]
- De Maio, N.; Shaw, L.P.; Hubbard, A.; George, S.; Sanderson, N.D.; Swann, J.; Wick, R.; AbuOun, M.; Stubberfield, E.; Hoosdally, S.J.; et al. Comparison of long-read sequencing technologies in the hybrid assembly of complex bacterial genomes. Microb. Genom. 2019, 5, e000294. [Google Scholar] [CrossRef]
- Petersen, L.M.; Martin, I.W.; Moschetti, W.E.; Kershaw, C.M.; Tsongalis, G.J. Third-generation sequencing in the clinical laboratory: Exploring the advantages and challenges of nanopore sequencing. J. Clin. Microbiol. 2019, 58, e01315-19. [Google Scholar] [CrossRef]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef]
- Feldgarden, M.; Brover, V.; Haft, D.H.; Prasad, A.B.; Slotta, D.J.; Tolstoy, I.; Tyson, G.H.; Zhao, S.; Hsu, C.H.; McDermott, P.F.; et al. Validating the AMRFinder tool and resistance gene database by using antimicrobial resistance genotype-phenotype correlations in a collection of isolates. Antimicrob. Agents Chemother. 2019, 63, e00483-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papp, M.; Solymosi, N. Review and Comparison of Antimicrobial Resistance Gene Databases. Antibiotics 2022, 11, 339. [Google Scholar] [CrossRef]
- Williams, L.E.; Detter, C.; Barry, K.; Lapidus, A.; Summers, A.O. Facile recovery of individual high-molecular-weight, low-copy-number natural plasmids for genomic sequencing. Appl. Environ. Microbiol. 2006, 72, 4899–4906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef] [PubMed]
- Truong, D.T.; Franzosa, E.A.; Tickle, T.L.; Scholz, M.; Weingart, G.; Pasolli, E.; Tett, A.; Huttenhower, C.; Segata, N. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 2015, 12, 902–903. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Antipov, D.; Hartwick, N.; Shen, M.; Raiko, M.; Lapidus, A.; Pevzner, P.A. plasmidSPAdes: Assembling plasmids from whole genome sequencing data. bioRxiv 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [Green Version]
- Redondo-Salvo, S.; Bartomeus-Peñalver, R.; Vielva, L.; Tagg, K.A.; Webb, H.E.; Fernández-López, R.; de la Cruz, F. COPLA, a taxonomic classifier of plasmids. BMC Bioinform. 2021, 22, 390. [Google Scholar] [CrossRef] [PubMed]
- Pasquali, F.; Valle, I.D.; Palma, F.; Remondini, D.; Manfreda, G.; Castellani, G.; Hendriksen, R.; De Cesare, A. Application of different DNA extraction procedures, library preparation protocols and sequencing platforms: Impact on sequencing results. Heliyon 2019, 5, e02745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogaerts, B.; Delcourt, T.; Soetaert, K.; Boarbi, S.; Ceyssens, P.J.; Winand, R.; Van Braekel, J.; De Keersmaecker, S.C.; Roosens, N.H.; Marchal, K.; et al. A Bioinformatics Whole-Genome Sequencing Workflow for Clinical Mycobacterium tuberculosis Complex Isolate Analysis, Validated Using a Reference Collection Extensively Characterized with Conventional Methods and In Silico Approaches. J. Clin. Microbiol. 2021, 59, e00202-21. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Antipov, D.; Korobeynikov, A.; McLean, J.S.; Pevzner, P.A. hybridSPAdes: An algorithm for hybrid assembly of short and long reads. Bioinformatics 2016, 32, 1009–1015. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [Green Version]
- Oliveros, J.C. VENNY. An Interactive Tool for Comparing Lists with Venn Diagrams. 2007. Available online: https://bioinfogp.cnb.csic.es/tools/venny (accessed on 10 July 2021).




{kind=link}
{kind=link}
{kind=link}
| Strain | Plasmid Size (kb) a | Nanopore- Only b Contig (bp) | Nanopore- Polished c Contig (bp) | Illumina-Only d Contig (bp) | Illumina-Hybrid e Contig (bp) |
|---|---|---|---|---|---|
| Pure Plasmid DNA | |||||
| E. coli DU1040 (NR1) | 94.289 | Several contigs f | 93,868 | Several contigs | 94,308 |
| K. pneumoniae | 232 | 313,736 | 313,014 | Several contigs | Several |
| LST1504-C2 | 153 | 265,010 | 265,397 | Several contigs | Several |
| 71 | 58,149 | 58,214 | Several contigs | Several | |
| 6 | 3380 | 3378 | 3003 | 6261 | |
| E. ludwigii LST1391B | 158.6 | 133,397 | 133,378 | 130,070 | 130,070 |
| 7 | 5187 | 5152 | 5283 | 5152 | |
| Total Genomic DNA | |||||
| E. coli DU1040 (NR1) | 94.289 | 94,296 | 94,410 | Several contigs | 93,656 |
| K. pneumoniae | 232 | 313,645 | 312,971 | Several contigs | 314,384 |
| LST1504-C2 | 153 | 264,823 | 264,679 | Several contigs | Several |
| 71 | 58,157 | 58,118 | Several contigs | Several | |
| 6 | 3350 | 3326 | 2930 | 2930 | |
| E. ludwigii LST1391B | 158.6 | 133,990 | 133,333 | 130,070 | 130,070 |
| 7 | 6174 | 6174 | Several contigs | 5033 | |
| ResFinder | AMRFinder | ||
|---|---|---|---|
| Total Genomic | Pure Plasmid | Total Genomic | Pure Plasmid |
| Escherichia coli DU1040 (NR1) | |||
| aadA1 | aadA1 | aadA1 | aadA1 |
| catA1 | catA1 | catA1 | catA1 |
| sul1 | sul1 | sul1 | sul1 |
| tet(B) | tet(B) | tet(B) | tet(B) |
| mdfA | qacEdelta1 | qacEdelta1 | |
| blaEC | |||
| Enterobacter ludwigii LST1391B | |||
| blaACT-12 | blaACT-12 | blaACT | blaACT |
| fosA2 | fosA2 | fosA2 | fosA2 |
| oqxA | oqxA | ||
| oqxB | oqxB | ||
| Klebsiella pneumoniae LST1504-C2 | |||
| blaSHV-40 | Not identified a | blaSHV | Not identified b |
| fosA | fosA | ||
| aadA1 | |||
| qacEdelta1 | |||
| Antimicrobial | E. coli DU1040 (NR1) | E. ludwigii LST1391B | K. pneumoniae LST1504-C2 | |||
|---|---|---|---|---|---|---|
| MIC Vitek-2 | MIC Sensititre | MIC Vitek-2 | MIC Sensititre | MIC Vitek-2 | MIC Sensititre | |
| Ampicillin | 8 (S) | ≤8 (S) | NI | >16 (R) | 16 (R) | >16 (R) |
| Amox./Clavulanic Acid | 4 (S) | 4 (S) | ≥32 (R) | 2 (R) | ≤2 S | >8 (R) |
| Piperacillin/Tazobactam | ≤4 (S) | ≤8 (S) | ≤4 (S) | ≤8 (S) | ≤4 (S) | ≤8 (S) |
| Cephalexin | 16 (R) | 16 (R) | ≥64 (R) | 8 (R) | ≥16 (R) | >16 (R) |
| Ceftriaxone | ≤1 (S) | ≤0.5 (S) | ≤1 (S) | ≤0.5 (S) | ≤1 (S) | ≤0.5 (S) |
| Cefazolin | ≤4 (S) | 4 (S) | ≥64 (R) | 2 (R) | 32 (R) | >32 (R) |
| Cefepime | ≤1 (S) | ≤4 (S) | ≤1 (S) | ≤4 (S) | ≤1 (S) | ≤4 (S) |
| Ceftazidime | ≤1 (S) | ≤1 (S) | ≤1 (S) | ≤1 (S) | ≤1 (S) | ≤1 (S) |
| Ciprofloxacin | ≤0.25 (S) | ≤0.5 (S) | ≤0.25 (S) | ≤0.5 (S) | ≤0.25 (S) | ≤0.5 (S) |
| Levofloxacin | ≤0.12 (S) | ≤1 (S) | ≤0.12 (S) | ≤1 (S) | ≤0.12 (S) | ≤1 (S) |
| Enrofloxacin | 0.5 (S) | 0.25 (S) | ≤0.12 (S) | ≤0.125 (S) | ≤0.12 (S) | ≤0.125 (S) |
| Gentamicin | ≤1 (S) | ≤0.5 (S) | ≤1 (S) | ≤0.5 (S) | ≤1 (S) | ≤1 (S) |
| Amikacin | ≤2 (S) | ≤4 (S) | ≤2 (S) | ≤4 (S) | ≤2 (S) | ≤4 (S) |
| Doxycycline | ≥16 (R) | ≥8 (R) | 4 (S) | 2 (S) | 1 (S) | 4 (S) |
| Tetracycline | ≥16 (R) | ≥16 (R) | ≤4 (S) | ≤4 (S) | 4 (S) | ≤4 (S) |
| Chloramphenicol | ≥64 (R) | ≥32 (R) | 16 (I) | 4 (S) | ≤2 (S) | 2 (S) |
| Nitrofurantoin | ≤16 (S) | ≤32 (S) | 32 (S) | ≤32 (S) | ≤16 (S) | ≤32 (S) |
| Trim./Sulfamethoxazole | ≤2 (S) | ≤0.5 (S) | ≤20 (S) | ≤0.5 (S) | ≤20 (S) | ≤0.5 (S) |
| Ertapenem | ≤0.5 (S) | ≤0.25 (S) | ≤0.5 (S) | ≤0.25 (S) | ≤5 (S) | ≤0.25 (S) |
| Imipenem | ≤0.25 (S) | ≤0.5 (S) | 0.5 (S) | ≤0.5 (S) | ≤0.25 (S) | ≤0.5 (S) |
| Meropenem | ≤0.25 (S) | ≤0.5 (S) | ≤0.25 (S) | ≤0.5 (S) | ≤0.25 (S) | ≤0.5 (S) |
| Strain | Resistance Phenotype b | Antibiotic Class | Genes Detected by | ||
|---|---|---|---|---|---|
| ResFinder | AMRFinder | CARD | |||
| E. coli DU1040 (NR1) | Cefalexin | Beta-lactam | ND | blaEC | Amp-C and Amp-H Beta-lactamases |
| Doxycycline | Tetracycline | tetB, mdf(A) | tetB | tetB | |
| Tetracycline | Tetracycline | tetB, mdf(A) | tetB | tetB, mdf(A) | |
| Chloramphenicol | Phenicol | catA1, mdf(A) | catA1 | catA1, mdf(A) | |
| E. ludwigiia LST1391B | Ampicillin | Beta-lactam | blaACT-12 | blaACT | blaACT-12 and Amp-H Beta-lactamases |
| Amoxicillin/clavulanic acid | Beta-lactam | blaACT-12 | blaACT | blaACT-12 and Amp-H Beta-lactamases | |
| Cefazolin | Beta-lactam | blaACT-12 | blaACT | blaACT-12 and Amp-H Beta-lactamases | |
| Cefalexin | Beta-lactam | blaACT-12 | blaACT-12 | blaACT-12 and Amp-H Beta-lactamases | |
| Chloramphenicol | Phenicol | ND | oqxA, oqxB | ND | |
| K. pneumoniae a LST1504-C2 | Ampicillin | Beta-lactam | blaSHV-40 | blaSHV | blaSHV-40 and Amp-H Beta-lactamases |
| Amoxicillin/clavulanic acid | Beta-lactam | blaSHV-40 | blaSHV | blaSHV-40 and Amp-H Beta-lactamases | |
| Cefazolin | Beta-lactam | blaSHV-40 | blaSHV | blaSHV-40 and Amp-H Beta-lactamases | |
| Cephalexin | Beta-lactam | blaSHV-40 | blaSHV | blaSHV-40 and Amp-H Beta-lactamases | |
| ASSEMBLY a > | Nanopore-Only | Nanopore-Polished | Illumina-Only | Illumina-Hybrid | Nanopore-Only | Nanopore-Polished | Illumina-Only | Illumina-Hybrid | |
|---|---|---|---|---|---|---|---|---|---|
| DATABASE> | ResFinder database | AMRFinder database | |||||||
| AMR GENE b | BACTERIUM | ||||||||
| E. coli DU1040 (control, NR1) | |||||||||
| aadA1 | + | + | + | + | + | + | + | + | |
| blaEC | ND | ND | ND | ND | + | + | + | + | |
| catA1 | + | + | + | + | + | + | + | + | |
| mdf(A) | + | + | + | + | ND | ND | ND | ND | |
| qacEΔ1 | NA | NA | NA | NA | + | + | + | + | |
| sul1 | + | + | + | + | ND | + | + | + | |
| tet(B) | + | + | + | + | ND | + | + | + | |
| E. ludwigii LST1391B | |||||||||
| blaACT-12 | + | + | + | + | + | + | + | + | |
| fosA | + | + | + | + | + | + | + | + | |
| oqxA | ND | ND | ND | ND | ND | + | + | + | |
| oqxB | ND | ND | ND | ND | ND | + | + | + | |
| K. pneumoniae LST1504-C2 | |||||||||
| aadA1 | ND | ND | ND | ND | ND | ND | + | + | |
| blaSHV | + | + | + | + | + | + | + | + | |
| fosA | + | + | + | + | + | + | + | + | |
| qacEΔ1 | NA | NA | NA | NA | ND | ND | + | + | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maboni, G.; Baptista, R.d.P.; Wireman, J.; Framst, I.; Summers, A.O.; Sanchez, S. Three Distinct Annotation Platforms Differ in Detection of Antimicrobial Resistance Genes in Long-Read, Short-Read, and Hybrid Sequences Derived from Total Genomic DNA or from Purified Plasmid DNA. Antibiotics 2022, 11, 1400. https://doi.org/10.3390/antibiotics11101400
Maboni G, Baptista RdP, Wireman J, Framst I, Summers AO, Sanchez S. Three Distinct Annotation Platforms Differ in Detection of Antimicrobial Resistance Genes in Long-Read, Short-Read, and Hybrid Sequences Derived from Total Genomic DNA or from Purified Plasmid DNA. Antibiotics. 2022; 11(10):1400. https://doi.org/10.3390/antibiotics11101400
Chicago/Turabian StyleMaboni, Grazieli, Rodrigo de Paula Baptista, Joy Wireman, Isaac Framst, Anne O. Summers, and Susan Sanchez. 2022. "Three Distinct Annotation Platforms Differ in Detection of Antimicrobial Resistance Genes in Long-Read, Short-Read, and Hybrid Sequences Derived from Total Genomic DNA or from Purified Plasmid DNA" Antibiotics 11, no. 10: 1400. https://doi.org/10.3390/antibiotics11101400