Molecular Mechanisms and Clinical Impact of Acquired and Intrinsic Fosfomycin Resistance

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
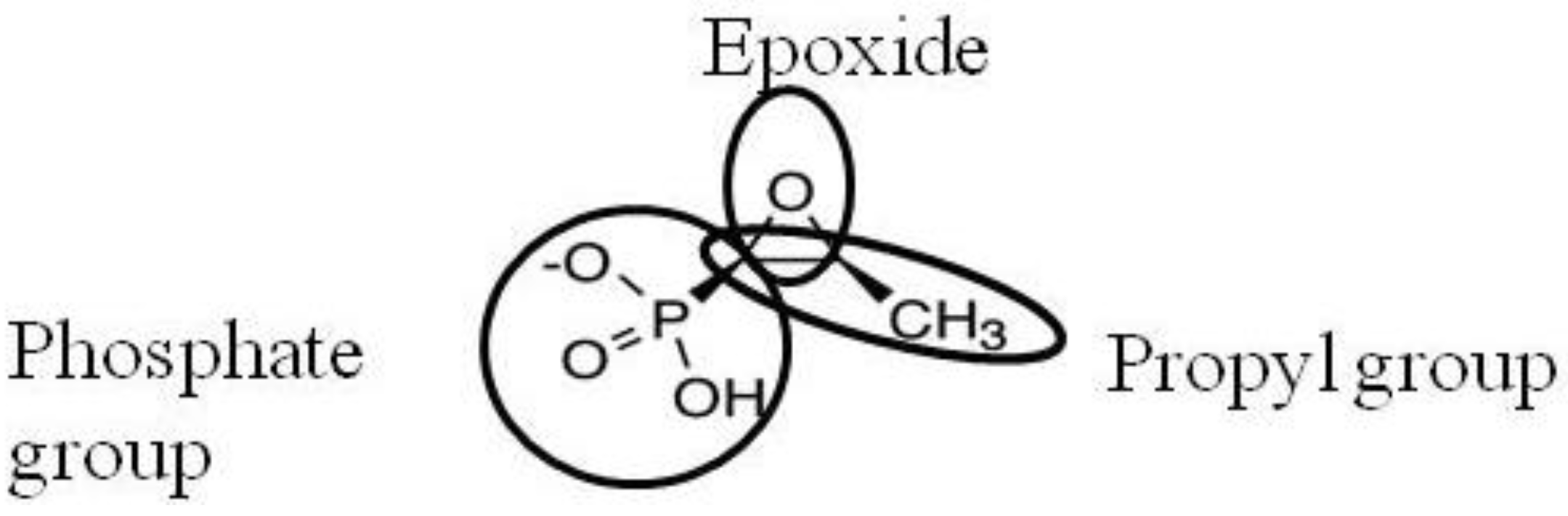
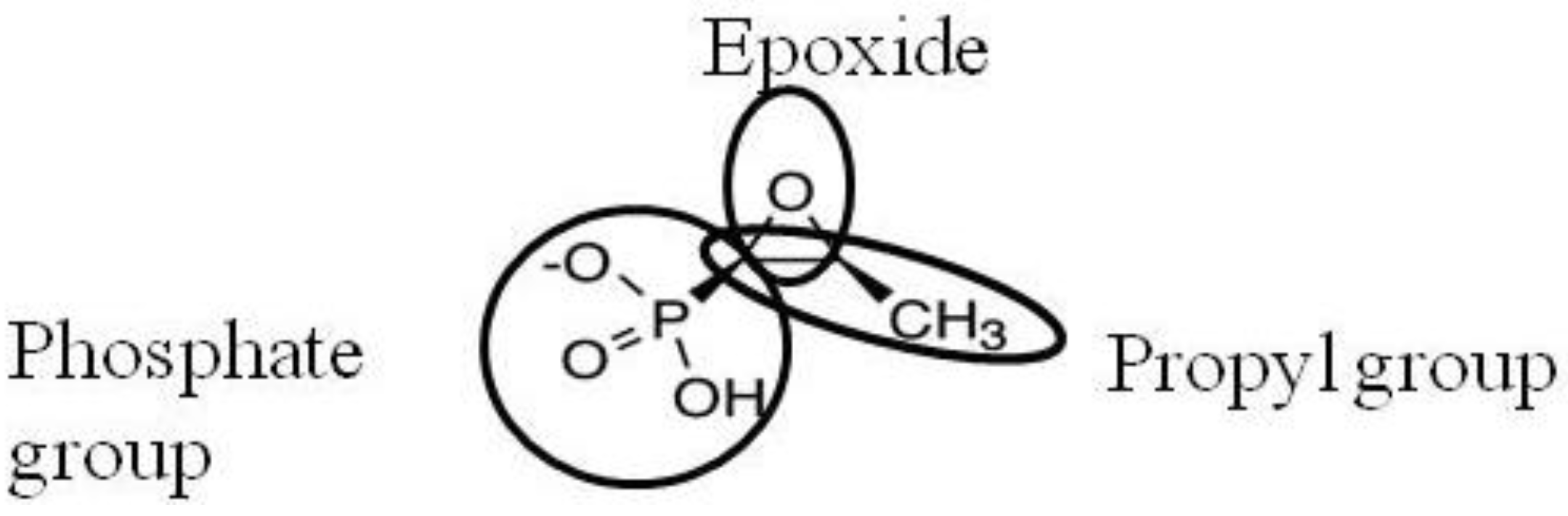
2. Activity
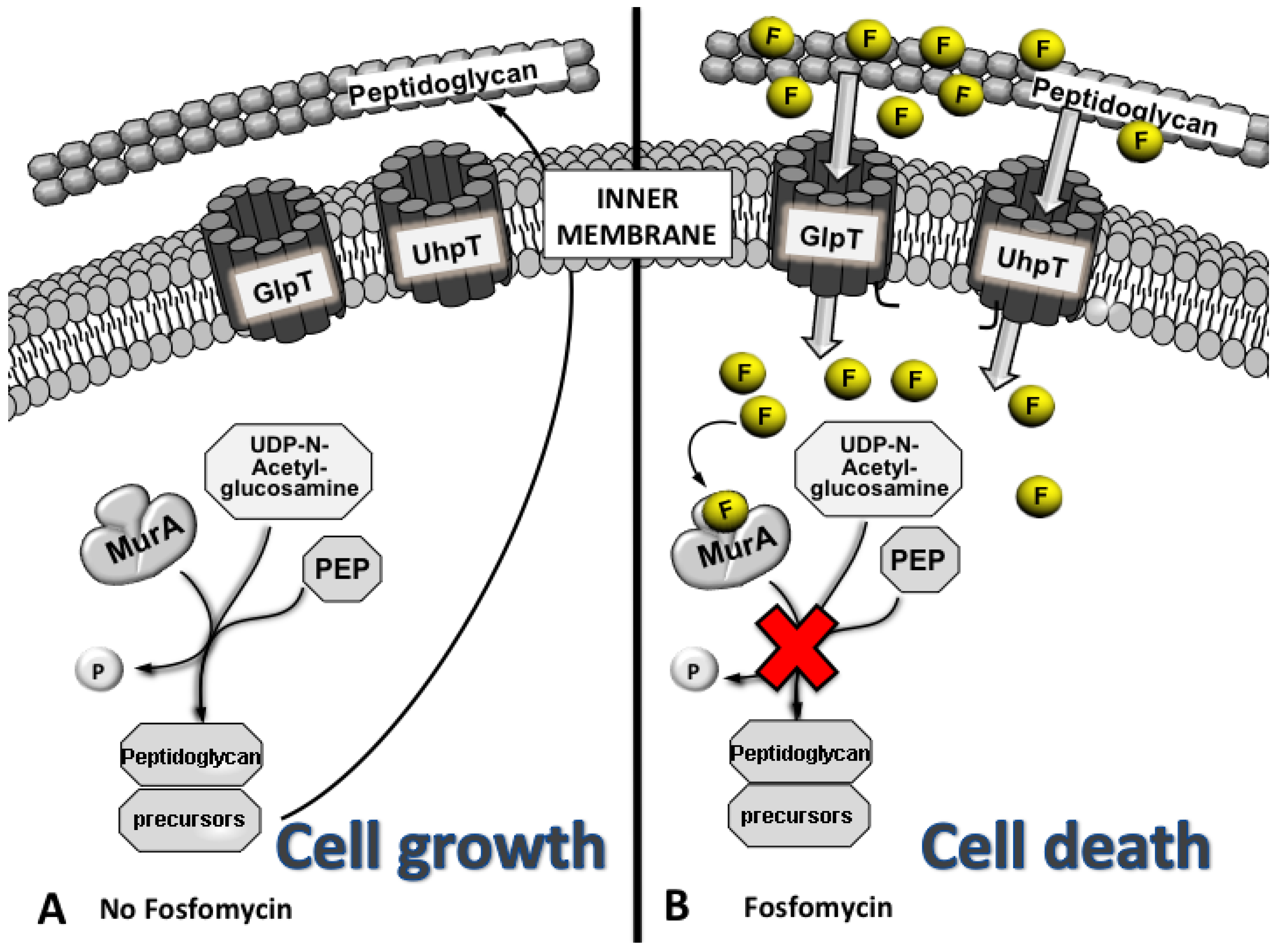
3. Mechanism of Action
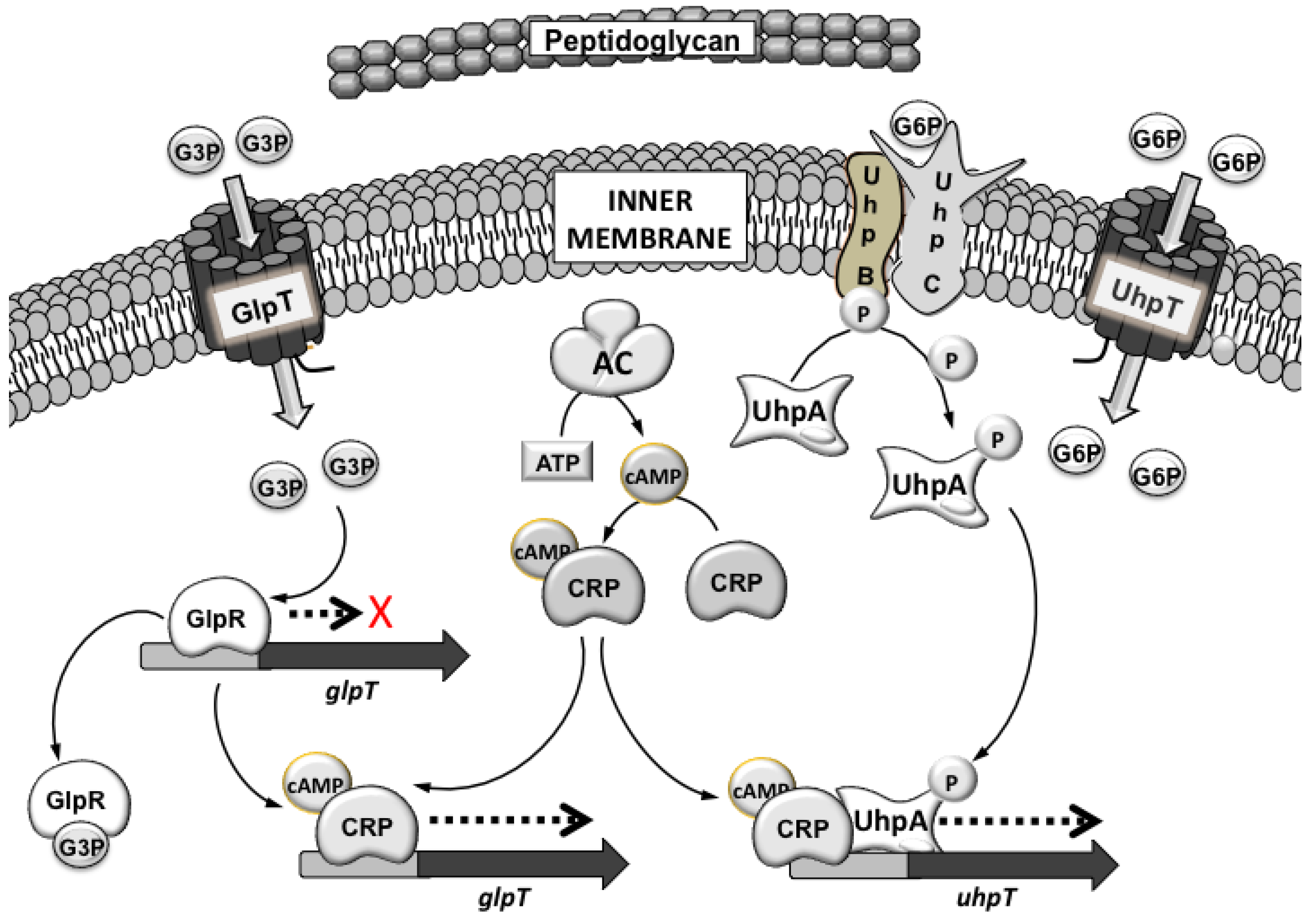
3.1. Mechanisms of Fosfomycin Resistance
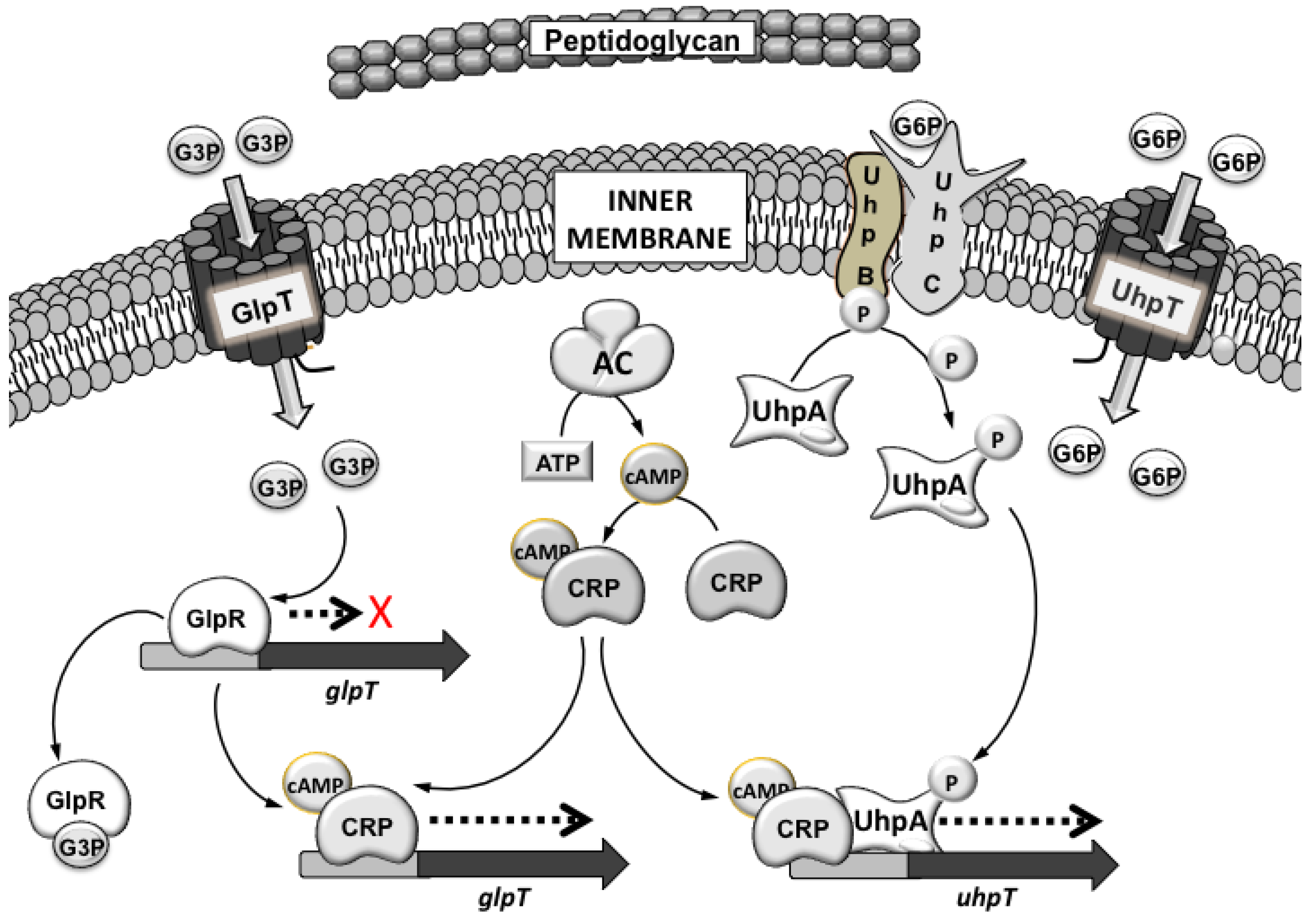

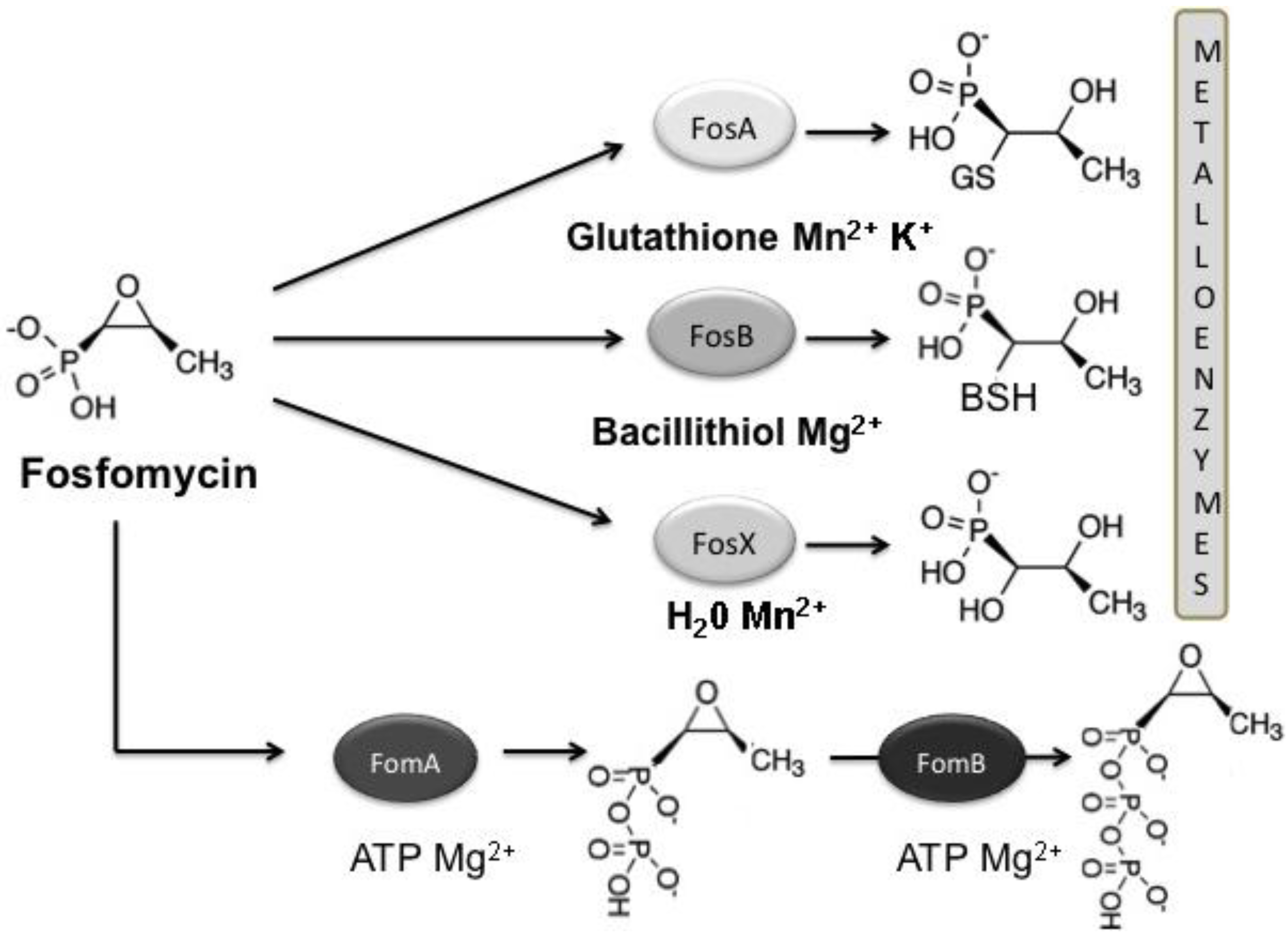
4. Clinical Impact
5. Conclusions
Acknowledgements
Conflict of Interest
References and Notes
- Nathan, C. Antibiotics at the crossroads. Nature 2004, 431, 899–902. [Google Scholar] [CrossRef]
- Livermore, D.M. Has the era of untreatable infections arrived? J. Antimicrob. Chemother. 2009, 64 (Suppl. 1), 29–36. [Google Scholar] [CrossRef]
- Falagas, M.E.; Grammatikos, A.P.; Michalopoulos, A. Potential of old-generation antibiotics to address current need for new antibiotics. Expert Rev. Anti. Infect. Ther. 2008, 6, 593–600. [Google Scholar]
- Hendlin, D.; Stapley, E.O.; Jackson, M.; Wallick, H.; Miller, A.K.; Wolf, F.J.; Miller, T.W.; Chaiet, L.; Kahan, F.M.; Foltz, E.L.; et al. Phosphonomycin, a new antibiotic produced by strains of Streptomyces. Science 1969, 166, 122–123. [Google Scholar]
- Kahan, F.M.; Kahan, J.S.; Cassidy, P.J.; Kropp, H. The mechanism of action of fosfomycin (phosphonomycin). Ann. NY Acad. Sci. 1974, 235, 364–386. [Google Scholar]
- Wagenlehner, F.M.; Hoyme, U.; Kaase, M.; Funfstuck, R.; Naber, K.G.; Schmiemann, G. Uncomplicated urinary tract infections. Dtsch. Arztebl. Int. 2011, 108, 415–423. [Google Scholar]
- Schito, G.C. Why fosfomycin trometamol as first line therapy for uncomplicated UTI? Int. J. Antimicrob. Agents 2003, 22 (Suppl. 2), 79–83. [Google Scholar] [CrossRef]
- Falagas, M.E.; Giannopoulou, K.P.; Kokolakis, G.N.; Rafailidis, P.I. Fosfomycin: Use beyond urinary tract and gastrointestinal infections. Clin. Infect. Dis. 2008, 46, 1069–1077. [Google Scholar]
- Barry, A.L.; Brown, S.D. Antibacterial spectrum of fosfomycin trometamol. J. Antimicrob. Chemother. 1995, 35, 228–230. [Google Scholar] [CrossRef]
- Lu, C.L.; Liu, C.Y.; Huang, Y.T.; Liao, C.H.; Teng, L.J.; Turnidge, J.D.; Hsueh, P.R. Antimicrobial susceptibilities of commonly encountered bacterial isolates to fosfomycin determined by agar dilution and disk diffusion methods. Antimicrob. Agents Chemother. 2011, 55, 4295–4301. [Google Scholar] [CrossRef]
- Falagas, M.E.; Kastoris, A.C.; Karageorgopoulos, D.E.; Rafailidis, P.I. Fosfomycin for the treatment of infections caused by multidrug-resistant non-fermenting gram-negative bacilli: A systematic review of microbiological, animal and clinical studies. Int. J. Antimicrob. Agents 2009, 34, 111–120. [Google Scholar] [CrossRef]
- Neuner, E.A.; Sekeres, J.; Hall, G.S.; van Duin, D. Experience with fosfomycin for treatment of urinary tract infections due to multidrug-resistant organisms. Antimicrob. Agents Chemother. 2012, 56, 5744–5748. [Google Scholar]
- Falagas, M.E.; Kastoris, A.C.; Kapaskelis, A.M.; Karageorgopoulos, D.E. Fosfomycin for the treatment of multidrug-resistant, including extended-spectrum beta-lactamase producing, Enterobacteriaceae infections: A systematic review. Lancet Infect. Dis. 2010, 10, 43–50. [Google Scholar] [CrossRef]
- Brown, E.D.; Vivas, E.I.; Walsh, C.T.; Kolter, R. MurA (MurZ), the enzyme that catalyzes the first committed step in peptidoglycan biosynthesis, is essential in Escherichia coli. J. Bacteriol. 1995, 177, 4194–4197. [Google Scholar]
- Marquardt, J.L.; Brown, E.D.; Lane, W.S.; Haley, T.M.; Ichikawa, Y.; Wong, C.H.; Walsh, C.T. Kinetics, stoichiometry, and identification of the reactive thiolate in the inactivation of UDP-GlcNAc enolpyruvoyl transferase by the antibiotic fosfomycin. Biochemistry 1994, 33, 10646–10651. [Google Scholar]
- Skarzynski, T.; Mistry, A.; Wonacott, A.; Hutchinson, S.E.; Kelly, V.A.; Duncan, K. Structure of UDP-N-acetylglucosamine enolpyruvyl transferase, an enzyme essential for the synthesis of bacterial peptidoglycan, complexed with substrate UDP-N-acetylglucosamine and the drug fosfomycin. Structure 1996, 4, 1465–1474. [Google Scholar] [CrossRef]
- Kadner, R.J.; Winkler, H.H. Isolation and characterization of mutations affecting the transport of hexose phosphates in Escherichia coli. J. Bacteriol. 1973, 113, 895–900. [Google Scholar]
- Tsuruoka, T.; Yamada, Y. Charactertization of spontaneous fosfomycin (phosphonomycin)-resistant cells of Escherichia coli B in vitro. J. Antibiot. (Tokyo) 1975, 28, 906–911. [Google Scholar] [CrossRef]
- Grimm, H. In vitro investigations with fosfomycin on Mueller-Hinton agar with and without glucose-6-phosphate. Infection 1979, 7, 256–259. [Google Scholar]
- Castaneda-Garcia, A.; Rodriguez-Rojas, A.; Guelfo, J.R.; Blazquez, J. The glycerol-3-phosphate permease GlpT is the only fosfomycin transporter in Pseudomonas aeruginosa. J. Bacteriol. 2009, 191, 6968–6974. [Google Scholar] [CrossRef]
- Schweizer, H.P.; Po, C. Regulation of glycerol metabolism in Pseudomonas aeruginosa: Characterization of the glpR repressor gene. J. Bacteriol. 1996, 178, 5215–5221. [Google Scholar]
- Scortti, M.; Lacharme-Lora, L.; Wagner, M.; Chico-Calero, I.; Losito, P.; Vazquez-Boland, J.A. Coexpression of virulence and fosfomycin susceptibility in Listeria: Molecular basis of an antimicrobial in vitro-in vivo paradox. Nat. Med. 2006, 12, 515–517. [Google Scholar] [CrossRef]
- Chico-Calero, I.; Suarez, M.; Gonzalez-Zorn, B.; Scortti, M.; Slaghuis, J.; Goebel, W.; Vazquez-Boland, J.A. Hpt, a bacterial homolog of the microsomal glucose-6-phosphate translocase, mediates rapid intracellular proliferation in Listeria. Proc. Natl. Acad. Sci. USA 2002, 99, 431–436. [Google Scholar]
- Lemieux, M.J.; Huang, Y.; Wang, D.N. Glycerol-3-phosphate transporter of Escherichia coli: Structure, function and regulation. Res. Microbiol. 2004, 155, 623–629. [Google Scholar] [CrossRef]
- Huang, Y.; Lemieux, M.J.; Song, J.; Auer, M.; Wang, D.N. Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science 2003, 301, 616–620. [Google Scholar] [CrossRef]
- Lemieux, M.J.; Huang, Y.; Wang, D.N. The structural basis of substrate translocation by the Escherichia coli glycerol-3-phosphate transporter: A member of the major facilitator superfamily. Curr. Opin. Struct. Biol. 2004, 14, 405–412. [Google Scholar] [CrossRef]
- Elvin, C.M.; Hardy, C.M.; Rosenberg, H. Pi exchange mediated by the glpt-dependent sn-glycerol-3-phosphate transport system in Escherichia coli. J. Bacteriol. 1985, 161, 1054–1058. [Google Scholar]
- Hardisson, C.; Llaneza, J. The action of fosfomycin on the growth of Pseudomonas aeruginosa. Chemotherapy 1977, 23 (Suppl. 1), 37–44. [Google Scholar] [CrossRef]
- Lindgren, V. Mapping of a genetic locus that affects glycerol 3-phosphate transport in Bacillus subtilis. J. Bacteriol. 1978, 133, 667–670. [Google Scholar]
- Santoro, A.; Cappello, A.R.; Madeo, M.; Martello, E.; Iacopetta, D.; Dolce, V. Interaction of fosfomycin with the glycerol 3-phosphate transporter of Escherichia coli. Biochim. Biophys. Acta 2011, 1810, 1323–1329. [Google Scholar] [CrossRef]
- Larson, T.J.; Ye, S.Z.; Weissenborn, D.L.; Hoffmann, H.J.; Schweizer, H. Purification and characterization of the repressor for the sn-glycerol 3-phosphate regulon of Escherichia coli K-12. J. Biol. Chem. 1987, 262, 15869–15874. [Google Scholar]
- Yang, B.; Gerhardt, S.G.; Larson, T.J. Action at a distance for glp repressor control of glpTQ transcription in Escherichia coli K-12. Mol. Microbiol. 1997, 24, 511–521. [Google Scholar] [CrossRef]
- Zeng, G.; Ye, S.; Larson, T.J. Repressor for the sn-glycerol 3-phosphate regulon of Escherichia coli K-12: Primary structure and identification of the DNA-binding domain. J. Bacteriol. 1996, 178, 7080–7089. [Google Scholar]
- Kadner, R.J.; Shattuck-Eidens, D.M. Genetic control of the hexose phosphate transport system of Escherichia coli: Mapping of deletion and insertion mutations in the uhp region. J. Bacteriol. 1983, 155, 1052–1061. [Google Scholar]
- Sonna, L.A.; Ambudkar, S.V.; Maloney, P.C. The mechanism of glucose 6-phosphate transport by Escherichia coli. J. Biol. Chem. 1988, 263, 6625–6630. [Google Scholar]
- Eiglmeier, K.; Boos, W.; Cole, S.T. Nucleotide sequence and transcriptional startpoint of the glpT gene of Escherichia coli: Extensive sequence homology of the glycerol-3-phosphate transport protein with components of the hexose-6-phosphate transport system. Mol. Microbiol. 1987, 1, 251–258. [Google Scholar] [CrossRef]
- Ambudkar, S.V.; Anantharam, V.; Maloney, P.C. UhpT, the sugar phosphate antiporter of Escherichia coli, functions as a monomer. J. Biol. Chem. 1990, 265, 12287–12292. [Google Scholar]
- Lloyd, A.D.; Kadner, R.J. Topology of the Escherichia coli uhpT sugar-phosphate transporter analyzed by using TnphoA fusions. J. Bacteriol. 1990, 172, 1688–1693. [Google Scholar]
- Island, M.D.; Kadner, R.J. Interplay between the membrane-associated UhpB and UhpC regulatory proteins. J. Bacteriol. 1993, 175, 5028–5034. [Google Scholar]
- Chen, Q.; Kadner, R.J. Effect of altered spacing between uhpT promoter elements on transcription activation. J. Bacteriol. 2000, 182, 4430–4436. [Google Scholar] [CrossRef]
- Dahl, J.L.; Wei, B.Y.; Kadner, R.J. Protein phosphorylation affects binding of the Escherichia coli transcription activator UhpA to the uhpT promoter. J. Biol. Chem. 1997, 272, 1910–1919. [Google Scholar]
- Olekhnovich, I.N.; Kadner, R.J. Mutational scanning and affinity cleavage analysis of UhpA-binding sites in the Escherichia coli uhpT promoter. J. Bacteriol. 2002, 184, 2682–2691. [Google Scholar] [CrossRef]
- Alper, M.D.; Ames, B.N. Transport of antibiotics and metabolite analogs by systems under cyclic AMP control: Positive selection of Salmonella typhimurium cya and crp mutants. J. Bacteriol. 1978, 133, 149–157. [Google Scholar]
- Cordaro, J.C.; Melton, T.; Stratis, J.P.; Atagun, M.; Gladding, C.; Hartman, P.E.; Roseman, S. Fosfomycin resistance: Selection method for internal and extended deletions of the phosphoenolpyruvate: Sugar phosphotransferase genes of Salmonella typhimurium. J. Bacteriol. 1976, 128, 785–793. [Google Scholar]
- Sakamoto, Y.; Furukawa, S.; Ogihara, H.; Yamasaki, M. Fosmidomycin resistance in adenylate cyclase deficient (cya) mutants of Escherichia coli. Biosci. Biotechnol. Biochem. 2003, 67, 2030–2033. [Google Scholar] [CrossRef]
- Tsuruoka, T.; Miyata, A.; Yamada, Y. Two kinds of mutants defective in multiple carbohydrate utilization isolated from in vitro fosfomycin-resistant strains of Escherichia coli K-12. J. Antibiot. (Tokyo) 1978, 31, 192–201. [Google Scholar] [CrossRef]
- Larson, T.J.; Cantwell, J.S.; van Loo-Bhattacharya, A.T. Interaction at a distance between multiple operators controls the adjacent, divergently transcribed glpTQ-glpACB operons of Escherichia coli K-12. J. Biol. Chem. 1992, 267, 6114–6121. [Google Scholar]
- Merkel, T.J.; Dahl, J.L.; Ebright, R.H.; Kadner, R.J. Transcription activation at the Escherichia coli uhpT promoter by the catabolite gene activator protein. J. Bacteriol. 1995, 177, 1712–1718. [Google Scholar]
- Olekhnovich, I.N.; Dahl, J.L.; Kadner, R.J. Separate contributions of UhpA and CAP to activation of transcription of the uhpT promoter of Escherichia coli. J. Mol. Biol. 1999, 292, 973–986. [Google Scholar] [CrossRef]
- Kim, D.H.; Lees, W.J.; Kempsell, K.E.; Lane, W.S.; Duncan, K.; Walsh, C.T. Characterization of a Cys115 to Asp substitution in the Escherichia coli cell wall biosynthetic enzyme UDP-GlcNAc enolpyruvyl transferase (MurA) that confers resistance to inactivation by the antibiotic fosfomycin. Biochemistry 1996, 35, 4923–4928. [Google Scholar]
- Brown, E.D.; Marquardt, J.L.; Lee, J.P.; Walsh, C.T.; Anderson, K.S. Detection and characterization of a phospholactoyl-enzyme adduct in the reaction catalyzed by UDP-N-acetylglucosamine enolpyruvoyl transferase, MurZ. Biochemistry 1994, 33, 10638–10645. [Google Scholar]
- Ramilo, C.; Appleyard, R.J.; Wanke, C.; Krekel, F.; Amrhein, N.; Evans, J.N. Detection of the covalent intermediate of UDP-N-acetylglucosamine enolpyruvyl transferase by solution-state and time-resolved solid-state NMR spectroscopy. Biochemistry 1994, 33, 15071–15079. [Google Scholar]
- Wanke, C.; Amrhein, N. Evidence that the reaction of the UDP-N-acetylglucosamine 1-carboxyvinyltransferase proceeds through the O-phosphothioketal of pyruvic acid bound to Cys115 of the enzyme. Eur. J. Biochem. 1993, 218, 861–870. [Google Scholar] [CrossRef]
- Eschenburg, S.; Priestman, M.; Schonbrunn, E. Evidence that the fosfomycin target Cys115 in UDP-N-acetylglucosamine enolpyruvyl transferase (MurA) is essential for product release. J. Biol. Chem. 2005, 280, 3757–3763. [Google Scholar] [CrossRef]
- De Smet, K.A.; Kempsell, K.E.; Gallagher, A.; Duncan, K.; Young, D.B. Alteration of a single amino acid residue reverses fosfomycin resistance of recombinant MurA from Mycobacterium tuberculosis. Microbiology 1999, 145, 3177–3184. [Google Scholar]
- Jiang, S.; Gilpin, M.E.; Attia, M.; Ting, Y.L.; Berti, P.J. Lyme disease enolpyruvyl-udp-glcnac synthase: Fosfomycin-resistant MurA from Borrelia burgdorferi, a fosfomycin-sensitive mutant, and the catalytic role of the active site Asp. Biochemistry 2011, 50, 2205–2212. [Google Scholar] [CrossRef]
- McCoy, A.J.; Sandlin, R.C.; Maurelli, A.T. In vitro and in vivo functional activity of Chlamydia MurA, a UDP-N-acetylglucosamine enolpyruvyl transferase involved in peptidoglycan synthesis and fosfomycin resistance. J. Bacteriol. 2003, 185, 1218–1228. [Google Scholar] [CrossRef]
- Venkateswaran, P.S.; Wu, H.C. Isolation and characterization of a phosphonomycin-resistant mutant of Escherichia coli K-12. J. Bacteriol. 1972, 110, 935–944. [Google Scholar]
- Takahata, S.; Ida, T.; Hiraishi, T.; Sakakibara, S.; Maebashi, K.; Terada, S.; Muratani, T.; Matsumoto, T.; Nakahama, C.; Tomono, K. Molecular mechanisms of fosfomycin resistance in clinical isolates of Escherichia coli. Int. J. Antimicrob. Agents 2010, 35, 333–337. [Google Scholar] [CrossRef]
- Marquardt, J.L.; Siegele, D.A.; Kolter, R.; Walsh, C.T. Cloning and sequencing of Escherichia coli MurZ and purification of its product, a UDP-N-acetylglucosamine enolpyruvyl transferase. J. Bacteriol. 1992, 174, 5748–5752. [Google Scholar]
- Couce, A.; Briales, A.; Rodriguez-Rojas, A.; Costas, C.; Pascual, A.; Blazquez, J. Genomewide overexpression screen for fosfomycin resistance in Escherichia coli: MurA confers clinical resistance at low fitness cost. Antimicrob. Agents Chemother. 2012, 56, 2767–2769. [Google Scholar] [CrossRef]
- Horii, T.; Kimura, T.; Sato, K.; Shibayama, K.; Ohta, M. Emergence of fosfomycin-resistant isolates of shiga-like toxin-producing Escherichia coli O26. Antimicrob. Agents Chemother. 1999, 43, 789–793. [Google Scholar]
- Rigsby, R.E.; Fillgrove, K.L.; Beihoffer, L.A.; Armstrong, R.N. Fosfomycin resistance proteins: A nexus of glutathione transferases and epoxide hydrolases in a metalloenzyme superfamily. Meth. Enzymol. 2005, 401, 367–379. [Google Scholar]
- Brown, D.W.; Schaab, M.R.; Birmingham, W.R.; Armstrong, R.N. Evolution of the antibiotic resistance protein, FosA, is linked to a catalytically promiscuous progenitor. Biochemistry 2009, 48, 1847–1849. [Google Scholar]
- Armstrong, R.N. Mechanistic imperatives for the evolution of glutathione transferases. Curr. Opin. Chem. Biol. 1998, 2, 618–623. [Google Scholar] [CrossRef]
- Armstrong, R.N. Mechanistic diversity in a metalloenzyme superfamily. Biochemistry 2000, 39, 13625–13632. [Google Scholar] [CrossRef]
- Leon, J.; Garcia-Lobo, J.M.; Navas, J.; Ortiz, J.M. Fosfomycin-resistance plasmids determine an intracellular modification of fosfomycin. J. Gen. Microbiol. 1985, 131, 1649–1655. [Google Scholar]
- Llaneza, J.; Villar, C.J.; Salas, J.A.; Suarez, J.E.; Mendoza, M.C.; Hardisson, C. Plasmid-mediated fosfomycin resistance is due to enzymatic modification of the antibiotic. Antimicrob. Agents Chemother. 1985, 28, 163–164. [Google Scholar] [CrossRef]
- Mendoza, C.; Garcia, J.M.; Llaneza, J.; Mendez, F.J.; Hardisson, C.; Ortiz, J.M. Plasmid-determined resistance to fosfomycin in Serratia marcescens. Antimicrob. Agents Chemother. 1980, 18, 215–219. [Google Scholar] [CrossRef]
- Garcia-Lobo, J.M.; Ortiz, J.M. Tn292l, a transposon encoding fosfomycin resistance. J. Bacteriol. 1982, 151, 477–479. [Google Scholar]
- Seoane, A.; Sangari, F.J.; Lobo, J.M. Complete nucleotide sequence of the fosfomycin resistance transposon Tn2921. Int. J. Antimicrob. Agents 2010, 35, 413–414. [Google Scholar] [CrossRef] [Green Version]
- Rife, C.L.; Pharris, R.E.; Newcomer, M.E.; Armstrong, R.N. Crystal structure of a genomically encoded fosfomycin resistance protein (FosA) at 1.19 A resolution by MAD phasing off the L-III edge of Tl(+). J. Am. Chem. Soc. 2002, 124, 11001–11003. [Google Scholar] [CrossRef]
- Arca, P.; Hardisson, C.; Suarez, J.E. Purification of a glutathione S-transferase that mediates fosfomycin resistance in bacteria. Antimicrob. Agents Chemother. 1990, 34, 844–848. [Google Scholar] [CrossRef]
- Arca, P.; Rico, M.; Brana, A.F.; Villar, C.J.; Hardisson, C.; Suarez, J.E. Formation of an adduct between fosfomycin and glutathione: A new mechanism of antibiotic resistance in bacteria. Antimicrob. Agents Chemother. 1988, 32, 1552–1556. [Google Scholar] [CrossRef]
- Bernat, B.A.; Laughlin, L.T.; Armstrong, R.N. Fosfomycin resistance protein (FosA) is a manganese metalloglutathione transferase related to glyoxalase I and the extradiol dioxygenases. Biochemistry 1997, 36, 3050–3055. [Google Scholar] [CrossRef]
- Bernat, B.A.; Armstrong, R.N. Elementary steps in the acquisition of Mn2+ by the fosfomycin resistance protein (FosA). Biochemistry 2001, 40, 12712–12718. [Google Scholar] [CrossRef]
- Bernat, B.A.; Laughlin, L.T.; Armstrong, R.N. Elucidation of a monovalent cation dependence and characterization of the divalent cation binding site of the fosfomycin resistance protein (FosA). Biochemistry 1999, 38, 7462–7469. [Google Scholar] [CrossRef]
- Beharry, Z.; Palzkill, T. Functional analysis of active site residues of the fosfomycin resistance enzyme FosA from Pseudomonas aeruginosa. J. Biol. Chem. 2005, 280, 17786–17791. [Google Scholar] [CrossRef]
- Etienne, J.; Gerbaud, G.; Fleurette, J.; Courvalin, P. Characterization of staphylococcal plasmids hybridizing with the fosfomycin resistance gene fosB. FEMS Microbiol. Lett. 1991, 68, 119–122. [Google Scholar]
- Zilhao, R.; Courvalin, P. Nucleotide sequence of the fosB gene conferring fosfomycin resistance in Staphylococcus epidermidis. FEMS Microbiol. Lett. 1990, 56, 267–272. [Google Scholar]
- Cao, M.; Bernat, B.A.; Wang, Z.; Armstrong, R.N.; Helmann, J.D. FosB, a cysteine-dependent fosfomycin resistance protein under the control of sigma(W), an extracytoplasmic-function sigma factor in Bacillus subtilis. J. Bacteriol. 2001, 183, 2380–2383. [Google Scholar] [CrossRef]
- Butcher, B.G.; Helmann, J.D. Identification of Bacillus subtilis sigma-dependent genes that provide intrinsic resistance to antimicrobial compounds produced by Bacilli. Mol. Microbiol. 2006, 60, 765–782. [Google Scholar] [CrossRef]
- Gaballa, A.; Newton, G.L.; Antelmann, H.; Parsonage, D.; Upton, H.; Rawat, M.; Claiborne, A.; Fahey, R.C.; Helmann, J.D. Biosynthesis and functions of bacillithiol, a major low-molecular-weight thiol in Bacilli. Proc. Natl. Acad. Sci. USA 2010, 107, 6482–6486. [Google Scholar] [CrossRef]
- Parsonage, D.; Newton, G.L.; Holder, R.C.; Wallace, B.D.; Paige, C.; Hamilton, C.J.; Dos Santos, P.C.; Redinbo, M.R.; Reid, S.D.; Claiborne, A. Characterization of the N-acetyl-alpha-d-glucosaminyl l-malate synthase and deacetylase functions for bacillithiol biosynthesis in Bacillus anthracis. Biochemistry 2010, 49, 8398–8414. [Google Scholar] [CrossRef]
- Roberts, A.A.; Sharma, S.V.; Strankman, A.W.; Duran, S.R.; Rawat, M.; Hamilton, C.J. Mechanistic studies of FosB: A divalent metal-dependent bacillithiol-S-transferase that mediates fosfomycin resistance in Staphylococcus aureus. Biochem. J. 2013, 451, 69–79. [Google Scholar] [CrossRef]
- Fillgrove, K.L.; Pakhomova, S.; Newcomer, M.E.; Armstrong, R.N. Mechanistic diversity of fosfomycin resistance in pathogenic microorganisms. J. Am. Chem. Soc. 2003, 125, 15730–15731. [Google Scholar] [CrossRef]
- Fillgrove, K.L.; Pakhomova, S.; Schaab, M.R.; Newcomer, M.E.; Armstrong, R.N. Structure and mechanism of the genomically encoded fosfomycin resistance protein, FosX, from Listeria monocytogene. Biochemistry 2007, 46, 8110–8120. [Google Scholar] [CrossRef]
- Kobayashi, S.; Kuzuyama, T.; Seto, H. Characterization of the fomA and fomB gene products from Streptomyces wedmorensis, which confer fosfomycin resistance on Escherichia coli. Antimicrob. Agents Chemother. 2000, 44, 647–650. [Google Scholar] [CrossRef]
- Kuzuyama, T.; Kobayashi, S.; O'Hara, K.; Hidaka, T.; Seto, H. Fosfomycin monophosphate and fosfomycin diphosphate, two inactivated fosfomycin derivates formed by gene products of fomA and fomB from a fosfomycin producing organism Streptomyces wedmorensis. J. Antibiot. (Tokyo) 1996, 49, 502–504. [Google Scholar] [CrossRef]
- Pakhomova, S.; Bartlett, S.G.; Augustus, A.; Kuzuyama, T.; Newcomer, M.E. Crystal structure of fosfomycin resistance kinase FomA from Streptomyces wedmorensis. J. Biol. Chem. 2008, 283, 28518–28526. [Google Scholar]
- Garcia, P.; Arca, P.; Evaristo Suarez, J. Product of fosC, a gene from Pseudomonas syringae, mediates fosfomycin resistance by using ATP as cosubstrate. Antimicrob. Agents Chemother. 1995, 39, 1569–1573. [Google Scholar] [CrossRef]
- Kim, S.Y.; Ju, K.S.; Metcalf, W.W.; Evans, B.S.; Kuzuyama, T.; van der Donk, W.A. Different biosynthetic pathways to fosfomycin in Pseudomonas syringae and Streptomyces species. Antimicrob. Agents Chemother. 2012, 56, 4175–4183. [Google Scholar] [CrossRef]
- Arca, P.; Reguera, G.; Hardisson, C. Plasmid-encoded fosfomycin resistance in bacteria isolated from the urinary tract in a multicentre survey. J. Antimicrob. Chemother. 1997, 40, 393–399. [Google Scholar] [CrossRef]
- O’Hara, K. Two different types of fosfomycin resistance in clinical isolates of Klebsiella pneumoniae. FEMS Microbiol. Lett. 1993, 114, 9–16. [Google Scholar] [CrossRef]
- Shimizu, M.; Shigeobu, F.; Miyakozawa, I.; Nakamura, A.; Suzuki, M.; Mizukoshi, S.; O’Hara, K.; Sawai, T. Novel fosfomycin resistance of Pseudomonas aeruginosa clinical isolates recovered in Japan in 1996. Antimicrob. Agents Chemother. 2000, 44, 2007–2008. [Google Scholar] [CrossRef]
- Wachino, J.; Yamane, K.; Suzuki, S.; Kimura, K.; Arakawa, Y. Prevalence of fosfomycin resistance among CTX-M-producing Escherichia coli clinical isolates in Japan and identification of novel plasmid-mediated fosfomycin-modifying enzymes. Antimicrob. Agents Chemother. 2010, 54, 3061–3064. [Google Scholar] [CrossRef]
- Ho, P.L.; Chan, J.; Lo, W.U.; Law, P.Y.; Chow, K.H. Plasmid-mediated fosfomycin resistance in Escherichia coli isolated from pig. Vet. Microbiol. 2013, 162, 964–967. [Google Scholar] [CrossRef] [Green Version]
- Ho, P.L.; Chan, J.; Lo, W.U.; Law, P.Y.; Li, Z.; Lai, E.L.; Chow, K.H. Dissemination of plasmid-mediated fosfomycin resistance fosA3 among multidrug-resistant Escherichia coli from livestock and other animals. J. Appl. Microbiol. 2013, 114, 695–702. [Google Scholar] [CrossRef]
- Hou, J.; Huang, X.; Deng, Y.; He, L.; Yang, T.; Zeng, Z.; Chen, Z.; Liu, J.H. Dissemination of the fosfomycin resistance gene fosA3 with CTX-M beta-lactamase genes and rmtB carried on IncFII plasmids among Escherichia coli isolates from pets in China. Antimicrob. Agents Chemother. 2012, 56, 2135–2138. [Google Scholar] [CrossRef]
- He, L.; Partridge, S.R.; Yang, X.; Hou, J.; Deng, Y.; Yao, Q.; Zeng, Z.; Chen, Z.; Liu, J.H. Complete nucleotide sequence of pHN7A8, an F33:A-:B-type epidemic plasmid carrying blaCTX-M-65, fosA3 and rmtB from China. J. Antimicrob. Chemother. 2013, 68, 46–50. [Google Scholar] [CrossRef]
- Shen, P.; Jiang, Y.; Zhou, Z.; Zhang, J.; Yu, Y.; Li, L. Complete nucleotide sequence of pKP96, a 67 850 bp multiresistance plasmid encoding qnrA1, aac(6')-Ib-cr and blaCTX-M-24 from Klebsiella pneumonia. J. Antimicrob. Chemother. 2008, 62, 1252–1256. [Google Scholar]
- Lee, S.Y.; Park, Y.J.; Yu, J.K.; Jung, S.; Kim, Y.; Jeong, S.H.; Arakawa, Y. Prevalence of acquired fosfomycin resistance among extended-spectrum beta-lactamase-producing Escherichia coli and Klebsiella pneumoniae clinical isolates in Korea and IS26-composite transposon surrounding fosA3. J. Antimicrob. Chemother. 2012, 60, 329–336. [Google Scholar]
- De Groote, V.N.; Fauvart, M.; Kint, C.I.; Verstraeten, N.; Jans, A.; Cornelis, P.; Michiels, J. Pseudomonas aeruginosa fosfomycin resistance mechanisms affect non-inherited fluoroquinolone tolerance. J. Med. Microbiol. 2011, 60, 329–336. [Google Scholar]
- Nilsson, A.I.; Berg, O.G.; Aspevall, O.; Kahlmeter, G.; Andersson, D.I. Biological costs and mechanisms of fosfomycin resistance in Escherichia coli. Antimicrob. Agents Chemother. 2003, 47, 2850–2858. [Google Scholar]
- Oteo, J.; Orden, B.; Bautista, V.; Cuevas, O.; Arroyo, M.; Martinez-Ruiz, R.; Perez-Vazquez, M.; Alcaraz, M.; Garcia-Cobos, S.; Campos, J. CTX-M-15-producing urinary Escherichia coli O25b-ST131-phylogroup B2 has acquired resistance to fosfomycin. J. Antimicrob. Chemother. 2009, 64, 712–717. [Google Scholar]
- Rodriguez-Rojas, A.; Macia, M.D.; Couce, A.; Gomez, C.; Castaneda-Garcia, A.; Oliver, A.; Blazquez, J. Assesing the emergence of resistance: The absence of biological cost in vivo may compromise fosfomycin treatments for Pseudomonas aeruginosa infections. PLoS One 2010, 5, e10193. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Castañeda-García, A.; Blázquez, J.; Rodríguez-Rojas, A. Molecular Mechanisms and Clinical Impact of Acquired and Intrinsic Fosfomycin Resistance. Antibiotics 2013, 2, 217-236. https://doi.org/10.3390/antibiotics2020217
Castañeda-García A, Blázquez J, Rodríguez-Rojas A. Molecular Mechanisms and Clinical Impact of Acquired and Intrinsic Fosfomycin Resistance. Antibiotics. 2013; 2(2):217-236. https://doi.org/10.3390/antibiotics2020217
Chicago/Turabian StyleCastañeda-García, Alfredo, Jesús Blázquez, and Alexandro Rodríguez-Rojas. 2013. "Molecular Mechanisms and Clinical Impact of Acquired and Intrinsic Fosfomycin Resistance" Antibiotics 2, no. 2: 217-236. https://doi.org/10.3390/antibiotics2020217