Foreign Body Infection Models to Study Host-Pathogen Response and Antimicrobial Tolerance of Bacterial Biofilm


Abstract
:1. Introduction
2. Subcutaneous Catheter Model
2.1. General Aspects of the Subcutaneous Catheter Model
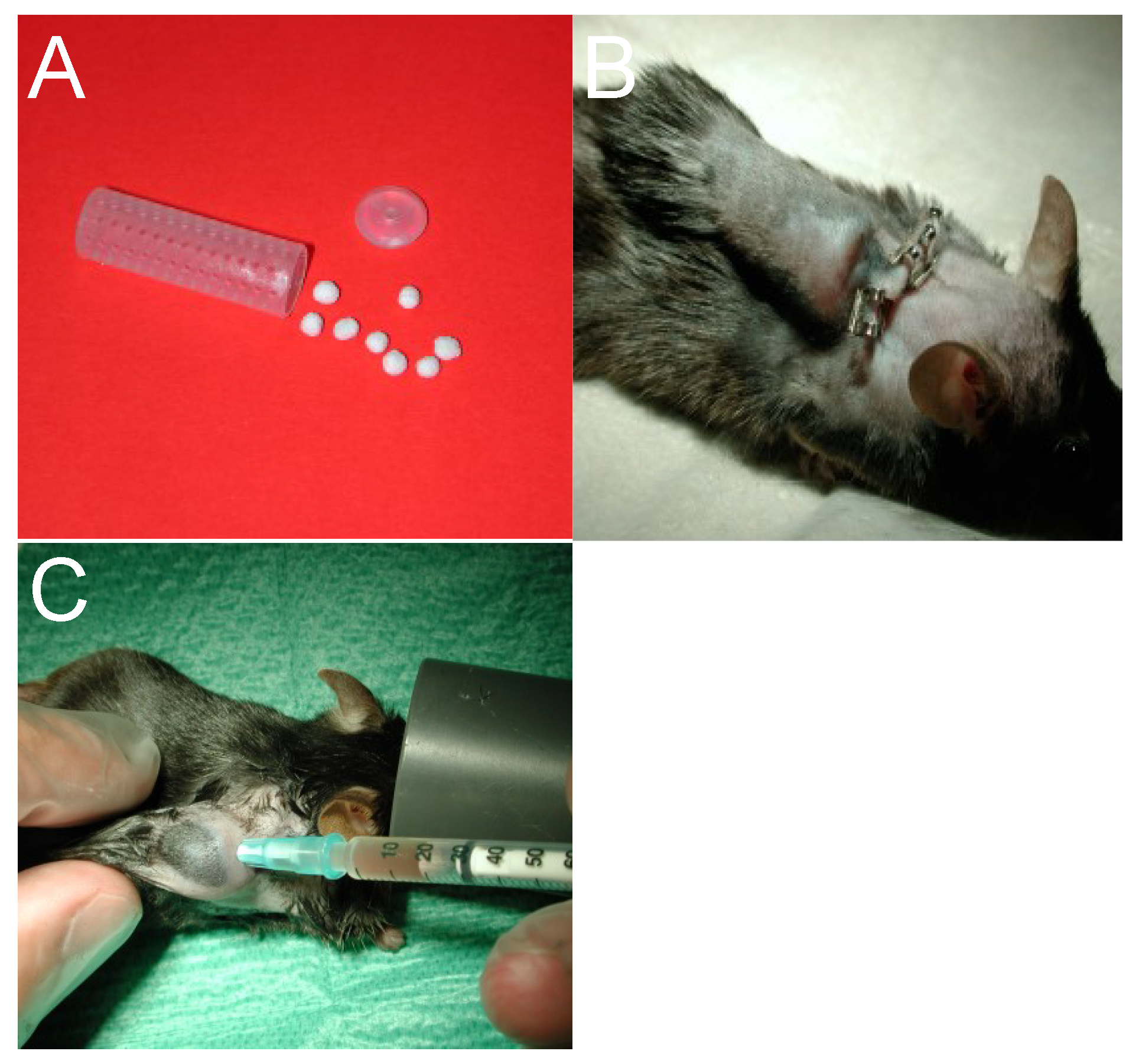
2.2. Catheter Infection Model in the Mouse
2.2.1. Technique
2.2.2. Assessment of the Host Response in the Catheter Infection Model

2.2.3. Assessment of Biofilm Formed by Pseudomonas aeruginosa
3. Tissue Cage Infection Model
3.1. Tissue Cage Model in Different Animal Species

{kind=link}
{kind=link}
{kind=link}
| Orthopaedic Models | Tissue Cage Model | Catheter Abscess Model | Ref. | ||||
|---|---|---|---|---|---|---|---|
| Animal Species | rabbit/sheep/rat/mouse/guinea pig/chicken/dog/pig/goat | guinea pig | Rat | mouse | mouse | [35,52] | |
| Labor intensity | +++ | +++ § | ++ | ++ | + | [50,53] | |
| Large scale experiments | no | nd | nd | yes | yes | [54,55] | |
| Localization | bone | sc | sc | sc | sc | [56] | |
| Susceptibility to staphylococcal infection | species-dependent | yes | no | yes | yes | [35,50,52] | |
| Antibiotic tolerance (long-term treatment) | species-dependent | no | yes | yes | yes | [50] | |
| Use of transgenic animals | nd | nd | nd | yes | yes | [57] | |
| Imaging | yes | nd | yes | yes | yes | [35,56,58,59,60,61,62,63,64,65] | |
| Bacterial virulence factors | only after sacrifice | yes | no | yes | yes | [18,27,35,44,57,66,67,68,69,70,71,72,73,74,75,76] | |
| Host immune response | yes | yes | yes | yes | yes | [21,36,37,54,58,62,63,77,78,79,80] | |
| Osseointegration | yes | no | no | no | no | [35] | |
| Various implant materials/coatings | yes | nd | nd | yes | yes | [38,81,82,83,84,85] | |
| Repeated assessment during experiment: | |||||||
| Cytotoxicity on eukaryotic cells | no | nd | nd | yes | no | [82,86] | |
| Pharmacokinetics (PK) at the infection site | no | yes | yes | yes | no | [9,30,50] | |
| Pharmacodynamics (PD) | no | yes | yes | yes | no | [30,32,35,86,87,88,89,90,91] | |
| Similarity to human disease: | |||||||
| Localized infection | yes | yes | yes | yes | no | [35,50] | |
3.2. Tissue Cage Infection Model in the Mouse
3.2.1. Technique

3.2.2. Assessment of Virulence of Bacterial Species
3.2.3. Assessment of Host Defense in the Tissue Cage

3.2.4. Assessment of Antibiotic Resistance in Vivo
3.2.5. Pharmacokinetic (PK) Studies, Pharmacodynamic (PD) Properties and Efficacy
3.2.6. Cytotoxicity of New Antimicrobial Compounds against Host Cells
3.2.7. Properties of Different Tissue Cage Materials
4. Orthopaedic Implant Infection Models
5. Disadvantages of Subcutaneous Animal Foreign Body Models
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tsaras, G.; Osmon, D.R.; Mabry, T.; Lahr, B.; St Sauveur, J.; Yawn, B.; Kurland, R.; Berbari, E.F. Incidence, secular trends, and outcomes of prosthetic joint infection: A population-based study, olmsted county, minnesota, 1969–2007. Infect. Control Hosp. Epidemiol. 2012, 33, 1207–1212. [Google Scholar] [CrossRef] [PubMed]
- Darouiche, R.O. Treatment of infections associated with surgical implants. N. Engl. J. Med. 2004, 350, 1422–1429. [Google Scholar] [CrossRef] [PubMed]
- Zappe, B.; Graf, S.; Ochsner, P.E.; Zimmerli, W.; Sendi, P. Propionibacterium spp. in prosthetic joint infections: A diagnostic challenge. Arch. Orthop. Trauma Surg. 2008, 128, 1039–1046. [Google Scholar]
- Zimmerli, W.; Trampuz, A.; Ochsner, P.E. Prosthetic-joint infections. N. Engl. J. Med. 2004, 351, 1645–1654. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.B.; Weber, D.J.; Goodrich, J.S.; Popowitch, E.B.; Poe, M.D.; Nyugen, V.; Shope, T.R.; Foster, D.T.; Miller, J.R.; Kotch, J. Prevalence and risk factor analysis for methicillin-resistant Staphylococcus aureus nasal colonization in children attending child care centers. J. Clin. Microbiol. 2011, 49, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Safdar, N.; Bradley, E.A. The risk of infection after nasal colonization with Staphylococcus aureus. Am. J. Med. 2008, 121, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.J.; Arduino, J.M.; Reed, S.D.; Sexton, D.J.; Kaye, K.S.; Grussemeyer, C.A.; Peter, S.A.; Hardy, C.; Choi, Y.I.; Friedman, J.Y.; et al. Variation in the type and frequency of postoperative invasive Staphylococcus aureus infections according to type of surgical procedure. Infect. Control Hosp. Epidemiol. 2010, 31, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Elek, S.D.; Conen, P.E. The virulence of Staphylococcus pyogenes for man; a study of the problems of wound infection. Br. J. Exp. Pathol. 1957, 38, 573–586. [Google Scholar] [PubMed]
- Zimmerli, W.; Waldvogel, F.A.; Vaudaux, P.; Nydegger, U.E. Pathogenesis of foreign body infection: Description and characteristics of an animal model. J. Infect. Dis. 1982, 146, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Resch, A.; Leicht, S.; Saric, M.; Pasztor, L.; Jakob, A.; Gotz, F.; Nordheim, A. Comparative proteome analysis of Staphylococcus aureus biofilm and planktonic cells and correlation with transcriptome profiling. Proteomics 2006, 6, 1867–1877. [Google Scholar] [CrossRef] [PubMed]
- Mack, D.; Davies, A.P.; Harris, L.G.; Rohde, H.; Horstkotte, M.A.; Knobloch, J.K. Microbial interactions in Staphylococcus epidermidis biofilms. Anal. Bioanal. Chem. 2007, 387, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Arciola, C.R.; Campoccia, D.; Speziale, P.; Montanaro, L.; Costerton, J.W. Biofilm formation in Staphylococcus implant infections. A review of molecular mechanisms and implications for biofilm-resistant materials. Biomaterials 2012, 33, 5967–5982. [Google Scholar]
- Geoghegan, J.A.; Monk, I.R.; O’Gara, J.P.; Foster, T.J. Subdomains N2N3 of fibronectin binding protein a mediate Staphylococcus aureus biofilm formation and adherence to fibrinogen using distinct mechanisms. J. Bacteriol. 2013, 195, 2675–2683. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, E.; Pozzi, C.; Houston, P.; Humphreys, H.; Robinson, D.A.; Loughman, A.; Foster, T.J.; O’Gara, J.P. A novel Staphylococcus aureus biofilm phenotype mediated by the fibronectin-binding proteins, FnBPA and FnBPB. J. Bacteriol. 2008, 190, 3835–3850. [Google Scholar]
- Gotz, F. Staphylococcus and biofilms. Mol. Microbiol. 2002, 43, 1367–1378. [Google Scholar] [CrossRef] [PubMed]
- Otto, M. Staphylococcal infections: Mechanisms of biofilm maturation and detachment as critical determinants of pathogenicity. Ann. Rev. Med. 2013, 64, 175–188. [Google Scholar] [PubMed]
- Otto, M. Staphylococcal biofilms. Curr. Topics Microbiol. Immunol. 2008, 322, 207–228. [Google Scholar]
- Wang, R.; Khan, B.A.; Cheung, G.Y.; Bach, T.H.; Jameson-Lee, M.; Kong, K.F.; Queck, S.Y.; Otto, M. Staphylococcus epidermidis surfactant peptides promote biofilm maturation and dissemination of biofilm-associated infection in mice. J. Clin. Invest. 2011, 121, 238–248. [Google Scholar] [PubMed]
- Chatterjee, S.S.; Joo, H.S.; Duong, A.C.; Dieringer, T.D.; Tan, V.Y.; Song, Y.; Fischer, E.R.; Cheung, G.Y.; Li, M.; Otto, M. Essential staphylococcus aureus toxin export system. Nat. Med. 2013, 19, 364–367. [Google Scholar] [CrossRef] [PubMed]
- Periasamy, S.; Joo, H.S.; Duong, A.C.; Bach, T.H.; Tan, V.Y.; Chatterjee, S.S.; Cheung, G.Y.; Otto, M. How Staphylococcus aureus biofilms develop their characteristic structure. Proc. Natl. Acad. Sci. USA 2012, 109, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Hanke, M.L.; Heim, C.E.; Angle, A.; Sanderson, S.D.; Kielian, T. Targeting macrophage activation for the prevention and treatment of Staphylococcus aureus biofilm infections. J. Immunol. 2013, 190, 2159–2168. [Google Scholar] [CrossRef] [PubMed]
- Hanke, M.L.; Kielian, T. Deciphering mechanisms of staphylococcal biofilm evasion of host immunity. Front. Cell. Infect. Microbiol. 2012, 2, e62. [Google Scholar] [CrossRef]
- Prabhakara, R.; Harro, J.M.; Leid, J.G.; Harris, M.; Shirtliff, M.E. Murine immune response to a chronic Staphylococcus aureus biofilm infection. Infect. Immun. 2011, 79, 1789–1796. [Google Scholar] [CrossRef] [PubMed]
- Prabhakara, R.; Harro, J.M.; Leid, J.G.; Keegan, A.D.; Prior, M.L.; Shirtliff, M.E. Suppression of the inflammatory immune response prevents the development of chronic biofilm infection due to methicillin-resistant Staphylococcus aureus. Infect. Immun. 2011, 79, 5010–5018. [Google Scholar] [CrossRef] [PubMed]
- Scherr, T.D.; Roux, C.M.; Hanke, M.L.; Angle, A.; Dunman, P.M.; Kielian, T. Global transcriptome analysis of Staphylococcus aureus biofilms in response to innate immune cells. Infect. Immun. 2013, 81, 4363–4376. [Google Scholar] [CrossRef] [PubMed]
- Thurlow, L.R.; Hanke, M.L.; Fritz, T.; Angle, A.; Aldrich, A.; Williams, S.H.; Engebretsen, I.L.; Bayles, K.W.; Horswill, A.R.; Kielian, T. Staphylococcus aureus biofilms prevent macrophage phagocytosis and attenuate inflammation in vivo. J. Immunol. 2011, 186, 6585–6596. [Google Scholar] [CrossRef] [PubMed]
- Fluckiger, U.; Ulrich, M.; Steinhuber, A.; Doring, G.; Mack, D.; Landmann, R.; Goerke, C.; Wolz, C. Biofilm formation, icaADBC transcription, and polysaccharide intercellular adhesin synthesis by Staphylococci in a device-related infection model. Infect. Immun. 2005, 73, 1811–1819. [Google Scholar] [CrossRef] [PubMed]
- Pragman, A.A.; Schlievert, P.M. Virulence regulation in Staphylococcus aureus: The need for in vivo analysis of virulence factor regulation. FEMS Immunol. Med. Microbiol. 2004, 42, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Voyich, J.M.; Braughton, K.R.; Sturdevant, D.E.; Whitney, A.R.; Said-Salim, B.; Porcella, S.F.; Long, R.D.; Dorward, D.W.; Gardner, D.J.; Kreiswirth, B.N.; et al. Insights into mechanisms used by Staphylococcus aureus to avoid destruction by human neutrophils. J. Immunol. 2005, 175, 3907–3919. [Google Scholar]
- John, A.K.; Schmaler, M.; Khanna, N.; Landmann, R. Reversible daptomycin tolerance of adherent Staphylococci in an implant infection model. Antimicrob. Agents Chemother. 2011, 55, 3510–3516. [Google Scholar] [CrossRef] [PubMed]
- Zimmerli, W.; Widmer, A.F.; Blatter, M.; Frei, R.; Ochsner, P.E. Role of rifampin for treatment of orthopedic implant-related staphylococcal infections: A randomized controlled trial. Foreign-Body Infection (FBI) study group. JAMA 1998, 279, 1537–1541. [Google Scholar]
- John, A.K.; Baldoni, D.; Haschke, M.; Rentsch, K.; Schaerli, P.; Zimmerli, W.; Trampuz, A. Efficacy of daptomycin in implant-associated infection due to methicillin-resistant Staphylococcus aureus: Importance of combination with rifampin. Antimicrob. Agents Chemother. 2009, 53, 2719–2724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osmon, D.R.; Berbari, E.F.; Berendt, A.R.; Lew, D.; Zimmerli, W.; Steckelberg, J.M.; Rao, N.; Hanssen, A.; Wilson, W.R. Diagnosis and management of prosthetic joint infection: Clinical practice guidelines by the infectious diseases society of America. Clin. Infect. Dis. 2013, 56, e1–e25. [Google Scholar] [CrossRef] [PubMed]
- Conlon, B.P.; Nakayasu, E.S.; Fleck, L.E.; LaFleur, M.D.; Isabella, V.M.; Coleman, K.; Leonard, S.N.; Smith, R.D.; Adkins, J.N.; Lewis, K. Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature 2013, 503, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Calabro, L.; Seif, E.L.; Din, A.F.; Richards, R.G.; Moriarty, T.F. Animal models of orthopedic implant-related infection. In Biomaterials Associated Infection: Immunological Aspects and Antimicrobial Strategies; Moriarty, T.F., Zaat, S.A.J., Busscher, H.J., Eds.; Springer Science+Business Media: New York, NY, USA, 2013; pp. 273–304. [Google Scholar]
- Kristian, S.A.; Birkenstock, T.A.; Sauder, U.; Mack, D.; Gotz, F.; Landmann, R. Biofilm formation induces C3a release and protects Staphylococcus epidermidis from IgG and complement deposition and from neutrophil-dependent killing. J. Infect. Dis. 2008, 197, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Zimmerli, W.; Lew, P.D.; Waldvogel, F.A. Pathogenesis of foreign body infection. Evidence for a local granulocyte defect. J. Clin. Invest. 1984, 73, 1191–1200. [Google Scholar]
- Zimmerli, W.; Sendi, P. Pathogenesis of implant-associated infection: The role of the host. Semin. Immunopathol. 2011, 33, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.; Kaksa, A.; Muller, W.; Denefleh, B.; Heppert, V.; Wentzensen, A.; Hansch, G.M. Polymorphonuclear neutrophils in posttraumatic osteomyelitis: Cells recovered from the inflamed site lack chemotactic activity but generate superoxides. Shock 2004, 22, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.; Kondella, K.; Bernschneider, T.; Heppert, V.; Wentzensen, A.; Hansch, G.M. Post-traumatic osteomyelitis: Analysis of inflammatory cells recruited into the site of infection. Shock 2003, 20, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Heim, C.E.; Vidlak, D.; Scherr, T.D.; Kozel, J.A.; Holzapfel, M.; Muirhead, D.E.; Kielian, T. Myeloid-derived suppressor cells contribute to Staphylococcus aureus orthopedic biofilm infection. J. Immunol. 2014, 192, 3778–3792. [Google Scholar] [CrossRef] [PubMed]
- Rupp, M.E.; Ulphani, J.S.; Fey, P.D.; Bartscht, K.; Mack, D. Characterization of the importance of polysaccharide intercellular adhesin/hemagglutinin of Staphylococcus epidermidis in the pathogenesis of biomaterial-based infection in a mouse foreign body infection model. Infect. Immun. 1999, 67, 2627–2632. [Google Scholar] [PubMed]
- Kadurugamuwa, J.L.; Sin, L.V.; Yu, J.; Francis, K.P.; Kimura, R.; Purchio, T.; Contag, P.R. Rapid direct method for monitoring antibiotics in a mouse model of bacterial biofilm infection. Antimicrob. Agents Chemother. 2003, 47, 3130–3137. [Google Scholar] [CrossRef] [PubMed]
- Malone, J.G.; Jaeger, T.; Spangler, C.; Ritz, D.; Spang, A.; Arrieumerlou, C.; Kaever, V.; Landmann, R.; Jenal, U. YfiBNR mediates cyclic di-GMP dependent small colony variant formation and persistence in Pseudomonas aeruginosa. PLoS Pathog. 2010, 6, e1000804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widmer, A.F.; Frei, R.; Rajacic, Z.; Zimmerli, W. Correlation between in vivo and in vitro efficacy of antimicrobial agents against foreign body infections. J. Infect. Dis. 1990, 162, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Zimmerli, W.; Frei, R.; Widmer, A.F.; Rajacic, Z. Microbiological tests to predict treatment outcome in experimental device-related infections due to Staphylococcus aureus. J. Antimicrob. Chemother. 1994, 33, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Drancourt, M.; Stein, A.; Argenson, J.N.; Zannier, A.; Curvale, G.; Raoult, D. Oral rifampin plus ofloxacin for treatment of Staphylococcus-infected orthopedic implants. Antimicrob. Agents Chemother. 1993, 37, 1214–1218. [Google Scholar] [CrossRef] [PubMed]
- Tafin, U.F.; Corvec, S.; Betrisey, B.; Zimmerli, W.; Trampuz, A. Role of rifampin against Propionibacterium acnes biofilm in vitro and in an experimental foreign-body infection model. Antimicrob. Agents Chemother. 2012, 56, 1885–1891. [Google Scholar] [CrossRef] [PubMed]
- Olson, M.E.; Slater, S.R.; Rupp, M.E.; Fey, P.D. Rifampicin enhances activity of daptomycin and vancomycin against both a polysaccharide intercellular adhesin (pia)-dependent and -independent Staphylococcus epidermidis biofilm. J. Antimicrob. Chemother. 2010, 65, 2164–2171. [Google Scholar] [CrossRef] [PubMed]
- Zimmerli, W. Tissue cage infection model. In Handbook of Animal Models of Infection. Experimental Models in Antimicrobial Chemotherapy; Zak, O.S., Merle, A., Eds.; Academic Press: London, UK, 1999. [Google Scholar]
- Lucet, J.C.; Herrmann, M.; Rohner, P.; Auckenthaler, R.; Waldvogel, F.A.; Lew, D.P. Treatment of experimental foreign body infection caused by methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 1990, 34, 2312–2317. [Google Scholar] [CrossRef] [PubMed]
- An, Y.H.; Kang, Q.K.; Arciola, C.R. Animal models of osteomyelitis. Int. J. Artif. Organs 2006, 29, 407–420. [Google Scholar] [PubMed]
- Auer, J.A.; Goodship, A.; Arnoczky, S.; Pearce, S.; Price, J.; Claes, L.; von Rechenberg, B.; Hofmann-Amtenbrinck, M.; Schneider, E.; Muller-Terpitz, R.; et al. Refining animal models in fracture research: Seeking consensus in optimising both animal welfare and scientific validity for appropriate biomedical use. BMC Musculoskelet. Disord. 2007, 8. [Google Scholar] [CrossRef] [Green Version]
- Odekerken, J.C.; Arts, J.J.; Surtel, D.A.; Walenkamp, G.H.; Welting, T.J. A rabbit osteomyelitis model for the longitudinal assessment of early post-operative implant infections. J. Orthop. Surg. Res. 2013, 8. [Google Scholar] [CrossRef]
- Schimandle, J.H.; Boden, S.D. Spine update. Animal use in spinal research. Spine 1994, 19, 2474–2477. [Google Scholar]
- Pearce, A.I.; Richards, R.G.; Milz, S.; Schneider, E.; Pearce, S.G. Animal models for implant biomaterial research in bone: A review. Eur. Cell. Mater. 2007, 13, 1–10. [Google Scholar] [PubMed]
- Kristian, S.A.; Lauth, X.; Nizet, V.; Goetz, F.; Neumeister, B.; Peschel, A.; Landmann, R. Alanylation of teichoic acids protects Staphylococcus aureus against toll-like receptor 2-dependent host defense in a mouse tissue cage infection model. J. Infect. Dis. 2003, 188, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Bernthal, N.M.; Stavrakis, A.I.; Billi, F.; Cho, J.S.; Kremen, T.J.; Simon, S.I.; Cheung, A.L.; Finerman, G.A.; Lieberman, J.R.; Adams, J.S.; et al. A mouse model of post-arthroplasty Staphylococcus aureus joint infection to evaluate in vivo the efficacy of antimicrobial implant coatings. PLoS One 2010, 5, e12580. [Google Scholar] [CrossRef] [PubMed]
- Broekhuizen, C.A.; Sta, M.; Vandenbroucke-Grauls, C.M.; Zaat, S.A. Microscopic detection of viable Staphylococcus epidermidis in peri-implant tissue in experimental biomaterial-associated infection, identified by bromodeoxyuridine incorporation. Infect. Immun. 2010, 78, 954–962. [Google Scholar] [CrossRef] [PubMed]
- Kuklin, N.A.; Pancari, G.D.; Tobery, T.W.; Cope, L.; Jackson, J.; Gill, C.; Overbye, K.; Francis, K.P.; Yu, J.; Montgomery, D.; et al. Real-time monitoring of bacterial infection in vivo: Development of bioluminescent staphylococcal foreign-body and deep-thigh-wound mouse infection models. Antimicrob. Agents Chemother. 2003, 47, 2740–2748. [Google Scholar] [CrossRef] [PubMed]
- Lankinen, P.; Lehtimaki, K.; Hakanen, A.J.; Roivainen, A.; Aro, H.T. A comparative 18F-FDG PET/CT imaging of experimental Staphylococcus aureus osteomyelitis and Staphylococcus epidermidis foreign-body-associated infection in the rabbit tibia. EJNMMI Res. 2012, 2, e41. [Google Scholar] [CrossRef]
- Li, D.; Gromov, K.; Soballe, K.; Puzas, J.E.; O’Keefe, R.J.; Awad, H.; Drissi, H.; Schwarz, E.M. Quantitative mouse model of implant-associated osteomyelitis and the kinetics of microbial growth, osteolysis, and humoral immunity. J. Orthop. Res. 2008, 26, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Niska, J.A.; Meganck, J.A.; Pribaz, J.R.; Shahbazian, J.H.; Lim, E.; Zhang, N.; Rice, B.W.; Akin, A.; Ramos, R.I.; Bernthal, N.M.; et al. Monitoring bacterial burden, inflammation and bone damage longitudinally using optical and muct imaging in an orthopaedic implant infection in mice. PLoS One 2012, 7, e47397. [Google Scholar] [CrossRef] [PubMed]
- Steinhuber, A.; Landmann, R.; Goerke, C.; Wolz, C.; Fluckiger, U. Bioluminescence imaging to study the promoter activity of HLA of Staphylococcus aureus in vitro and in vivo. Int. J. Med. Microbiol. 2008, 298, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Stoodley, P.; Kathju, S.; Hu, F.Z.; Erdos, G.; Levenson, J.E.; Mehta, N.; Dice, B.; Johnson, S.; Hall-Stoodley, L.; Nistico, L.; et al. Molecular and imaging techniques for bacterial biofilms in joint arthroplasty infections. Clin. Orthop. Relat. Res. 2005, 31–40. [Google Scholar] [CrossRef]
- Beenken, K.E.; Dunman, P.M.; McAleese, F.; Macapagal, D.; Murphy, E.; Projan, S.J.; Blevins, J.S.; Smeltzer, M.S. Global gene expression in Staphylococcus aureus biofilms. J. Bacteriol. 2004, 186, 4665–4684. [Google Scholar] [CrossRef] [PubMed]
- Christensen, G.D.; Simpson, W.A.; Bisno, A.L.; Beachey, E.H. Experimental foreign body infections in mice challenged with slime-producing Staphylococcus epidermidis. Infect. Immun. 1983, 40, 407–410. [Google Scholar] [PubMed]
- Espersen, F.; Frimodt-Moller, N.; Corneliussen, L.; Thamdrup Rosdahl, V.; Skinhoj, P. Experimental foreign body infection in mice. J. Antimicrob. Chemother. 1993, 31, D103–D111. [Google Scholar] [CrossRef]
- Francois, P.; Tu Quoc, P.H.; Bisognano, C.; Kelley, W.L.; Lew, D.P.; Schrenzel, J.; Cramton, S.E.; Gotz, F.; Vaudaux, P. Lack of biofilm contribution to bacterial colonisation in an experimental model of foreign body infection by Staphylococcus aureus and Staphylococcus epidermidis. FEMS Immunol. Med. Microbiol. 2003, 35, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Kristian, S.A.; Golda, T.; Ferracin, F.; Cramton, S.E.; Neumeister, B.; Peschel, A.; Gotz, F.; Landmann, R. The ability of biofilm formation does not influence virulence of Staphylococcus aureus and host response in a mouse tissue cage infection model. Microb. Pathog. 2004, 36, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Laughton, J.M.; Devillard, E.; Heinrichs, D.E.; Reid, G.; McCormick, J.K. Inhibition of expression of a staphylococcal superantigen-like protein by a soluble factor from Lactobacillus reuteri. Microbiology 2006, 152, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Fan, J.; Niu, C.; Wang, D.; Wang, J.; Wang, X.; Villaruz, A.E.; Li, M.; Otto, M.; Gao, Q. The eukaryotic-type serine/threonine protein kinase STK is required for biofilm formation and virulence in Staphylococcus epidermidis. PLoS One 2011, 6, e25380. [Google Scholar] [CrossRef] [PubMed]
- Luong, T.T.; Lei, M.G.; Lee, C.Y. Staphylococcus aureus RBF activates biofilm formation in vitro and promotes virulence in a murine foreign body infection model. Infect. Immun. 2009, 77, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Sander, G.; Borner, T.; Kriegeskorte, A.; von Eiff, C.; Becker, K.; Mahabir, E. Catheter colonization and abscess formation due to Staphylococcus epidermidis with normal and small-colony-variant phenotype is mouse strain dependent. PLoS One 2012, 7, e36602. [Google Scholar] [CrossRef] [PubMed]
- Taj, Y.; Abdullah, F.E.; Aziz, F.; Kazmi, S.U. Temporal expression of extracellular products of Staphylococcus aureus in vivo mouse cage model. JPMA 2012, 62, 539–545. [Google Scholar]
- Rupp, M.E.; Ulphani, J.S.; Fey, P.D.; Mack, D. Characterization of Staphylococcus epidermidis polysaccharide intercellular adhesin/hemagglutinin in the pathogenesis of intravascular catheter-associated infection in a rat model. Infect. Immun. 1999, 67, 2656–2659. [Google Scholar] [PubMed]
- Falcieri, E.; Vaudaux, P.; Huggler, E.; Lew, D.; Waldvogel, F. Role of bacterial exopolymers and host factors on adherence and phagocytosis of Staphylococcus aureus in foreign body infection. J. Infect. Dis. 1987, 155, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Marriott, I.; Gray, D.L.; Tranguch, S.L.; Fowler, V.G., Jr.; Stryjewski, M.; Scott Levin, L.; Hudson, M.C.; Bost, K.L. Osteoblasts express the inflammatory cytokine interleukin-6 in a murine model of Staphylococcus aureus osteomyelitis and infected human bone tissue. Am. J. Pathol. 2004, 164, 1399–1406. [Google Scholar] [CrossRef] [PubMed]
- Shahrooei, M.; Hira, V.; Khodaparast, L.; Khodaparast, L.; Stijlemans, B.; Kucharikova, S.; Burghout, P.; Hermans, P.W.; van Eldere, J. Vaccination with sesc decreases Staphylococcus epidermidis biofilm formation. Infect. Immun. 2012, 80, 3660–3668. [Google Scholar] [CrossRef] [PubMed]
- Vaudaux, P.; Grau, G.E.; Huggler, E.; Schumacher-Perdreau, F.; Fiedler, F.; Waldvogel, F.A.; Lew, D.P. Contribution of tumor necrosis factor to host defense against Staphylococci in a guinea pig model of foreign body infections. J. Infect. Dis. 1992, 166, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Cobrado, L.; Silva-Dias, A.; Azevedo, M.M.; Pina-Vaz, C.; Rodrigues, A.G. In vivo antibiofilm effect of cerium, chitosan and hamamelitannin against usual agents of catheter-related bloodstream infections. J. Antimicrob. Chemother. 2013, 68, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Gordon, O.; Vig Slenters, T.; Brunetto, P.S.; Villaruz, A.E.; Sturdevant, D.E.; Otto, M.; Landmann, R.; Fromm, K.M. Silver coordination polymers for prevention of implant infection: Thiol interaction, impact on respiratory chain enzymes, and hydroxyl radical induction. Antimicrob. Agents Chemother. 2010, 54, 4208–4218. [Google Scholar] [CrossRef] [PubMed]
- Hudetz, D.; Ursic Hudetz, S.; Harris, L.G.; Luginbuhl, R.; Friederich, N.F.; Landmann, R. Weak effect of metal type and ica genes on staphylococcal infection of titanium and stainless steel implants. Clin. Microbiol. Infect. 2008, 14, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Moriarty, T.F.; Schlegel, U.; Perren, S.; Richards, R.G. Infection in fracture fixation: Can we influence infection rates through implant design? J. Mater. Sci. 2010, 21, 1031–1035. [Google Scholar]
- Zimmerli, W. Implanted devices: Biocompatibility, infection and tissue engineering. Semin. Immunopathol. 2011, 33, 219–220. [Google Scholar] [CrossRef] [PubMed]
- Nowakowska, J.; Griesser, H.J.; Textor, M.; Landmann, R.; Khanna, N. Antimicrobial properties of 8-hydroxyserrulat-14-en-19-oic acid for treatment of implant-associated infections. Antimicrob. Agents Chemother. 2013, 57, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Chambers, H.F. Ceftobiprole: In-vivo profile of a bactericidal cephalosporin. Clin. Microbiol. Infect. 2006, 12, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Garrigos, C.; Murillo, O.; Lora-Tamayo, J.; Verdaguer, R.; Tubau, F.; Cabellos, C.; Cabo, J.; Ariza, J. Fosfomycin-daptomycin and other fosfomycin combinations as alternative therapies in experimental foreign-body infection by methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2013, 57, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Murillo, O.; Garrigos, C.; Pachon, M.E.; Euba, G.; Verdaguer, R.; Cabellos, C.; Cabo, J.; Gudiol, F.; Ariza, J. Efficacy of high doses of daptomycin versus alternative therapies against experimental foreign-body infection by methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2009, 53, 4252–4257. [Google Scholar] [CrossRef] [PubMed]
- Van Wijngaerden, E.; Peetermans, W.E.; Vandersmissen, J.; van Lierde, S.; Bobbaers, H.; van Eldere, J. Foreign body infection: A new rat model for prophylaxis and treatment. J. Antimicrob. Chemother. 1999, 44, 669–674. [Google Scholar]
- Vaudaux, P.; Fleury, B.; Gjinovci, A.; Huggler, E.; Tangomo-Bento, M.; Lew, D.P. Comparison of tigecycline and vancomycin for treatment of experimental foreign-body infection due to methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2009, 53, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- John, A.K.; Landmann, R.; Khanna, N.; Infection Biology, Department of Biomedicine, University and University Hospital Basel, Basel, Switzerland. Unpublished data. 2014.
- Foster, T.J. Immune evasion by Staphylococci. Nat. Rev. Microbiol. 2005, 3, 948–958. [Google Scholar] [CrossRef] [PubMed]
- Wolz, C.; Goerke, C.; Landmann, R.; Zimmerli, W.; Fluckiger, U. Transcription of clumping factor a in attached and unattached Staphylococcus aureus in vitro and during device-related infection. Infect. Immun. 2002, 70, 2758–2762. [Google Scholar] [CrossRef] [PubMed]
- Mally, M.; Shin, H.; Paroz, C.; Landmann, R.; Cornelis, G.R. Capnocytophaga canimorsus: A human pathogen feeding at the surface of epithelial cells and phagocytes. PLoS Pathog. 2008, 4, e1000164. [Google Scholar] [CrossRef] [PubMed]
- Kadurugamuwa, J.L.; Sin, L.; Albert, E.; Yu, J.; Francis, K.; DeBoer, M.; Rubin, M.; Bellinger-Kawahara, C.; Parr, T.R., Jr.; Contag, P.R. Direct continuous method for monitoring biofilm infection in a mouse model. Infect. Immun. 2003, 71, 882–890. [Google Scholar] [CrossRef] [PubMed]
- McCallum, N.; Karauzum, H.; Getzmann, R.; Bischoff, M.; Majcherczyk, P.; Berger-Bachi, B.; Landmann, R. In vivo survival of teicoplanin-resistant Staphylococcus aureus and fitness cost of teicoplanin resistance. Antimicrob. Agents Chemother. 2006, 50, 2352–2360. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Herrera, J.; Docobo-Perez, F.; Lopez-Rojas, R.; Pichardo, C.; Ruiz-Valderas, R.; Lepe, J.A.; Pachon, J. Efficacy of daptomycin versus vancomycin in an experimental model of foreign-body and systemic infection caused by biofilm producers and methicillin-resistant Staphylococcus epidermidis. Antimicrob. Agents Chemother. 2012, 56, 613–617. [Google Scholar]
- John, A.K.; Rajacic, Z.; Landmann, R.; Khanna, N.; Infection Biology, Department of Biomedicine, University and University Hospital Basel, Basel, Switzerland. Unpublished data. 2014.
- Rajacic, Z.; Fromm, K.M.; Khanna, N.; Infection Biology, Department of Biomedicine, University and University Hospital Basel, Basel, Switzerland. Unpublished data. 2014.
- Chai, H.; Guo, L.; Wang, X.; Fu, Y.; Guan, J.; Tan, L.; Ren, L.; Yang, K. Antibacterial effect of 317L stainless steel contained copper in prevention of implant-related infection in vitro and in vivo. J. Mater. Sci. 2011, 22, 2525–2535. [Google Scholar] [CrossRef]
- Stewart, S.; Barr, S.; Engiles, J.; Hickok, N.J.; Shapiro, I.M.; Richardson, D.W.; Parvizi, J.; Schaer, T.P. Vancomycin-modified implant surface inhibits biofilm formation and supports bone-healing in an infected osteotomy model in sheep: A proof-of-concept study. J. Bone Joint Surg. 2012, 94, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nowakowska, J.; Landmann, R.; Khanna, N. Foreign Body Infection Models to Study Host-Pathogen Response and Antimicrobial Tolerance of Bacterial Biofilm. Antibiotics 2014, 3, 378-397. https://doi.org/10.3390/antibiotics3030378
Nowakowska J, Landmann R, Khanna N. Foreign Body Infection Models to Study Host-Pathogen Response and Antimicrobial Tolerance of Bacterial Biofilm. Antibiotics. 2014; 3(3):378-397. https://doi.org/10.3390/antibiotics3030378
Chicago/Turabian StyleNowakowska, Justyna, Regine Landmann, and Nina Khanna. 2014. "Foreign Body Infection Models to Study Host-Pathogen Response and Antimicrobial Tolerance of Bacterial Biofilm" Antibiotics 3, no. 3: 378-397. https://doi.org/10.3390/antibiotics3030378
APA StyleNowakowska, J., Landmann, R., & Khanna, N. (2014). Foreign Body Infection Models to Study Host-Pathogen Response and Antimicrobial Tolerance of Bacterial Biofilm. Antibiotics, 3(3), 378-397. https://doi.org/10.3390/antibiotics3030378