
Antimicrobial Use, Human Gut Microbiota and Clostridium difficile Colonization and Infection

Abstract
:1. Introduction
2. Impact of Antimicrobial Use on the Human Intestinal Microbiota
2.1. Clindamycin
2.2. Penicillins
2.3. Cephalosporins
2.4. Fluoroquinolones
3. Colonization Resistance against C. difficile
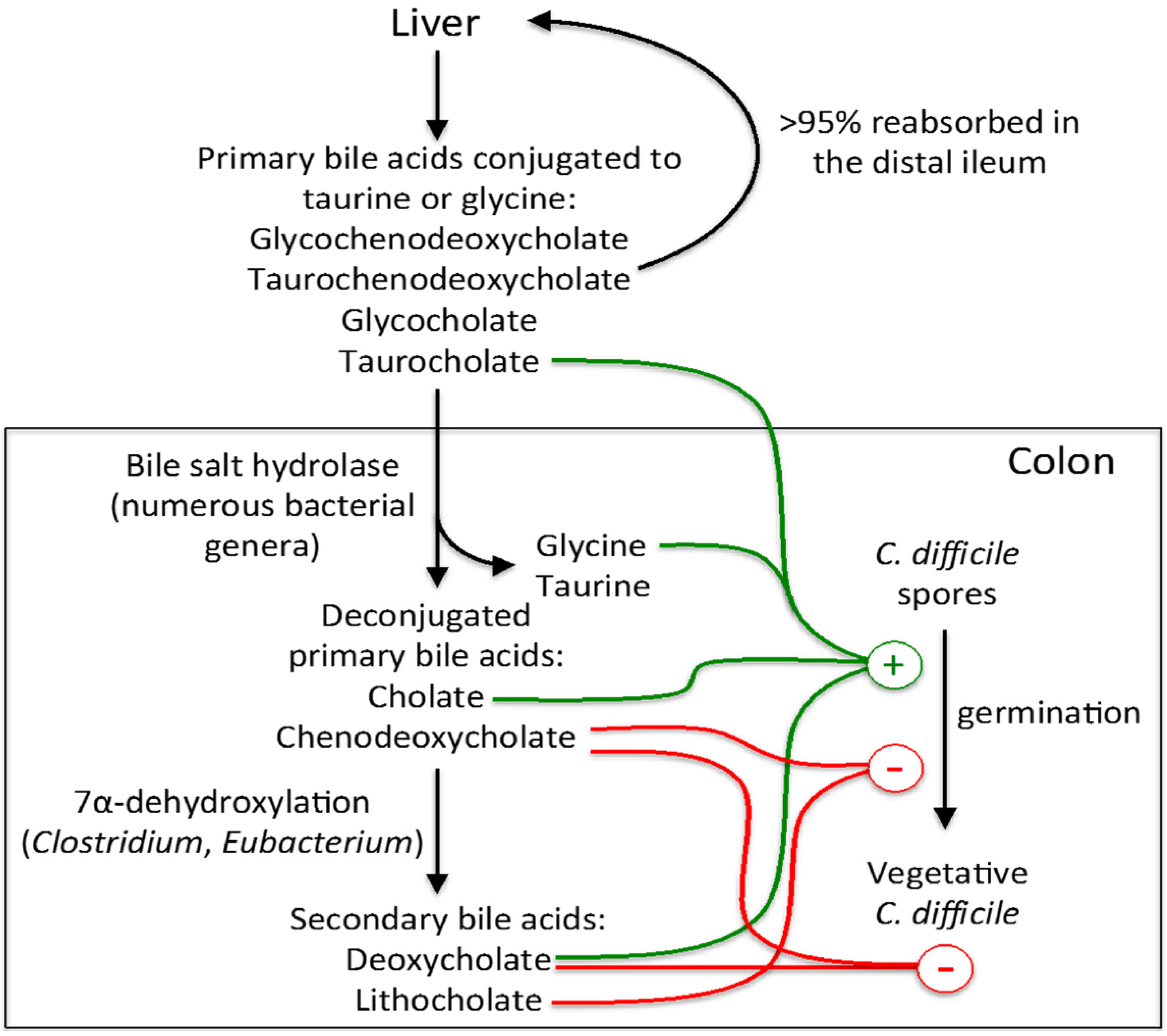
3.1. Influence of Bile Acids on C. difficile Spore Germination and Vegetative Growth
3.2. Competition for Resources

4. Human Gut Microbiota and C. difficile Colonization and Infection

{kind=link}
| Subjects | Fecal Samples Collected Prior to C. difficile Acquisition or CDI Diagnosis | Longitudinal Sample Collection | Inclusion of Epidemiologic Data in Analyses * | Method of Microbiota Measurement | Findings | Reference |
|---|---|---|---|---|---|---|
| Children (n = 10), young adults (n = 7), healthy elderly (n = 5) and geriatric patients diagnosed with CDI (n = 4) | No | No | No | Bacterial culture and in situ hybridization with 16S rRNA probes |
| [62,63,64] |
| Patients with an initial episode of CDI (n = 4), patients with recurrent CDI (n = 3) and healthy subjects (n = 3) | No | No | No | 16S rRNA gene clone libraries |
| [51] |
| Adult outpatients who acquired C. difficile (n = 11), or did not acquire C. difficile (n = 76) during antibiotic treatment | Yes | Yes | Yes | Temporal temperature gradient gel electrophoresis (TGGE) of the 16S rRNA gene (V6–V8 region) |
| [60] |
| Patients with CDI (n = 25) and hospitalized controls (n = 50) | Yes | No | Yes | 16S rRNA microarray |
| [59] |
| Patients with CDI (n = 2), asymptomatic C. difficile carriers (n = 20) and C. difficile-negative elderly subjects (n = 252) | No | No | No | 16S rRNA gene sequencing (V4 region) |
| [52] |
| C. difficile-positive patients (n = 105), C. difficile-negative patients (n = 66) and C. difficile-negative healthy subjects (n = 37) | No | No | No | Denaturing high-pressure liquid chromatography |
| [61] |
| Patients with CDI (n = 39), patients with C. difficile-negative diarrhea (n = 36) and healthy controls (n = 40) | No | No | No | 16S rRNA gene sequencing (V1–V3 region) |
| [53] |
| Patients with CDI (n = 25) and hospitalized controls (n = 25) | Yes | No | Yes | 16S rRNA gene sequencing (V1–V3 and V3–V5 regions) |
| [54] |
| Patients with CDI (n = 14), asymptomatic C. difficile carriers (n = 14) patients with C. difficile-negative diarrhea (n = 16), and patients without diarrhea and C. difficile-negative stool (controls, n = 15) | No | No | No | Quantitative real-time PCR |
| [65] |
| Patients with CDI (n = 10), patients under antibiotic therapy who did not develop CDI (n = 15) and healthy individuals without antibiotic therapy (n = 18) | No | No | No | 16S rRNA sequencing from DNA and RNA (V1–V2 region) |
| [55] |
| Patients with CDI (n = 94), patients with C. difficile-negative diarrhea (n = 89), and healthy controls (n = 155) | No | No | Yes | 16S rRNA gene sequencing (V3–V5 region) |
| [56] |
| Patients who acquired (n = 3) or did not acquire C. difficile (n = 5) during antibiotic treatment | Yes | Yes | No | 16S rRNA gene (V1–V3 region) and metagenomic sequencing |
| [57] |
| Patients with CDI (n = 8), asymptomatic C. difficile carriers (n = 8) and healthy individuals (n = 9) | No | No | No | 16S rRNA gene sequencing (V3–V4 region) |
| [58] |
| Increased Abundance | Decreased Abundance |
|---|---|
| Enterococcus | Bacteroidetes (Bacteroides and Prevotella) |
| Lactobacillus | Bifidobacterium |
| Erysipelotrichaceae | Alistipes |
| Proteobacteria (Enterobacteriaceae) | Roseburia |
| Clostridium clusters XI and XIVa | Coprococcus |
| Veillonella |
5. Fecal Microbiota Transplantation
5.1. Recovery of the Intestinal Microbiota after FMT
5.2. Elaboration of Bacterial Mixtures as a Stool Substitute for FMT
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lessa, F.C.; Mu, Y.; Bamberg, W.M.; Beldavs, Z.G.; Dumyati, G.K.; Dunn, J.R.; Farley, M.M.; Holzbauer, S.M.; Meek, J.I.; Phipps, E.C.; et al. Burden of Clostridium difficile infection in the united states. N. Engl. J. Med. 2015, 372, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Surawicz, C.M.; Brandt, L.J.; Binion, D.G.; Ananthakrishnan, A.N.; Curry, S.R.; Gilligan, P.H.; McFarland, L.V.; Mellow, M.; Zuckerbraun, B.S. Guidelines for diagnosis, treatment, and prevention of Clostridium difficile infections. Am. J. Gastroenterol. 2013, 108, 478–498; quiz 499. [Google Scholar] [CrossRef] [PubMed]
- Vesteinsdottir, I.; Gudlaugsdottir, S.; Einarsdottir, R.; Kalaitzakis, E.; Sigurdardottir, O.; Bjornsson, E.S. Risk factors for Clostridium difficile toxin-positive diarrhea: A population-based prospective case-control study. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 2601–2610. [Google Scholar] [CrossRef] [PubMed]
- Dethlefsen, L.; Huse, S.; Sogin, M.L.; Relman, D.A. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16s rRNA sequencing. PLoS Biol. 2008, 6, e280. [Google Scholar] [CrossRef] [PubMed]
- Dethlefsen, L.; Relman, D.A. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc. Natl. Acad. Sci. USA 2011, 108, 4554–4561. [Google Scholar] [CrossRef] [PubMed]
- Ananthakrishnan, A.N. Clostridium difficile infection: Epidemiology, risk factors and management. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Owens, R.C., Jr.; Donskey, C.J.; Gaynes, R.P.; Loo, V.G.; Muto, C.A. Antimicrobial-associated risk factors for Clostridium difficile infection. Clin. Infect. Dis. 2008, 4, S19–S31. [Google Scholar] [CrossRef] [PubMed]
- Pepin, J.; Saheb, N.; Coulombe, M.A.; Alary, M.E.; Corriveau, M.P.; Authier, S.; Leblanc, M.; Rivard, G.; Bettez, M.; Primeau, V.; et al. Emergence of fluoroquinolones as the predominant risk factor for Clostridium difficile-associated diarrhea: A cohort study during an epidemic in Quebec. Clin. Infect. Dis. 2005, 41, 1254–1260. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Gerding, D.N. Clostridium difficile-associated diarrhea. Clin. Infect. Dis. 1998, 26, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.K.; Johnson, S.; Samore, M.H.; Bliss, D.Z.; Gerding, D.N. Primary symptomless colonisation by Clostridium difficile and decreased risk of subsequent diarrhoea. Lancet 1998, 351, 633–636. [Google Scholar] [CrossRef]
- Zacharioudakis, I.M.; Zervou, F.N.; Pliakos, E.E.; Ziakas, P.D.; Mylonakis, E. Colonization with toxinogenic C. difficile upon hospital admission, and risk of infection: A systematic review and meta-analysis. Am. J. Gastroenterol. 2015, 110, 381–390; quiz 391. [Google Scholar] [CrossRef] [PubMed]
- Riggs, M.M.; Sethi, A.K.; Zabarsky, T.F.; Eckstein, E.C.; Jump, R.L.; Donskey, C.J. Asymptomatic carriers are a potential source for transmission of epidemic and nonepidemic Clostridium difficile strains among long-term care facility residents. Clin. Infect. Dis. 2007, 45, 992–998. [Google Scholar] [CrossRef] [PubMed]
- Eyre, D.W.; Cule, M.L.; Wilson, D.J.; Griffiths, D.; Vaughan, A.; O’Connor, L.; Ip, C.L.; Golubchik, T.; Batty, E.M.; Finney, J.M.; et al. Diverse sources of C. difficile infection identified on whole-genome sequencing. N. Engl. J. Med. 2013, 369, 1195–1205. [Google Scholar] [CrossRef] [PubMed]
- Brandt, L.J.; Aroniadis, O.C.; Mellow, M.; Kanatzar, A.; Kelly, C.; Park, T.; Stollman, N.; Rohlke, F.; Surawicz, C. Long-term follow-up of colonoscopic fecal microbiota transplant for recurrent Clostridium difficile infection. Am. J. Gastroenterol. 2012, 107, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.P.; LaMont, J.T. Clostridium difficile—More difficult than ever. N. Engl. J. Med. 2008, 359, 1932–1940. [Google Scholar] [CrossRef] [PubMed]
- Bakken, J.S.; Borody, T.; Brandt, L.J.; Brill, J.V.; Demarco, D.C.; Franzos, M.A.; Kelly, C.; Khoruts, A.; Louie, T.; Martinelli, L.P.; et al. Treating Clostridium difficile infection with fecal microbiota transplantation. Clin. Gastroenterol. Hepatol. 2011, 9, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- McFarland, L.V.; Elmer, G.W.; Surawicz, C.M. Breaking the cycle: Treatment strategies for 163 cases of recurrent Clostridium difficile disease. Am. J. Gastroenterol. 2002, 97, 1769–1775. [Google Scholar] [CrossRef] [PubMed]
- Van Nood, E.; Vrieze, A.; Nieuwdorp, M.; Fuentes, S.; Zoetendal, E.G.; de Vos, W.M.; Visser, C.E.; Kuijper, E.J.; Bartelsman, J.F.; Tijssen, J.G.; et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N. Engl. J. Med. 2013, 368, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Jernberg, C.; Lofmark, S.; Edlund, C.; Jansson, J.K. Long-term impacts of antibiotic exposure on the human intestinal microbiota. Microbiology 2010, 156, 3216–3223. [Google Scholar] [CrossRef] [PubMed]
- Britton, R.A.; Young, V.B. Role of the intestinal microbiota in resistance to colonization by Clostridium difficile. Gastroenterology 2014, 146, 1547–1553. [Google Scholar] [CrossRef] [PubMed]
- Willing, B.P.; Russell, S.L.; Finlay, B.B. Shifting the balance: Antibiotic effects on host-microbiota mutualism. Nat. Rev. Microbiol. 2011, 9, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, A.; Edlund, C.; Nord, C.E. Effect of antimicrobial agents on the ecological balance of human microflora. Lancet Infect. Dis. 2001, 1, 101–114. [Google Scholar] [CrossRef]
- Rafii, F.; Sutherland, J.B.; Cerniglia, C.E. Effects of treatment with antimicrobial agents on the human colonic microflora. Ther. Clin. Risk Manag. 2008, 4, 1343–1358. [Google Scholar] [PubMed]
- Dhawan, V.K.; Thadepalli, H. Clindamycin: A review of fifteen years of experience. Rev. Infect. Dis. 1982, 4, 1133–1153. [Google Scholar] [CrossRef] [PubMed]
- Orrhage, K.; Brismar, B.; Nord, C.E. Effect of supplements with bifidobacteriurn longum and lactobacillus acidophilus on the intestinal microbiota during administration of clindamycin. Microb. Ecol. Health Dis. 1994, 7, 17–25. [Google Scholar] [CrossRef]
- Kager, L.; Liljeqvist, L.; Malmborg, A.S.; Nord, C.E. Effect of clindamycin prophylaxis on the colonic microflora in patients undergoing colorectal surgery. Antimicrob. Agents Chemother. 1981, 20, 736–740. [Google Scholar] [CrossRef] [PubMed]
- Jernberg, C.; Lofmark, S.; Edlund, C.; Jansson, J.K. Long-term ecological impacts of antibiotic administration on the human intestinal microbiota. ISME J. 2007, 1, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Barc, M.C.; Bourlioux, F.; Rigottier-Gois, L.; Charrin-Sarnel, C.; Janoir, C.; Boureau, H.; Dore, J.; Collignon, A. Effect of amoxicillin-clavulanic acid on human fecal flora in a gnotobiotic mouse model assessed with fluorescence hybridization using group-specific 16s rRNA probes in combination with flow cytometry. Antimicrob. Agents Chemother. 2004, 48, 1365–1368. [Google Scholar] [CrossRef] [PubMed]
- Young, V.B.; Schmidt, T.M. Antibiotic-associated diarrhea accompanied by large-scale alterations in the composition of the fecal microbiota. J. Clin. Microbiol. 2004, 42, 1203–1206. [Google Scholar] [CrossRef] [PubMed]
- De La Cochetiere, M.F.; Durand, T.; Lepage, P.; Bourreille, A.; Galmiche, J.P.; Dore, J. Resilience of the dominant human fecal microbiota upon short-course antibiotic challenge. J. Clin. Microbiol. 2005, 43, 5588–5592. [Google Scholar] [CrossRef] [PubMed]
- Perez-Cobas, A.E.; Gosalbes, M.J.; Friedrichs, A.; Knecht, H.; Artacho, A.; Eismann, K.; Otto, W.; Rojo, D.; Bargiela, R.; von Bergen, M.; et al. Gut microbiota disturbance during antibiotic therapy: A multi-omic approach. Gut 2013, 62, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Vollaard, E.J.; Clasener, H.A. Colonization resistance. Antimicrob. Agents Chemother. 1994, 38, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Stecher, B.; Hardt, W.D. Mechanisms controlling pathogen colonization of the gut. Curr. Opin. Microbiol. 2011, 14, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Stecher, B.; Hardt, W.D. The role of microbiota in infectious disease. Trends Microbiol. 2008, 16, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Stecher, B.; Chaffron, S.; Kappeli, R.; Hapfelmeier, S.; Freedrich, S.; Weber, T.C.; Kirundi, J.; Suar, M.; McCoy, K.D.; von Mering, C.; et al. Like will to like: Abundances of closely related species can predict susceptibility to intestinal colonization by pathogenic and commensal bacteria. PLoS Pathog. 2010, 6, e1000711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guérard, P. Metabolism of cholesterol and bile acids by the gut microbiota. Pathogens 2014, 3, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.V.; Begley, M.; Hill, C.; Gahan, C.G.; Marchesi, J.R. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc. Natl. Acad. Sci. USA 2008, 105, 13580–13585. [Google Scholar] [CrossRef] [PubMed]
- Sorg, J.A.; Sonenshein, A.L. Bile salts and glycine as cogerminants for Clostridium difficile spores. J. Bacteriol. 2008, 190, 2505–2512. [Google Scholar] [CrossRef] [PubMed]
- Sorg, J.A.; Sonenshein, A.L. Chenodeoxycholate is an inhibitor of Clostridium difficile spore germination. J. Bacteriol. 2009, 191, 1115–1117. [Google Scholar] [CrossRef] [PubMed]
- Sorg, J.A.; Sonenshein, A.L. Inhibiting the initiation of Clostridium difficile spore germination using analogs of chenodeoxycholic acid, a bile acid. J. Bacteriol. 2010, 192, 4983–4990. [Google Scholar] [CrossRef] [PubMed]
- Buffie, C.G.; Bucci, V.; Stein, R.R.; McKenney, P.T.; Ling, L.; Gobourne, A.; No, D.; Liu, H.; Kinnebrew, M.; Viale, A.; et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 2015, 517, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.H.; Perini, F. Role of competition for nutrients in suppression of Clostridium difficile by the colonic microflora. Infect. Immun. 1988, 56, 2610–2614. [Google Scholar] [PubMed]
- Ng, K.M.; Ferreyra, J.A.; Higginbottom, S.K.; Lynch, J.B.; Kashyap, P.C.; Gopinath, S.; Naidu, N.; Choudhury, B.; Weimer, B.C.; Monack, D.M.; et al. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature 2013, 502, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Britton, R.A.; Young, V.B. Interaction between the intestinal microbiota and host in Clostridium difficile colonization resistance. Trends Microbiol. 2012, 20, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Sambol, S.P.; Merrigan, M.M.; Tang, J.K.; Johnson, S.; Gerding, D.N. Colonization for the prevention of Clostridium difficile disease in hamsters. J. Infect. Dis. 2002, 186, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Merrigan, M.M.; Sambol, S.P.; Johnson, S.; Gerding, D.N. Prevention of fatal Clostridium difficile-associated disease during continuous administration of clindamycin in hamsters. J. Infect. Dis. 2003, 188, 1922–1927. [Google Scholar] [CrossRef] [PubMed]
- Merrigan, M.M.; Sambol, S.P.; Johnson, S.; Gerding, D.N. New approach to the management of Clostridium difficile infection: Colonisation with non-toxigenic C. difficile during daily ampicillin or ceftriaxone administration. Int. J. Antimicrob. Agents 2009, 33, S46–S50. [Google Scholar] [CrossRef]
- Gerding, D.N.; Meyer, T.; Lee, C.; Cohen, S.H.; Murthy, U.K.; Poirier, A.; van Schooneveld, T.C.; Pardi, D.S.; Ramos, A.; Barron, M.A.; et al. Administration of spores of nontoxigenic Clostridium difficile strain m3 for prevention of recurrent c difficile infection: A randomized clinical trial. JAMA 2015, 313, 1719–1727. [Google Scholar] [CrossRef] [PubMed]
- Theriot, C.M.; Young, V.B. Microbial and metabolic interactions between the gastrointestinal tract and Clostridium difficile infection. Gut Microb. 2014, 5, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.Y.; Antonopoulos, D.A.; Kalra, A.; Tonelli, A.; Khalife, W.T.; Schmidt, T.M.; Young, V.B. Decreased diversity of the fecal microbiome in recurrent Clostridium difficile-associated diarrhea. J. Infect. Dis. 2008, 197, 435–438. [Google Scholar] [CrossRef] [PubMed]
- Rea, M.C.; O’Sullivan, O.; Shanahan, F.; O’Toole, P.W.; Stanton, C.; Ross, R.P.; Hill, C. Clostridium difficile carriage in elderly subjects and associated changes in the intestinal microbiota. J. Clin. Microbiol. 2012, 50, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Antharam, V.C.; Li, E.C.; Ishmael, A.; Sharma, A.; Mai, V.; Rand, K.H.; Wang, G.P. Intestinal dysbiosis and depletion of butyrogenic bacteria in Clostridium difficile infection and nosocomial diarrhea. J. Clin. Microbiol. 2013, 51, 2884–2892. [Google Scholar] [CrossRef] [PubMed]
- Vincent, C.; Stephens, D.A.; Loo, V.G.; Edens, T.J.; Behr, M.A.; Dewar, K.; Manges, A.R. Reductions in intestinal clostridiales precede the development of nosocomial Clostridium difficile infection. Microbiome 2013, 1, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Knecht, H.; Neulinger, S.C.; Heinsen, F.A.; Knecht, C.; Schilhabel, A.; Schmitz, R.A.; Zimmermann, A.; dos Santos, V.M.; Ferrer, M.; Rosenstiel, P.C.; et al. Effects of beta-lactam antibiotics and fluoroquinolones on human gut microbiota in relation to Clostridium difficile associated diarrhea. PLoS ONE 2014, 9, e89417. [Google Scholar] [CrossRef] [PubMed]
- Schubert, A.M.; Rogers, M.A.; Ring, C.; Mogle, J.; Petrosino, J.P.; Young, V.B.; Aronoff, D.M.; Schloss, P.D. Microbiome data distinguish patients with Clostridium difficile infection and non-C. difficile-associated diarrhea from healthy controls. MBIO 2014, 5, e01021-14. [Google Scholar] [CrossRef] [PubMed]
- Perez-Cobas, A.E.; Artacho, A.; Ott, S.J.; Moya, A.; Gosalbes, M.J.; Latorre, A. Structural and functional changes in the gut microbiota associated to Clostridium difficile infection. Front. microbiol. 2014, 5. [Google Scholar] [CrossRef]
- Zhang, L.; Dong, D.; Jiang, C.; Li, Z.; Wang, X.; Peng, Y. Insight into alteration of gut microbiota in Clostridium difficile infection and asymptomatic C. difficile colonization. Anaerobe 2015, 34, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Manges, A.R.; Labbe, A.; Loo, V.G.; Atherton, J.K.; Behr, M.A.; Masson, L.; Tellis, P.A.; Brousseau, R. Comparative metagenomic study of alterations to the intestinal microbiota and risk of nosocomial Clostridum difficile-associated disease. J. Infect. Dis. 2010, 202, 1877–1884. [Google Scholar] [CrossRef] [PubMed]
- De la Cochetière, M.F.; Durand, T.; Lalande, V.; Petit, J.C.; Potel, G.; Beaugerie, L. Effect of antibiotic therapy on human fecal microbiota and the relation to the development of Clostridium difficile. Microb. Ecol. 2008, 56, 395–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skraban, J.; Dzeroski, S.; Zenko, B.; Mongus, D.; Gangl, S.; Rupnik, M. Gut microbiota patterns associated with colonization of different Clostridium difficile ribotypes. PLoS ONE 2013, 8, e58005. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, M.J.; Sharp, R.; Macfarlane, G.T. Age and disease related changes in intestinal bacterial populations assessed by cell culture, 16s rrna abundance, and community cellular fatty acid profiles. Gut 2001, 48, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, M.J.; Macfarlane, G.T. Changes in predominant bacterial populations in human faeces with age and with Clostridium difficile infection. J. Med. Microbiol. 2002, 51, 448–454. [Google Scholar] [PubMed]
- Hopkins, M.J.; Sharp, R.; Macfarlane, G.T. Variation in human intestinal microbiota with age. Dig. Liver Dis. 2002, 34, S12–S18. [Google Scholar] [CrossRef]
- Goldberg, E.; Amir, I.; Zafran, M.; Gophna, U.; Samra, Z.; Pitlik, S.; Bishara, J. The correlation between Clostridium-difficile infection and human gut concentrations of bacteroidetes phylum and clostridial species. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Donskey, C.J.; Chowdhry, T.K.; Hecker, M.T.; Hoyen, C.K.; Hanrahan, J.A.; Hujer, A.M.; Hutton-Thomas, R.A.; Whalen, C.C.; Bonomo, R.A.; Rice, L.B. Effect of antibiotic therapy on the density of vancomycin-resistant enterococci in the stool of colonized patients. N. Engl. J. Med. 2000, 343, 1925–1932. [Google Scholar] [CrossRef] [PubMed]
- Ubeda, C.; Taur, Y.; Jenq, R.R.; Equinda, M.J.; Son, T.; Samstein, M.; Viale, A.; Socci, N.D.; van den Brink, M.R.; Kamboj, M.; et al. Vancomycin-resistant Enterococcus domination of intestinal microbiota is enabled by antibiotic treatment in mice and precedes bloodstream invasion in humans. J. Clin. Investig. 2010, 120, 4332–4341. [Google Scholar] [CrossRef] [PubMed]
- Lawley, T.D.; Clare, S.; Walker, A.W.; Goulding, D.; Stabler, R.A.; Croucher, N.; Mastroeni, P.; Scott, P.; Raisen, C.; Mottram, L.; et al. Antibiotic treatment of Clostridium difficile carrier mice triggers a supershedder state, spore-mediated transmission, and severe disease in immunocompromised hosts. Infect. Immun. 2009, 77, 3661–3669. [Google Scholar] [CrossRef] [PubMed]
- Mazmanian, S.K.; Round, J.L.; Kasper, D.L. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 2008, 453, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.B.; Gill, N.; Willing, B.P.; Antunes, L.C.; Russell, S.L.; Croxen, M.A.; Finlay, B.B. The intestinal microbiota plays a role in Salmonella-induced colitis independent of pathogen colonization. PLoS ONE 2011, 6, e20338. [Google Scholar] [CrossRef] [PubMed]
- Pryde, S.E.; Duncan, S.H.; Hold, G.L.; Stewart, C.S.; Flint, H.J. The microbiology of butyrate formation in the human colon. FEMS Microbiol. Lett. 2002, 217, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Guilloteau, P.; Martin, L.; Eeckhaut, V.; Ducatelle, R.; Zabielski, R.; van Immerseel, F. From the gut to the peripheral tissues: The multiple effects of butyrate. Nutr. Res. Rev. 2010, 23, 366–384. [Google Scholar] [CrossRef] [PubMed]
- Hamer, H.M.; Jonkers, D.; Venema, K.; Vanhoutvin, S.; Troost, F.J.; Brummer, R.J. Review article: The role of butyrate on colonic function. Aliment. Pharmacol. Ther. 2008, 27, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Rolfe, R.D. Role of volatile fatty acids in colonization resistance to Clostridium difficile. Infect. Immun. 1984, 45, 185–191. [Google Scholar] [PubMed]
- May, T.; Mackie, R.I.; Fahey, G.C., Jr.; Cremin, J.C.; Garleb, K.A. Effect of fiber source on short-chain fatty acid production and on the growth and toxin production by Clostridium difficile. Scand. J.Gastroenterol. 1994, 29, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.M.; de Souza, R.; Kendall, C.W.; Emam, A.; Jenkins, D.J. Colonic health: Fermentation and short chain fatty acids. J. Clin. Gastroenterol. 2006, 40, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Eiseman, B.; Silen, W.; Bascom, G.S.; Kauvar, A.J. Fecal enema as an adjunct in the treatment of pseudomembranous enterocolitis. Surgery 1958, 44, 854–859. [Google Scholar] [PubMed]
- Dodin, M.; Katz, D.E. Faecal microbiota transplantation for Clostridium difficile infection. Int. J. Clin. Pract. 2014, 68, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Gough, E.; Shaikh, H.; Manges, A.R. Systematic review of intestinal microbiota transplantation (fecal bacteriotherapy) for recurrent Clostridium difficile infection. Clin. Infect. Dis. 2011, 53, 994–1002. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, M.J.; Weingarden, A.R.; Unno, T.; Khoruts, A.; Sadowsky, M.J. High-throughput DNA sequence analysis reveals stable engraftment of gut microbiota following transplantation of previously frozen fecal bacteria. Gut Microb. 2013, 4, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Khoruts, A.; Dicksved, J.; Jansson, J.K.; Sadowsky, M.J. Changes in the composition of the human fecal microbiome after bacteriotherapy for recurrent Clostridium difficile-associated diarrhea. J. Clin. Gastroenterol. 2010, 44, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Shahinas, D.; Silverman, M.; Sittler, T.; Chiu, C.; Kim, P.; Allen-Vercoe, E.; Weese, S.; Wong, A.; Low, D.E.; Pillai, D.R. Toward an understanding of changes in diversity associated with fecal microbiome transplantation based on 16s rRNA gene deep sequencing. MBIO 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Lawley, T.D.; Clare, S.; Walker, A.W.; Stares, M.D.; Connor, T.R.; Raisen, C.; Goulding, D.; Rad, R.; Schreiber, F.; Brandt, C.; et al. Targeted restoration of the intestinal microbiota with a simple, defined bacteriotherapy resolves relapsing Clostridium difficile disease in mice. PLoS Pathog. 2012, 8, e1002995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tvede, M.; Rask-Madsen, J. Bacteriotherapy for chronic relapsing Clostridium difficile diarrhoea in six patients. Lancet 1989, 1, 1156–1160. [Google Scholar] [CrossRef]
- Petrof, E.O.; Gloor, G.B.; Vanner, S.J.; Weese, S.J.; Carter, D.; Daigneault, M.C.; Brown, E.M.; Schroeter, K.; Allen-Vercoe, E. Stool substitute transplant therapy for the eradication of Clostridium difficile infection: “Repoopulating” the gut. Microbiome 2013. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vincent, C.; Manges, A.R. Antimicrobial Use, Human Gut Microbiota and Clostridium difficile Colonization and Infection. Antibiotics 2015, 4, 230-253. https://doi.org/10.3390/antibiotics4030230
Vincent C, Manges AR. Antimicrobial Use, Human Gut Microbiota and Clostridium difficile Colonization and Infection. Antibiotics. 2015; 4(3):230-253. https://doi.org/10.3390/antibiotics4030230
Chicago/Turabian StyleVincent, Caroline, and Amee R. Manges. 2015. "Antimicrobial Use, Human Gut Microbiota and Clostridium difficile Colonization and Infection" Antibiotics 4, no. 3: 230-253. https://doi.org/10.3390/antibiotics4030230
APA StyleVincent, C., & Manges, A. R. (2015). Antimicrobial Use, Human Gut Microbiota and Clostridium difficile Colonization and Infection. Antibiotics, 4(3), 230-253. https://doi.org/10.3390/antibiotics4030230