Corrosion Resistance of Boronized, Aluminized, and Chromized Thermal Diffusion-Coated Steels in Simulated High-Temperature Recovery Boiler Conditions

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. Corrosion Testing
2.3. Materials Examination
3. Results and Discussion
3.1. Microstructural Analysis
3.1.1. Coating Microstructure before Corrosive Attack
3.1.2. Qualitative Comparison of Top Surface of Samples (Appearance) after Corrosive Attack
3.1.3. Microstructural Analysis after Corrosive Attack
Low-Temperature Corrosive Attack
High-Temperature Corrosive Attack
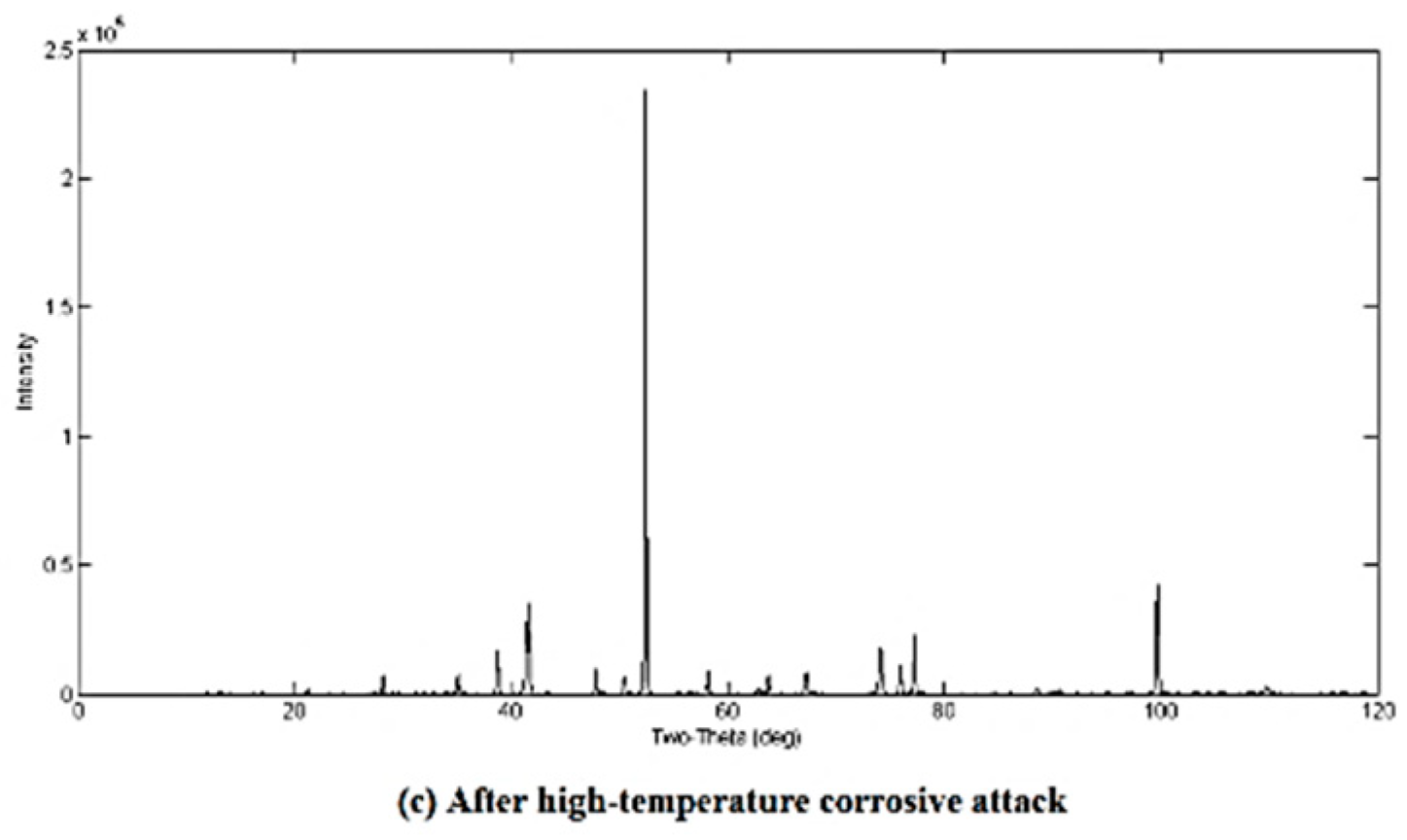
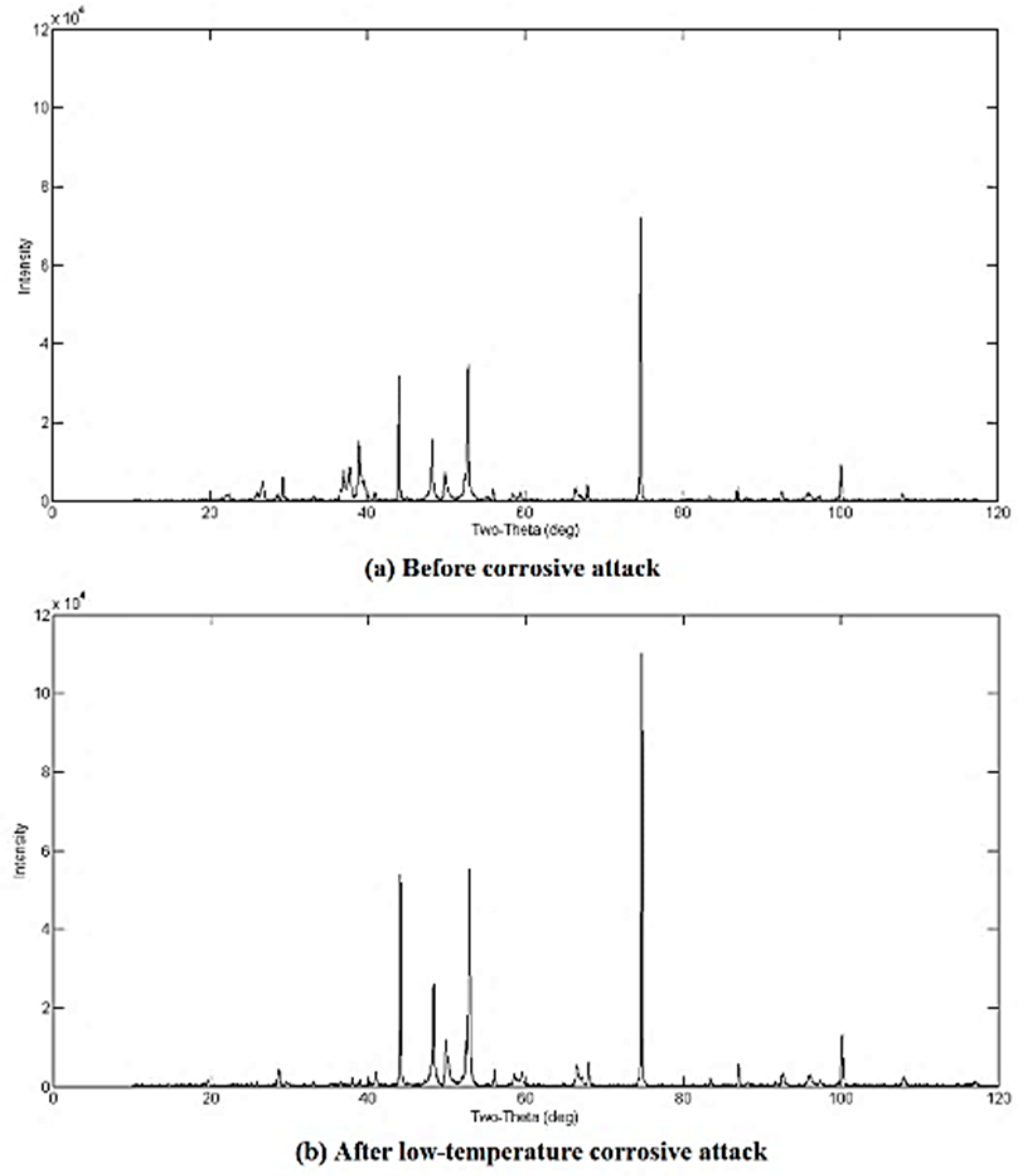
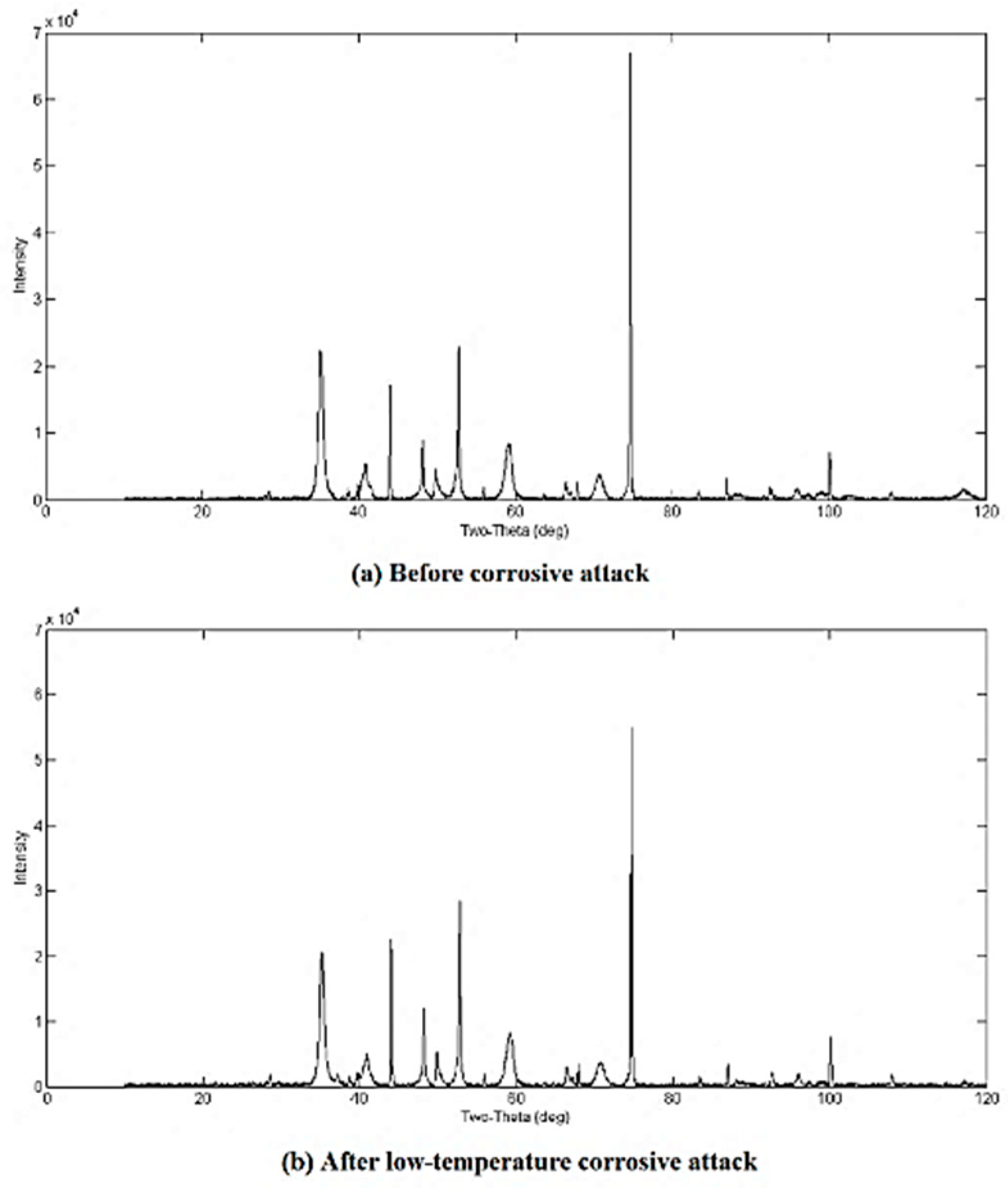
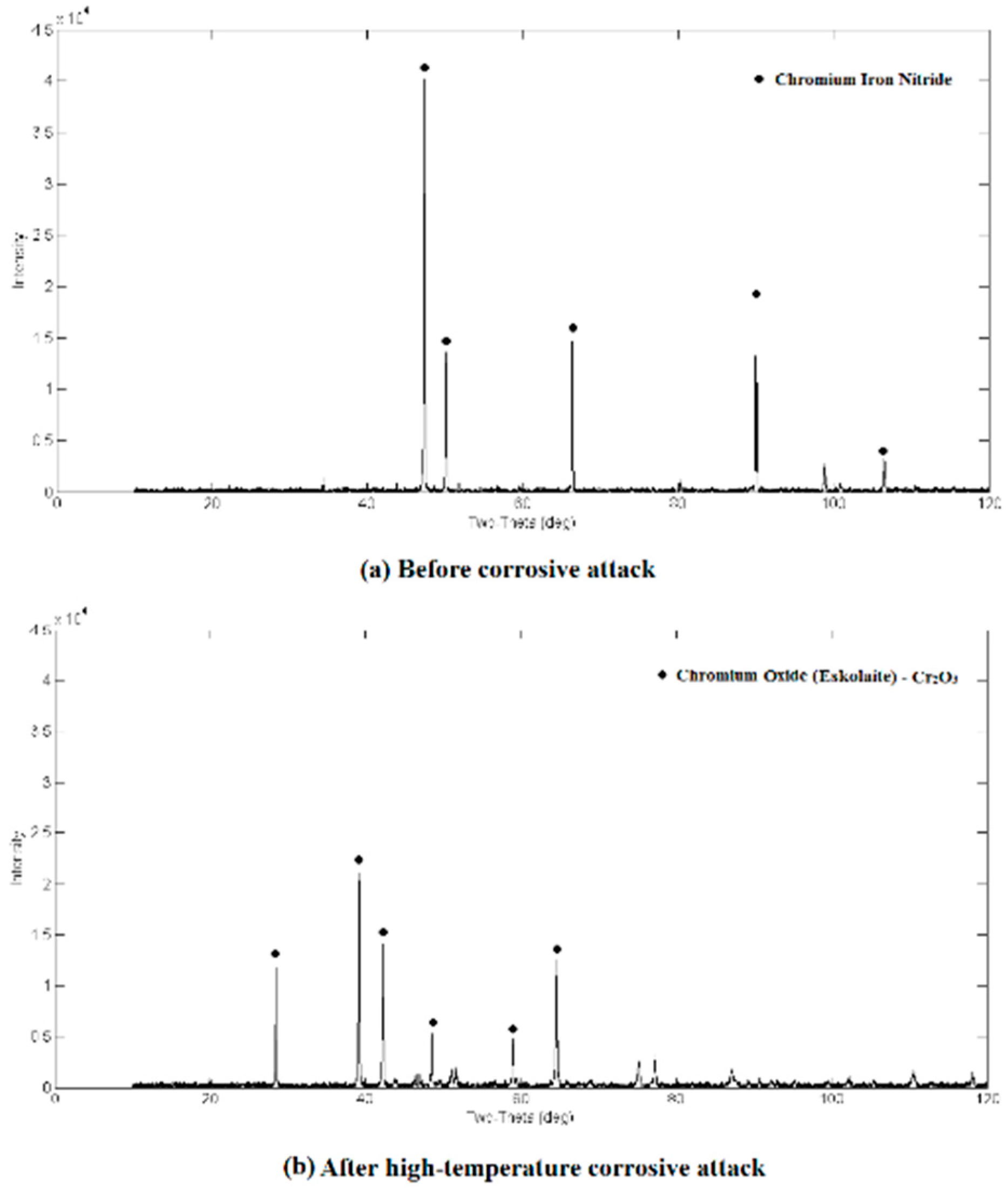
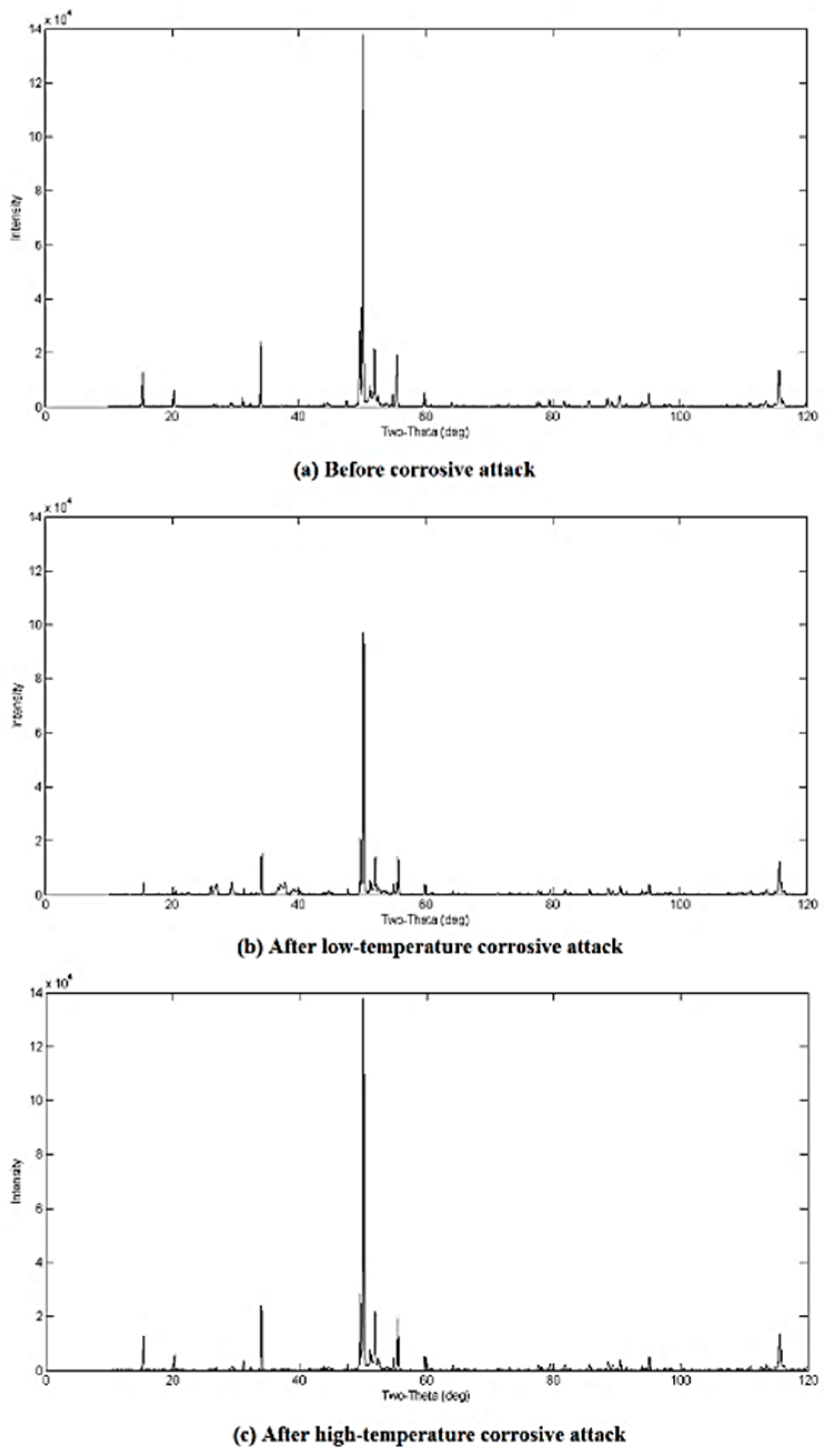
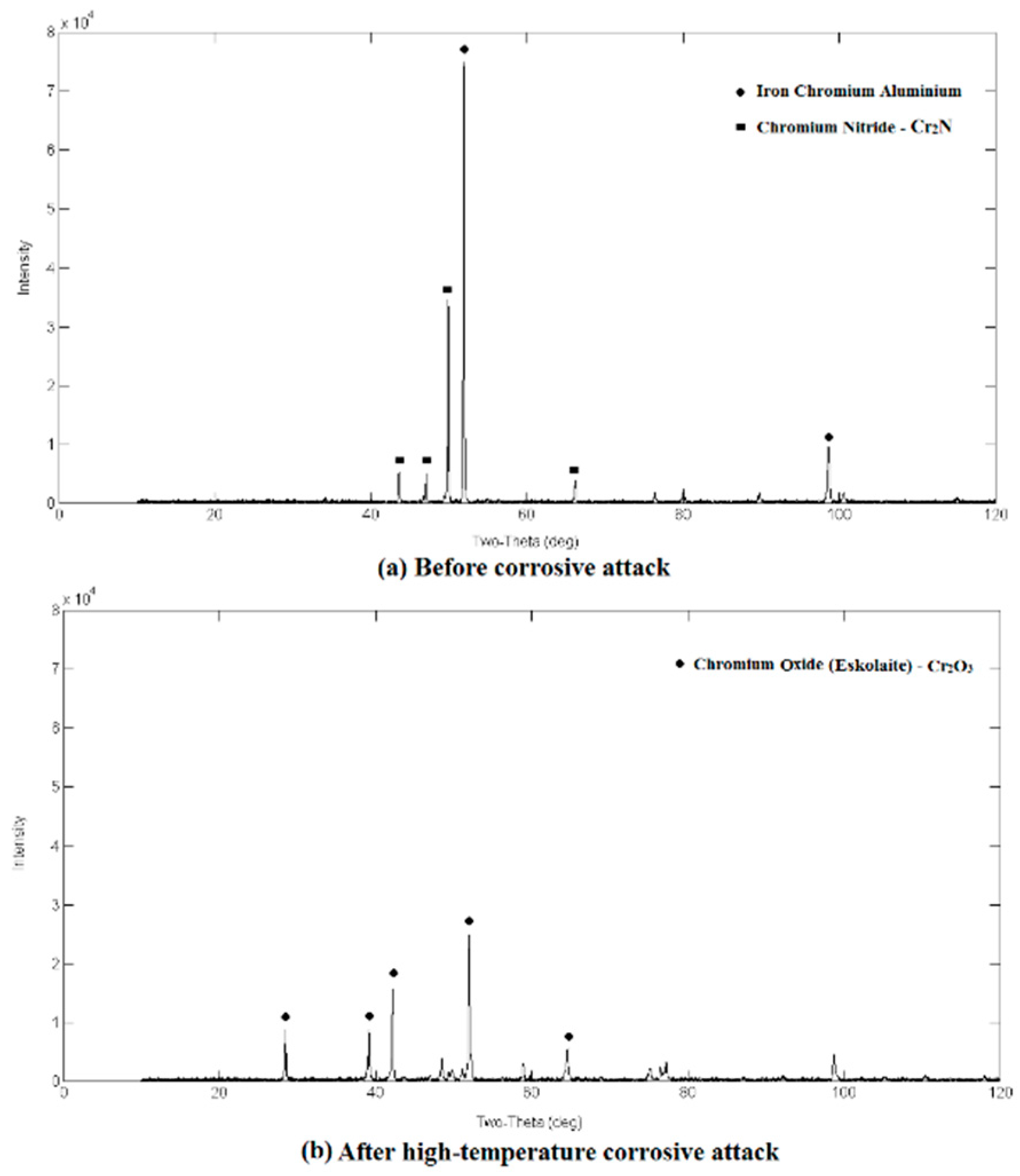
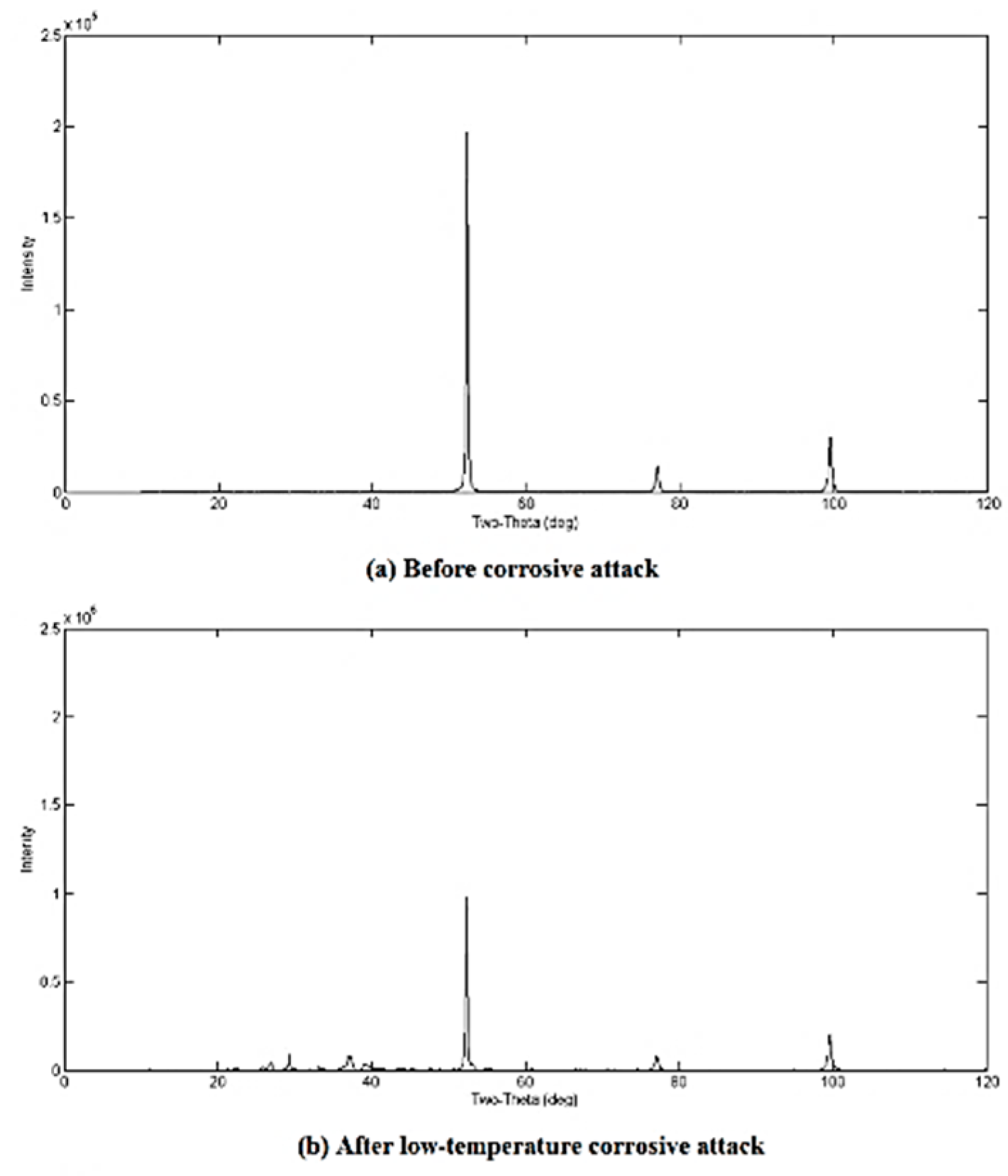
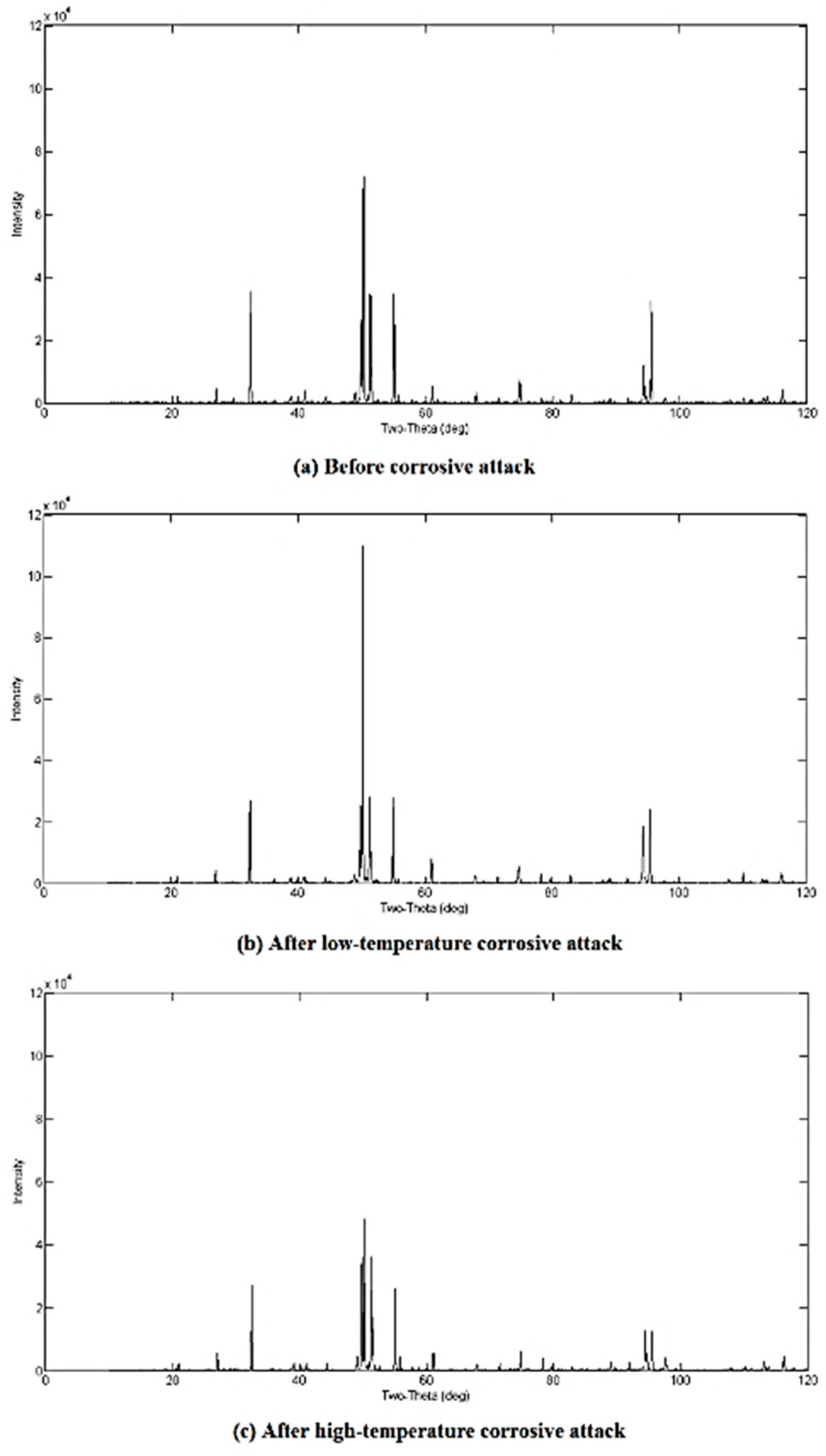
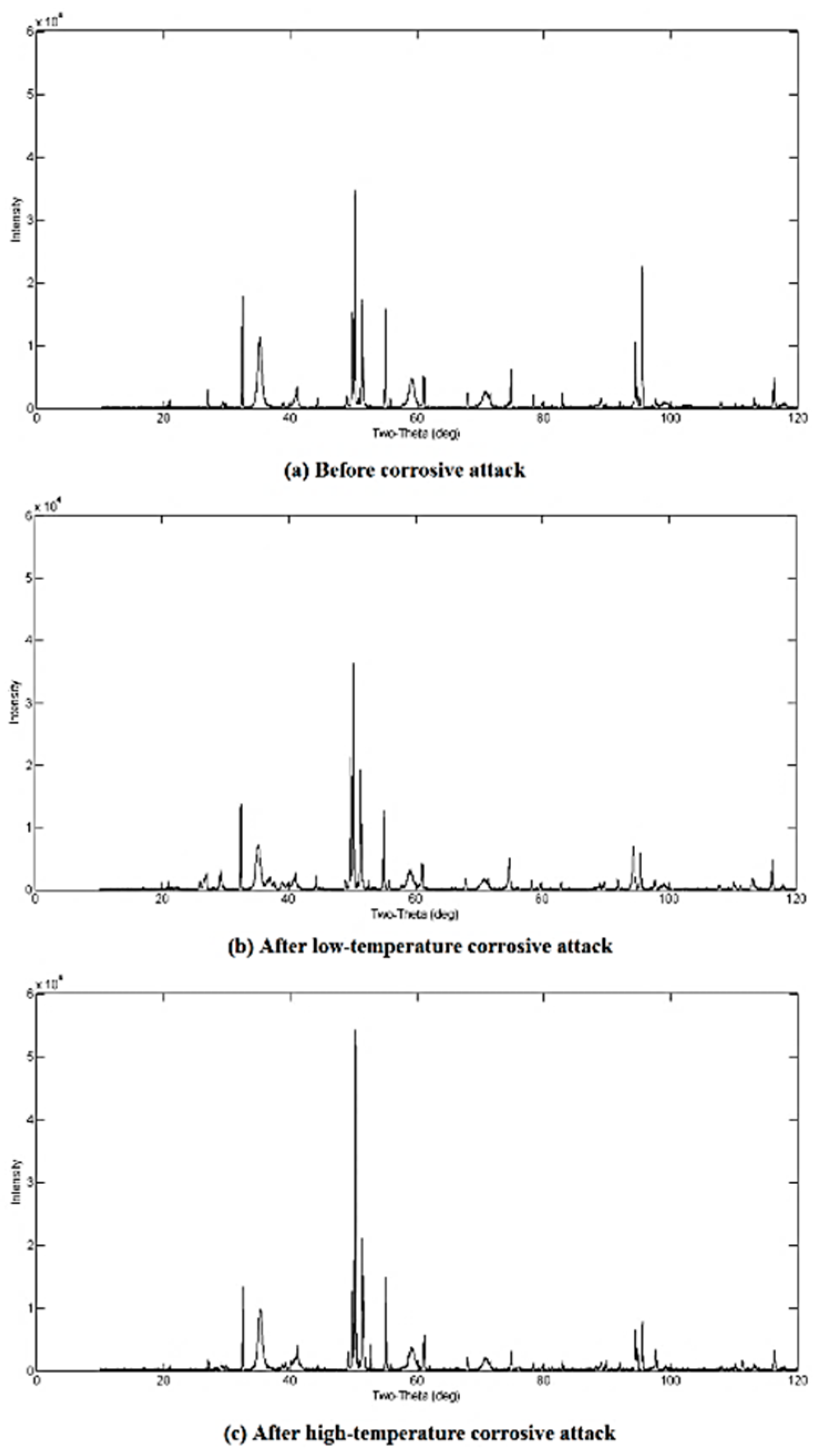
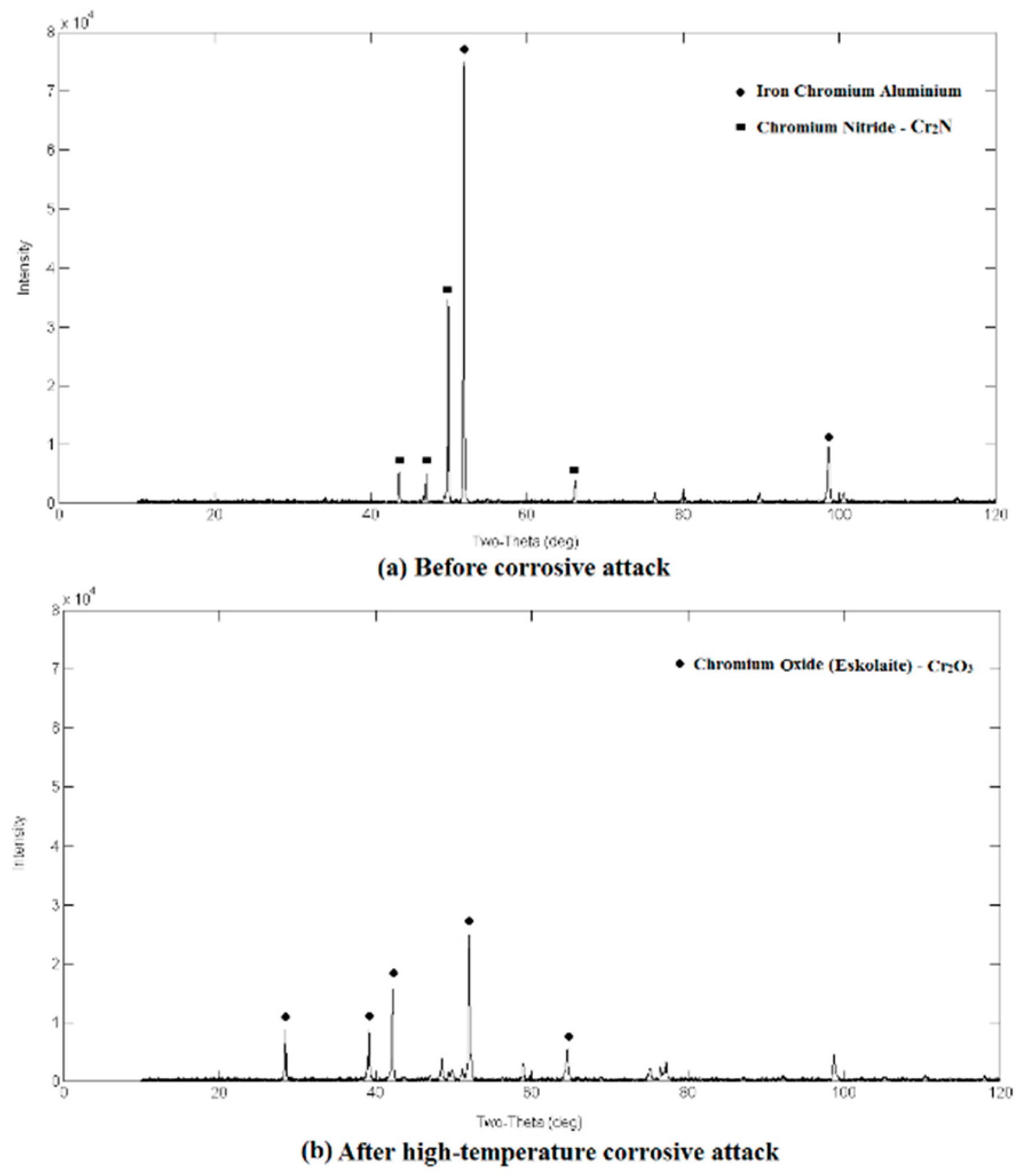
3.2. Qualitative XRD Analysis
3.3. Mass Change after Corrosive Attack
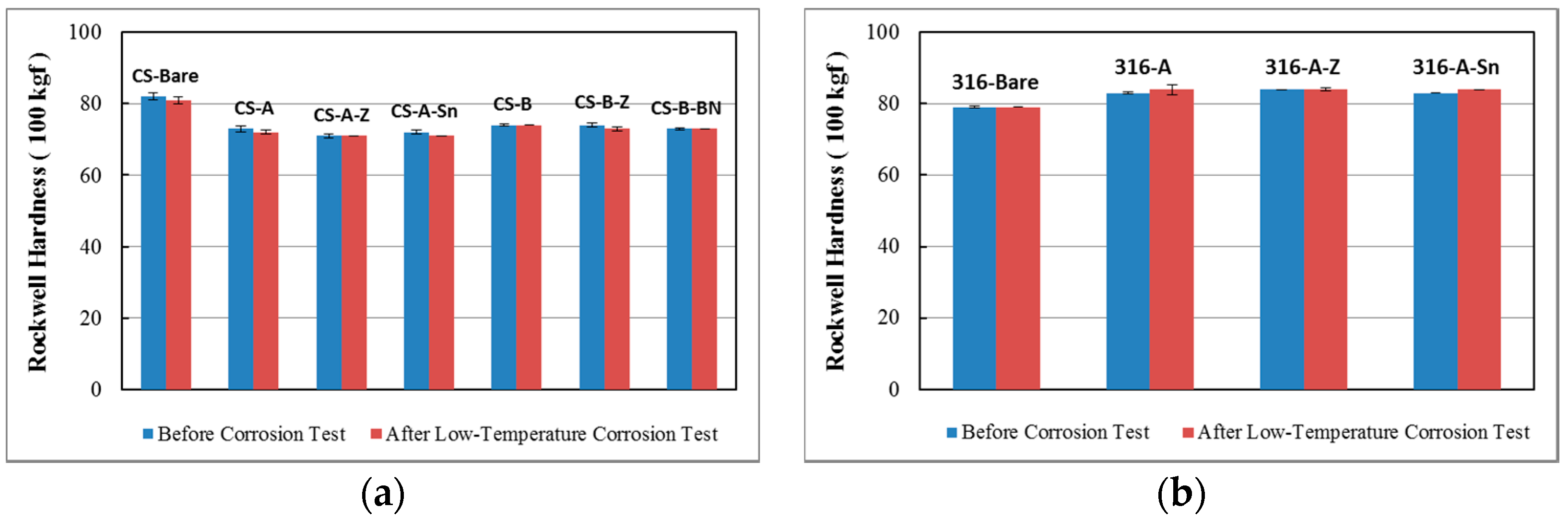
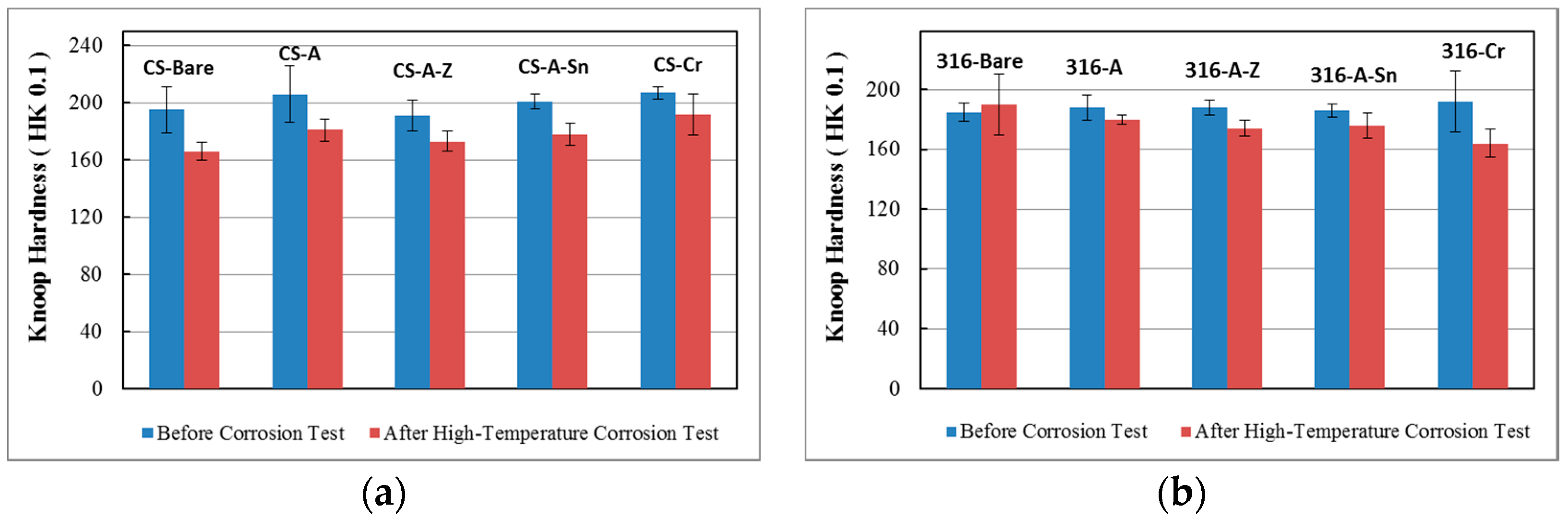
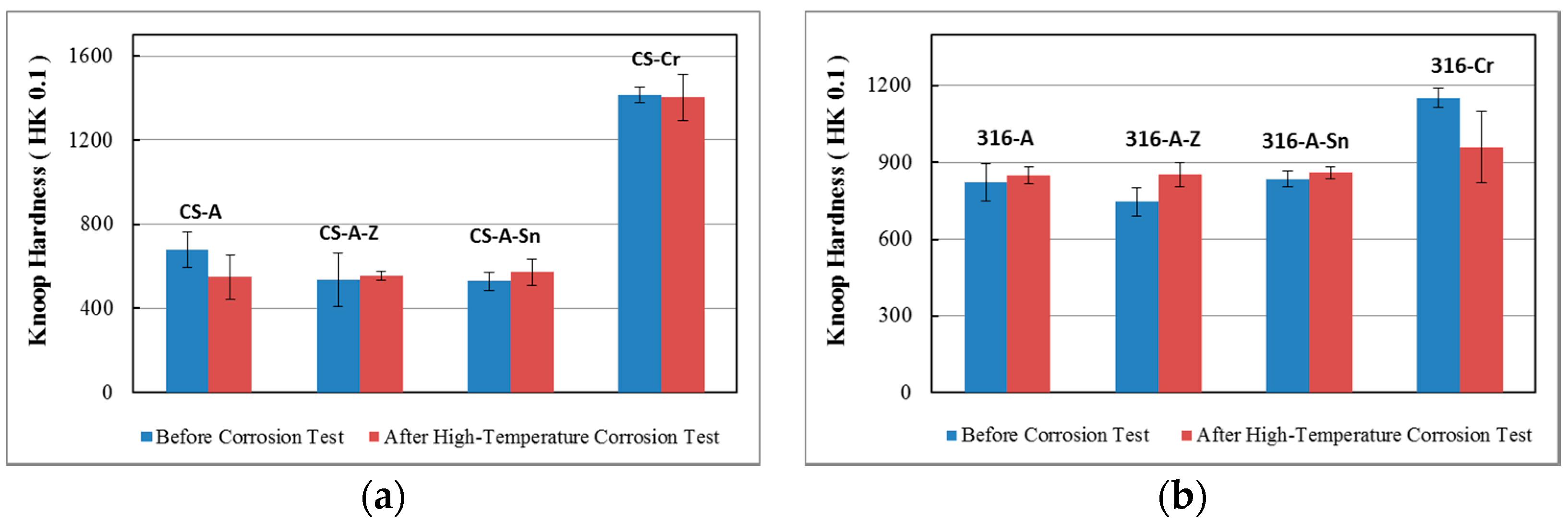
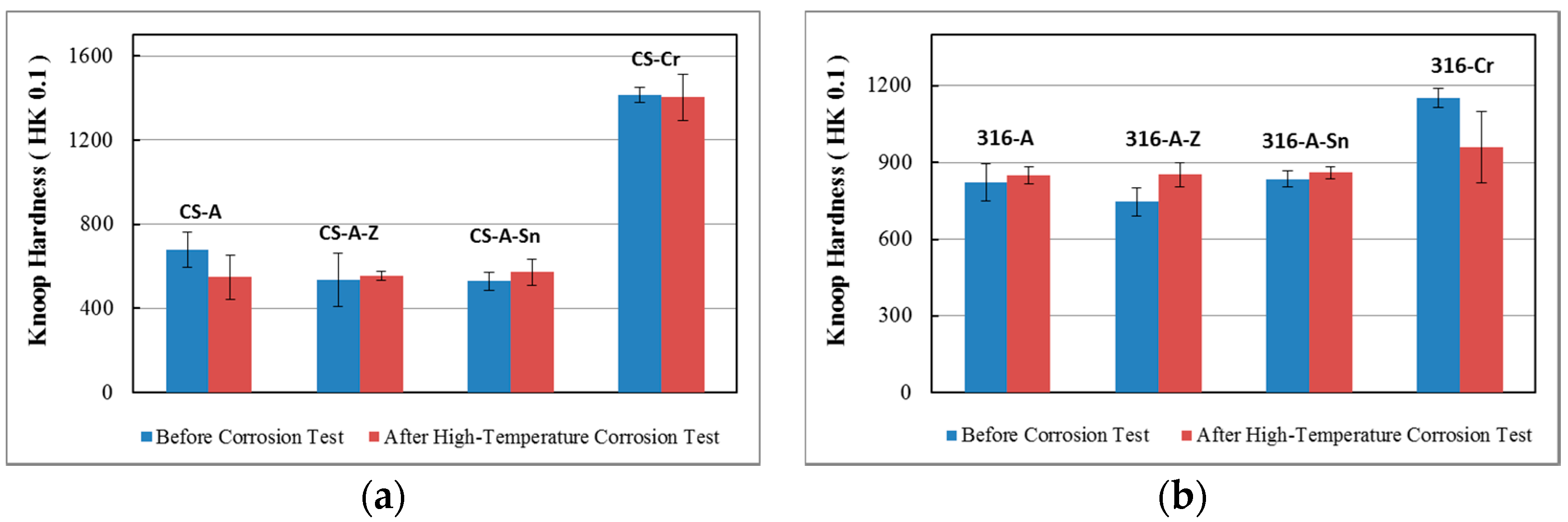
3.4. Hardness Evaluation
3.4.1. Hardness of the Substrates
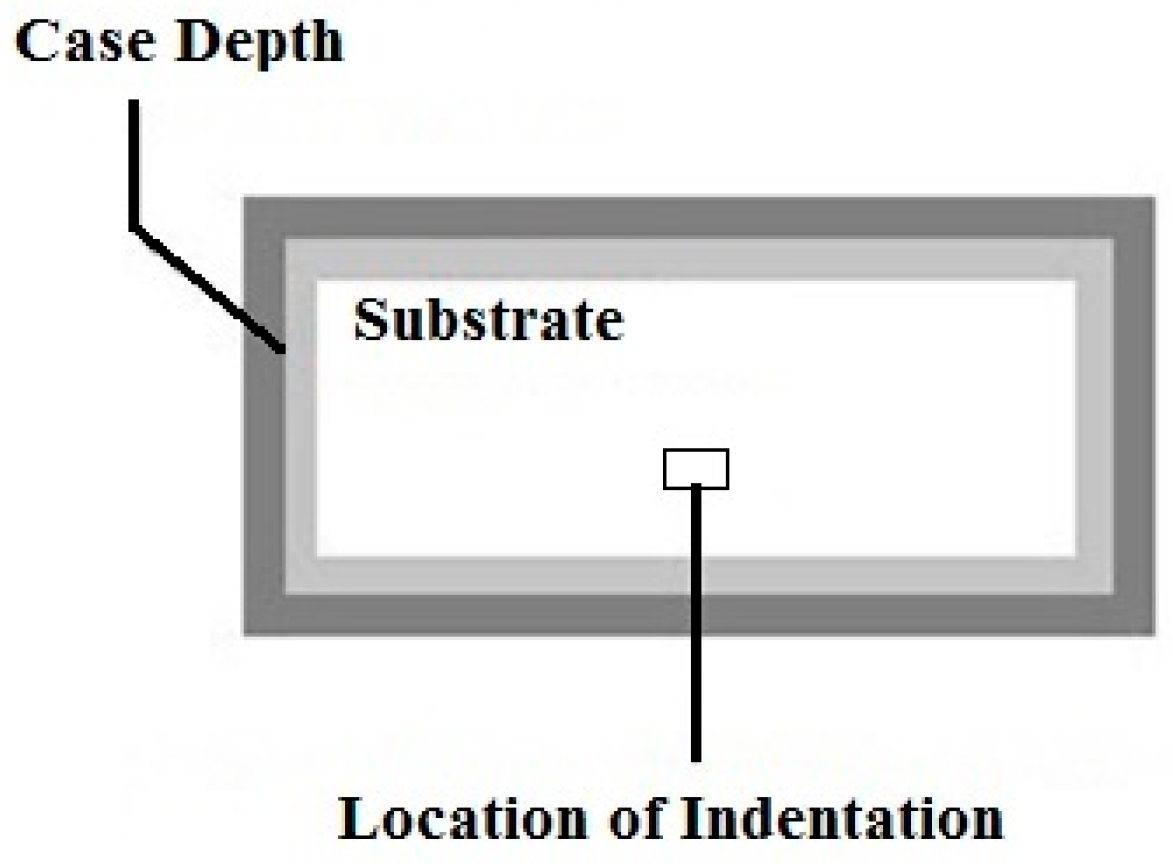
3.4.2. Hardness of the Case Depth
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A




Appendix B











Appendix C








Appendix D






References
- Darmawan, A.; Hardi, F.; Yoshikawa, K.; Aziz, M.; Tokimatsu, K. Enhanced process integration of black liquor evaporation, gasification, and combined cycle. Appl. Energy 2017, 204, 1035–1042. [Google Scholar] [CrossRef]
- Tran, H.N. Upper furnace deposition and plugging. In Kraft Recovery Boilers; Adams, T.N., Ed.; Tappi Press: Atlanta, GA, USA, 1997; pp. 245–282. [Google Scholar]
- Tran, H.N.; Barham, D.; Hupa, M. Fireside corrosion in Kraft recovery boilers—An overview. Mater. Preform. 1988, 27, 40–45. [Google Scholar]
- Naqvi, M.; Yan, J.; Dahlquist, E. Black liquor gasification integrated in pulp and paper mills: A critical review. Bioresour. Technol. 2010, 101, 8001–8015. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Bie, H.; Zhang, Y.; Chen, P.; Fang, W.; Bie, R. Release of K and Cl and emissions of NOx and SO2 during reed black liquor combustion in a fluidized bed. Energy Fuels 2017, 31, 1631–1637. [Google Scholar] [CrossRef]
- Li, S.; Themelis, N.; Castaldi, M. High-temperature corrosion in waste-to-energy boilers. J. Therm. Spray Technol. 2007, 16, 104–110. [Google Scholar]
- Riedl, R.; Dahl, J.; Obernberger, I.; Narodoslawsky, M. Corrosion in fire tube boilers of biomass combustion plants. In Proceedings of the China International Corrosion Control Conference, Beijing, China, 26–28 October 1999; p. 90129. [Google Scholar]
- Grabke, H.J.; Spigel, M.; Zahs, A. Role of alloying elements and carbides in the chlorine-induced corrosion of steels and alloys. Mater. Res. 2004, 7, 89–95. [Google Scholar] [CrossRef] [Green Version]
- Grabke, H.J.; Reese, E.; Spigel, M. The effects of chlorides, hydrogen chloride, and sulfur dioxide in the oxidation of steels below deposits. Corros. Sci. 1995, 37, 1023–1043. [Google Scholar] [CrossRef]
- Sorell, G. The role of chlorine in high temperature corrosion in waste-to-energy plants. Mater. High Temp. 1997, 14, 207–220. [Google Scholar] [CrossRef]
- Buscaglia, V.; Nanni, P.; Bottino, C. The mechanism of sodium sulphate-induced low temperature hot corrosion of pure iron. Corros. Sci. 1990, 311, 327–349. [Google Scholar] [CrossRef]
- He, J.; Xiong, W.; Zhang, W.; Li, W.; Long, K. Study on the high-temperature corrosion behavior of superheater steels of biomass-fired boiler in molten alkali salts’ mixtures. Adv. Mech. Eng. 2016, 8, 1687814016678163. [Google Scholar] [CrossRef]
- Xu, S.; Wang, C.; Wang, W. Failure analysis of stress corrosion cracking in heat exchanger tubes during start-up operation. Eng. Fail. Anal. 2015, 51, 1–8. [Google Scholar] [CrossRef]
- Barbosa, C.; De Barros, S.K.; De Cerqueira Abud, I.; Do Nascimento, J.L.; De Carvalho, S.S. Failure analysis of an aqua tubular boiler tube. J. Fail. Anal. Prev. 2012, 12, 654–659. [Google Scholar] [CrossRef]
- Ahmad, J.; Purbolaksono, J.; Beng, L.C. Thermal fatigue and corrosion fatigue in heat recovery area wall side tubes. Eng. Fail. Anal. 2010, 17, 334–343. [Google Scholar] [CrossRef]
- Roberge, P.R. Handbook of Corrosion Engineering, 2nd ed.; McGraw-Hill Education: New York, NY, USA, 2012; ISBN 9780071750370. [Google Scholar]
- Levy, A.V. The erosion-corrosion behavior of protective coatings. Surf. Coat. Technol. 1988, 36, 387–406. [Google Scholar] [CrossRef]
- Wang, D.; Bierwagen, G.P. Sol-gel coatings on metals for corrosion protection. Prog. Org. Coat. 2009, 64, 327–338. [Google Scholar] [CrossRef]
- Cha, S.C.; Gudenau, H.W.; Bayer, G.T. Comparison of corrosion behaviour of thermal sprayed and diffusion-coated materials. Mater. Corros. 2002, 53, 195–205. [Google Scholar] [CrossRef]
- Günthner, M.; Kraus, T.; Krenkel, W.; Motz, G.; Dierdorf, A.; Decker, D. Particle-filled PHPS silazane-based coatings on steel. Appl. Ceram. Technol. 2009, 6, 373–380. [Google Scholar] [CrossRef]
- Movchan, B.A.; Yu, Y.K. High-temperature protective coatings produced by EB-PVD. J. Coat. Sci. Technol. 2014, 1, 96–110. [Google Scholar]
- Drozdz, M.; Kyziol, K.; Grzesik, Z. Chromium-based oxidation-resistant coatings for the protection of engine valves in automotive vehicles. Mater. Technol. 2017, 51, 603–607. [Google Scholar]
- Panjan, P.; Drnovšek, A.; Kovač, J. Tribological aspects related to the morphology of PVD hard coatings. Surf. Coat. Technol. 2018, 343, 138–147. [Google Scholar] [CrossRef]
- Chicatun, F.; Cho, J.; Schaab, S.; Brusatin, G.; Colombo, P.; Roather, J.A.; Boccaccini, A.R. Carbon nanotube deposits and CNT/SiO2 composite coatings by electrophoretic deposition. Adv. Appl. Ceram. 2007, 106, 186–195. [Google Scholar] [CrossRef]
- Musil, J.; Vlcek, J.; Zeman, P. Hard amorphous nanocomposite coatings with oxidation resistance above 1000 °C. Adv. Appl. Ceram. 2008, 107, 149–154. [Google Scholar] [CrossRef]
- Tuthill, A.H. Stainless Steels and Specialty Alloys for Modern Pulp and Paper Mills; Nickel Development Institute (NiDI): Toronto, ON, Canada, 2002; pp. 47–50. [Google Scholar]
- Rao, S.; Frederick, L.; McDonald, A. Resistance of nanostructured environmental barrier coatings to the movement of molten salts. J. Therm. Spray Technol. 2012, 21, 887–899. [Google Scholar] [CrossRef]
- Heath, G.R.; Heimgartner, P.; Irons, G.; Miller, R.; Gustafsson, S. An assessment of thermal spray coating technologies for high temperature corrosion protection. Mater. Sci. Forum 1997, 251–254, 809–816. [Google Scholar] [CrossRef]
- Fauchais, P.; Montavon, G. Thermal and cold spray: Recent developments. Key Eng. Mater. 2008, 384, 1–59. [Google Scholar] [CrossRef]
- Davis, J. Handbook of Thermal Spray Technology; ASM International: Materials Park, OH, USA, 2004; pp. 1–36. [Google Scholar]
- Nelson, G.; Nychka, J.; McDonald, A.G. Flame spray deposition of titanium alloy-bio-active glass composite coatings. J. Therm. Spray Technol. 2011, 20, 1339–1351. [Google Scholar] [CrossRef]
- Davis, J. Surface Engineering for Corrosion and Wear Resistance, 1st ed.; ASM International: Materials Park, OH, USA, 2001; pp. 1–43. [Google Scholar]
- Dearnley, P. Surface engineering with diffusion technologies. In Introduction to Surface Engineering; Cambridge University Press: Cambridge, UK, 2017; pp. 35–115. [Google Scholar]
- Mittemeijer, E.J.; Somers, M.A.J. Thermochemical Surface Engineering of Steels, 1st ed.; Elsevier-Woodhead Publishing: Cambridge, UK, 2014. [Google Scholar]
- Choy, K.L. Chemical vapor deposition of coatings. Prog. Mater. Sci. 2003, 48, 57–170. [Google Scholar] [CrossRef]
- Budinski, K.G. Surface Engineering for Wear Resistance; Prentice Hall: Englewood Cliffs, NJ, USA, 1988. [Google Scholar]
- Takadoum, J. Materials and Surface Engineering in Tribology; Wiley: London, UK, 2010. [Google Scholar]
- Liu, X.; Wang, H.; Li, D.; Wu, Y. Study on kinetics of carbide growth by thermal diffusion Process. Surf. Coat. Technol. 2006, 201, 2414–2418. [Google Scholar] [CrossRef]
- Nicholls, J.R. Designing oxidation-resistant coatings. JOM 2000, 52, 28–35. [Google Scholar] [CrossRef]
- Medvedovski, E. Formation of corrosion-resistant thermal diffusion boride coatings. Adv. Eng. Mater. 2016, 18, 11–33. [Google Scholar] [CrossRef]
- Medvedovski, E.; Chinski, F.; Stewart, J. Wear- and corrosion-resistant boride-based coatings obtained through thermal diffusion CVD processing. Adv. Eng. Mater. 2014, 16, 713–728. [Google Scholar] [CrossRef]
- Medvedovski, E.; Jiang, J.; Robertson, M. Iron boride-based thermal diffusion coatings for tribo-corrosion oil production applications. Ceram. Int. 2016, 42, 3190–3211. [Google Scholar] [CrossRef]
- Bangaru, N.V.; Krutenat, R.C. Diffusion coatings of steels: Formation mechanism and microstructure of aluminized heat-resistant stainless steels. J. Vac. Sci. Technol. B 1984, 2, 806–815. [Google Scholar] [CrossRef]
- Fitzer, E.; Maurer, H.J. Diffusion and precipitation phenomena in aluminized and chromium-aluminized iron- and nickel-base alloys in materials and coatings to resist high temperature corrosion. In Materials and Coatings to Resist High Temperature Corrosion; Holmes, D.R., Rahmel, A., Eds.; Applied Science Publishers Ltd.: London, UK, 1978; pp. 253–269. [Google Scholar]
- Wang, D. Corrosion behavior of chromized and/or aluminized 214Cr-1Mo steel in medium—BTU coal gasifier environments. Surf. Coat. Technol. 1988, 36, 49–60. [Google Scholar] [CrossRef]
- Bai, C.; Ger, M.; Wu, M. Corrosion behaviors and contact resistances of the low-carbon steel bipolar plate with a chromized coating containing carbides and nitrides. Int. J. Hydrogen Energy 2009, 34, 6778–6789. [Google Scholar] [CrossRef]
- Meier, G.H.; Cheng, C.; Perlkins, R.A.; Bakker, W. Diffusion chromizing of ferrous alloys. Surf. Coat. Technol. 1989, 39–40, 53–64. [Google Scholar] [CrossRef]
- Cheetham, A.K.; Day, P. Solid State Chemistry: Techniques; Oxford Science Publications: Oxford, UK, 1991. [Google Scholar]
- Donald, H.; Jenkins, B. Thermodynamics of the relationship between lattice energy and lattice enthalpy. J. Chem. Educ. 2005, 82, 950–952. [Google Scholar]
- Ladd, M. Crystal Structures: Lattices and Solids in Stereoview; Horwood Series in Chemical Science; Elsevier: Chichester, UK, 1999. [Google Scholar]
- Grabke, H.J.; Schutze, M. Oxidation of Intermetallics; WILEY-VCH Verlag GmbH: Berlin, Germany, 1998. [Google Scholar]
- Callister, W.; Rethwisch, D. Fundamentals of Material Science and Engineering: An Integrated Approach, 5th ed.; John Wiley & Sons, Inc.: Marblehead, MA, USA, 2011; pp. 170–199. [Google Scholar]
- Shrier, L.; Burstein, G.; Jarman, R. Corrosion: Metal/Environment Reactions, 3rd ed.; Butterworth-Heinemann, Elsevier: Oxford, UK, 1994. [Google Scholar]
- Ehrburger, P.; Baranne, P.; Lahaye, J. Inhibition of the oxidation of carbon-carbon composite by boron oxide. Carbon 1986, 24, 495–499. [Google Scholar] [CrossRef]
- Fehlner, F.P. Low Temperature Oxidation, the Role of Vitreous Oxides; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1986. [Google Scholar]
- Seitz, F. On the porosity observed in the Kirkendal effect. Acta Metall. 1953, 1, 355–369. [Google Scholar] [CrossRef]
- Melendez, M.; McDonald, A. Development of WC-based metal matrix composite coatings using low-pressure cold gas dynamic spraying. Surf. Coat. Technol. 2013, 214, 101–109. [Google Scholar] [CrossRef]
- Pint, B.A.; Zhang, Y.; Tortorelli, P.F.; Haynes, J.A.; Wright, I.G. Evaluation of iron-aluminide CVD coatings for high temperature corrosion protection. Mater. High Temp. 2001, 18, 185–192. [Google Scholar] [CrossRef]
- Agüero, A.; Muelas, R.; Pastor, A.; Osgerby, S. Long exposure steam oxidation testing and mechanical properties of slurry aluminide coatings for steam turbine components. Surf. Coat. Technol. 2005, 200, 1219–1224. [Google Scholar] [CrossRef]
- Pérez, F.J.; Hierro, M.P.; Pedraza, F.; Carpintero, M.C.; Gómez, C.; Tarín, R. Effect of fluidized bed CVD aluminide coatings on the cyclic oxidation of austenitic AISI 304 stainless steel. Surf. Coat. Technol. 2001, 145, 1–7. [Google Scholar] [CrossRef]
- Busby, J.; Hash, M.; Was, G. The relationship between hardness and yield stress in irradiated austenitic and ferritic steels. J. Nucl. Mater. 2005, 336, 267–278. [Google Scholar] [CrossRef]
- Bahadur, A.; Mohanty, O. Structural studies of hot dip aluminized coatings on mild steel. Mater. Trans. 1991, 32, 1053–1061. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical | Weight Percent (%) |
|---|---|
| KCl | 10.2 |
| Na2CO3 | 11.5 |
| Na2SO4 | 73.9 |
| K2SO4 | 4.4 |
| Test Type | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| High Temperature Corrosion Test | Uncoated Samples (Control Sample) | Aluminized Samples | Boronized Samples | ||||||||
| CS-Bare | 316-Bare | CS-A | CS-A-Sn | CS-A-Z | 316-A | 316-A-Sn | 316-A-Z | CS-B | CS-B–BN | CS-B-Z | |
| High Temperature Corrosion Test | Uncoated Samples (Control Sample) | Aluminized Samples | Chromized Samples | ||||||||
| CS-Bare | 316-Bare | CS-A | CS-A-Sn | CS-A-Z | 316-A | 316-A-Sn | 316-A-Z | CS-Cr | 316-Cr | – | |
| Coating Information | CS-Bare | CS-A | CS-A-Sn | CS-A-Z | CS-Cr | 316-Bare | 316-A | 316-A-Sn | 316-A-Z | 316-Cr |
|---|---|---|---|---|---|---|---|---|---|---|
| Average roughness (Ra) (µm) | 2.62 | 6.4 | 6.22 | 6.32 | 3.05 | 1.41 | 1.26 | 1.37 | 1.36 | 3 |
| Standard deviation (n = 3) | ±0.2 | ±0.1 | ±0.28 | ±0.06 | ±0.06 | ±0.32 | ±0.07 | ±0.24 | ±0.18 | ±0.44 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahdavi, A.; Medvedovski, E.; Mendoza, G.L.; McDonald, A. Corrosion Resistance of Boronized, Aluminized, and Chromized Thermal Diffusion-Coated Steels in Simulated High-Temperature Recovery Boiler Conditions. Coatings 2018, 8, 257. https://doi.org/10.3390/coatings8080257
Mahdavi A, Medvedovski E, Mendoza GL, McDonald A. Corrosion Resistance of Boronized, Aluminized, and Chromized Thermal Diffusion-Coated Steels in Simulated High-Temperature Recovery Boiler Conditions. Coatings. 2018; 8(8):257. https://doi.org/10.3390/coatings8080257
Chicago/Turabian StyleMahdavi, Amirhossein, Eugene Medvedovski, Gerardo Leal Mendoza, and André McDonald. 2018. "Corrosion Resistance of Boronized, Aluminized, and Chromized Thermal Diffusion-Coated Steels in Simulated High-Temperature Recovery Boiler Conditions" Coatings 8, no. 8: 257. https://doi.org/10.3390/coatings8080257