HIV-1 Tat Binding to PCAF Bromodomain: Structural Determinants from Computational Methods

Abstract
:1. Introduction

2. Results and Discussion
2.1. Intermolecular Contacts

{kind=link}
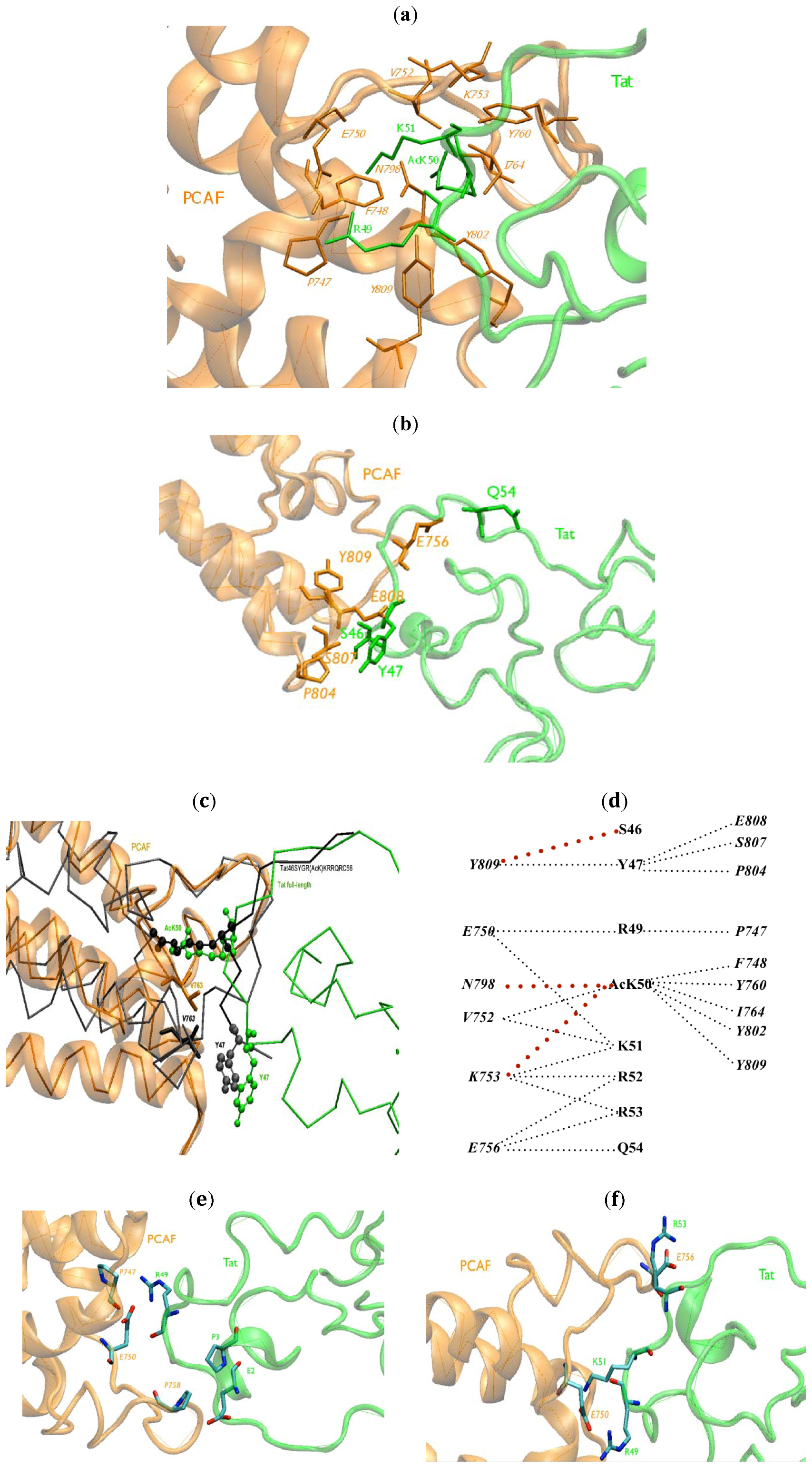{kind=link}
| Effect on Tat.PCAF binding | Mutants | In contact with | BRU Tat.PCAF model (docking) | BRU Tat.PCAF model (MD) | Coverage (%) |
|---|---|---|---|---|---|
| inhibiting binding [18] | AcK50A | F748, V752, Y760, I764 | F748, V752, Y760, I764 | F748, V752, Y760 | 100% |
| Y802 | Y802 | Y802 | 80% | ||
| Y809 | Y809 | Absent | |||
 |  | 100% | |||
| inhibiting binding [18] | Y47A | V763 | Absent | Absent | |
| V763 | Y47 | P804, S807 | N803 | 80% | |
| E808 | E808 | 100% | |||
| Y809 | 80% | ||||
| Q54A | E756 | E756 | E756 | 80% | |
| E756A | Q54 |  | | 80% | |
| strongly diminishing binding [18] | R53E | E756 |  | | 100% |
| F748A | Tat46SYGR(AcK)KRRQRC56 | AcK50 | AcK50 | 100% | |
| V752A | Tat46SYGR(AcK)KRRQRC56 | AcK50, R51 | AcK50, R51 | 100% | |
| Y802A | Tat46SYGR(AcK)KRRQRC56 | AcK50 | AcK50 | 100% | |
| Y809A | Tat46SYGR(AcK)KRRQRC56 | AcK50, S46 | S46 | 100% | |
| Diminishing binding [18] | R49A | PCAF BRD | P747, E750 | W746, P747, E750, Y802 | 100% |
| F748 | 80% | ||||
| K51A | PCAF BRD | E750, V752, K753 | F748, E750, V752, E756 | 100% | |
| K753 | 50% | ||||
| R52A | PCAF BRD | K753 | K753 | 25% | |
| E756 | E756 | 100% | |||
| Diminishing binding [52] | Y760D | AcK50 | AcK50 | AcK50 | 100% |
| Y761D | AcK50 | AcK50 | Absent | ||
| no effect [18] | W746A, D769A, C799A, N803A | R49 | 100% | ||
| E750A | R49 | R49 | 100% | ||
| T755A | R53 | R53 | 100% | ||
| I764A | AcK50 | AcK50 | 100% | ||
| N798A | AcK50 | AcK50 | 100% |

| Donors | Acceptors |
|---|---|
| AcK50 | V752 |
| AcK50 | F748 |
| AcK50 | Y809 |
| AcK50 | Y802 |
| N798 | AcK50 |
| R49 * | P747* |
| R49 * | E750* |
| R53 | E756 |
| R53 | K753 |
| R55 | E756 |
| Q54 | E756 |
| E2 * | P758* |
| P3 * | P758* |
2.2. Structural Rearrangements of Tat and PCAF upon Binding
| Systems | Backbone RMSD (Å):PCAF 722–781 | Backbone RMSD (Å): Tat47YGR(AcK)KRRQR55 | Backbone RMSD (Å): Full complex (same residues) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| min. | max. | avg. | min. | max. | avg. | min. | max. | avg. | |
| Tat47YGR(AcK)KRRQR55.PCAF*(1JM4) | 0.6 | 1.5 | 1.0 | 0.7 | 1.2 | 2.3 | 0.9 | 1.9 | 1.33 |
| sd. 0.2 | sd. 0.5 | sd. 0.26 | |||||||
| Tat47YGR(AcK)KRRQR55.PCAF (Prediction) | 1.3 | 0.9 | 1.7 | ||||||
| BRU Tat.PCAF (Prediction by docking) | 0.8 | 4.0 | 14.4 | ||||||
| BRU Tat.PCAF (Prediction by MD simulation, the last frame) | 2.9 3.3 3.1 sd. 0.2 | 2.8 3.4 2.6 sd. 0.4 | 24.7 25.8 24.5 sd. 1.2 | ||||||
2.3. Key Residues of PCAF BRD for Its Interaction with Tat
| Residues | Identity (%) | Residues | Identity (%) |
|---|---|---|---|
| P747 | 53% | T772 | 69% |
| F748 | 91% | Y782 | 97% |
| P751 | 68% | F788 | 63% |
| V752 | 71% | D791 | 89% |
| P758 | 71% | F796 | 60% |
| I764 | 76% | N798 | 96% |
| P767 | 97% | Y802 | 77% |
| D769 | 85% | N803 | 88% |
| L770 | 66% |
3. Computational Methods Section
4. Conclusions
Supplementary Files
Acknowledgments
References
- Flexner, C. HIV drug development: The next 25 years. Nat. Rev. Drug Discov. 2007, 6, 959–966. [Google Scholar] [CrossRef]
- Wainberg, M.A.; Zaharatos, G.J.; Brenner, B.G. Development of antiretroviral drug resistance. N. Engl. J. Med. 2011, 365, 637–646. [Google Scholar] [CrossRef]
- Detels, R. The search for protection against HIV infection. Ann. Epidemiol. 2009, 19, 250–252. [Google Scholar] [CrossRef]
- Al-Mawsawi, L.Q.; Al-Safi, R.I.; Neamati, N. Anti-infectives: Clinical progress of HIV-1 integrase inhibitors. Expert. Opin. Emerg. Drugs 2008, 13, 213–225. [Google Scholar] [CrossRef]
- Menendez-Arias, L. Molecular basis of human immunodeficiency virus drug resistance: An update. Antivir. Res. 2010, 85, 210–231. [Google Scholar]
- Menendez-Arias, L. Targeting HIV: Antiretroviral therapy and development of drug resistance. Trends Pharmacol. Sci. 2002, 23, 381–388. [Google Scholar] [CrossRef]
- Al-Mawsawi, L.Q.; Neamati, N. Blocking interactions between HIV-1 integrase and cellular cofactors: An emerging anti-retroviral strategy. Trends Pharmacol. Sci. 2007, 28, 526–535. [Google Scholar] [CrossRef]
- Adamson, C.S.; Freed, E.O. Novel approaches to inhibiting HIV-1 replication. Antivir. Res. 2010, 85, 119–141. [Google Scholar]
- Ensoli, B.; Fiorelli, V.; Ensoli, F.; Cafaro, A.; Titti, F.; Butto, S.; Monini, P.; Magnani, M.; Caputo, A.; Garaci, E. Candidate HIV-1 Tat vaccine development: From basic science to clinical trials. AIDS 2006, 20, 2245–2261. [Google Scholar]
- Gatignol, A. Transcription of HIV: Tat and cellular chromatin. Adv. Pharmacol. 2007, 55, 137–159. [Google Scholar] [CrossRef]
- Johri, M.K.; Mishra, R.; Chhatbar, C.; Unni, S.K.; Singh, S.K. Tits and bits of HIV Tat protein. Expert. Opin. Biol. Ther. 2011, 11, 269–283. [Google Scholar] [CrossRef]
- Pantano, S.; Carloni, P. Comparative analysis of HIV-1 Tat variants. Proteins 2005, 58, 638–643. [Google Scholar] [CrossRef]
- Peloponese, J.M., Jr.; Gregoire, C.; Opi, S.; Esquieu, D.; Sturgis, J.; Lebrun, E.; Meurs, E.; Collette, Y.; Olive, D.; Aubertin, A.M.; et al. 1H-13C nuclear magnetic resonance assignment and structural characterization of HIV-1 Tat protein. C. R. Acad. Sci. III 2000, 323, 883–894. [Google Scholar] [CrossRef]
- Gregoire, C.; Peloponese, J.M., Jr.; Esquieu, D.; Opi, S.; Campbell, G.; Solomiac, M.; Lebrun, E.; Lebreton, J.; Loret, E.P. Homonuclear (1)H-NMR assignment and structural characterization of human immunodeficiency virus type 1 Tat Mal protein. Biopolymers 2001, 62, 324–335. [Google Scholar] [CrossRef]
- Bayer, P.; Kraft, M.; Ejchart, A.; Westendorp, M.; Frank, R.; Rosch, P. Structural studies of HIV-1 Tat protein. J. Mol. Biol. 1995, 247, 529–535. [Google Scholar]
- Foucault, M.; Mayol, K.; Receveur-Brechot, V.; Bussat, M.C.; Klinguer-Hamour, C.; Verrier, B.; Beck, A.; Haser, R.; Gouet, P.; Guillon, C. UV and X-ray structural studies of a 101-residue long Tat protein from a HIV-1 primary isolate and of its mutated, detoxified, vaccine candidate. Proteins 2010, 78, 1441–1456. [Google Scholar]
- Tahirov, T.H.; Babayeva, N.D.; Varzavand, K.; Cooper, J.J.; Sedore, S.C.; Price, D.H. Crystal structure of HIV-1 Tat complexed with human P-TEFb. Nature 2010, 465, 747–751. [Google Scholar]
- Mujtaba, S.; He, Y.; Zeng, L.; Farooq, A.; Carlson, J.E.; Ott, M.; Verdin, E.; Zhou, M.M. Structural basis of lysine-acetylated HIV-1 Tat recognition by PCAF bromodomain. Mol. Cell 2002, 9, 575–586. [Google Scholar] [CrossRef]
- Opi, S.; Peloponese, J.M., Jr.; Esquieu, D.; Campbell, G.; de Mareuil, J.; Walburger, A.; Solomiac, M.; Gregoire, C.; Bouveret, E.; Yirrell, D.L.; et al. Tat HIV-1 primary and tertiary structures critical to immune response against non-homologous variants. J. Biol. Chem. 2002, 277, 35915–35919. [Google Scholar]
- Ott, M.; Schnolzer, M.; Garnica, J.; Fischle, W.; Emiliani, S.; Rackwitz, H.R.; Verdin, E. Acetylation of the HIV-1 Tat protein by p300 is important for its transcriptional activity. Curr. Biol. 1999, 9, 1489–1492. [Google Scholar]
- Kiernan, R.E.; Vanhulle, C.; Schiltz, L.; Adam, E.; Xiao, H.; Maudoux, F.; Calomme, C.; Burny, A.; Nakatani, Y.; Jeang, K.T.; et al. HIV-1 tat transcriptional activity is regulated by acetylation. EMBO J. 1999, 18, 6106–6118. [Google Scholar] [CrossRef]
- Nakatani, Y. HIV-1 transcription: Activation mediated by acetylation of Tat. Structure 2002, 10, 443–444. [Google Scholar] [CrossRef]
- Dhalluin, C.; Carlson, J.E.; Zeng, L.; He, C.; Aggarwal, A.K.; Zhou, M.M. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 399, 491–496. [Google Scholar]
- Shikama, N.; Chan, H.M.; Krstic-Demonacos, M.; Smith, L.; Lee, C.W.; Cairns, W.; La Thangue, N.B. Functional interaction between nucleosome assembly proteins and p300/CREB-binding protein family coactivators. Mol. Cell. Biol. 2000, 20, 8933–8943. [Google Scholar] [CrossRef]
- Teufel, D.P.; Freund, S.M.; Bycroft, M.; Fersht, A.R. Four domains of p300 each bind tightly to a sequence spanning both transactivation subdomains of p53. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 7009–7014. [Google Scholar]
- Deng, L.; de la Fuente, C.; Fu, P.; Wang, L.; Donnelly, R.; Wade, J.D.; Lambert, P.; Li, H.; Lee, C.G.; Kashanchi, F. Acetylation of HIV-1 Tat by CBP/P300 increases transcription of integrated HIV-1 genome and enhances binding to core histones. Virology 2000, 277, 278–295. [Google Scholar] [CrossRef]
- Benkirane, M.; Chun, R.F.; Xiao, H.; Ogryzko, V.V.; Howard, B.H.; Nakatani, Y.; Jeang, K.T. Activation of integrated provirus requires histone acetyltransferase. p300 and P/CAF are coactivators for HIV-1 Tat. J. Biol. Chem. 1998, 273, 24898–24905. [Google Scholar]
- Deng, L.; Wang, D.; de la Fuente, C.; Wang, L.; Li, H.; Lee, C.G.; Donnelly, R.; Wade, J.D.; Lambert, P.; Kashanchi, F. Enhancement of the p300 HAT activity by HIV-1 Tat on chromatin DNA. Virology 2001, 289, 312–326. [Google Scholar]
- Lusic, M.; Marcello, A.; Cereseto, A.; Giacca, M. Regulation of HIV-1 gene expression by histone acetylation and factor recruitment at the LTR promoter. EMBO J. 2003, 22, 6550–6561. [Google Scholar] [CrossRef]
- Zeng, L.; Zhou, M.M. Bromodomain: An acetyl-lysine binding domain. FEBS Lett. 2002, 513, 124–128. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Muller, S.; Pawson, T.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef]
- Zeng, L.; Zhang, Q.; Gerona-Navarro, G.; Moshkina, N.; Zhou, M.M. Structural basis of site-specific histone recognition by the bromodomains of human coactivators PCAF and CBP/p300. Structure 2008, 16, 643–652. [Google Scholar] [CrossRef]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef]
- Struhl, K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998, 12, 599–606. [Google Scholar] [CrossRef]
- Kuppuswamy, M.; Subramanian, T.; Srinivasan, A.; Chinnadurai, G. Multiple functional domains of Tat, the trans-activator of HIV-1, defined by mutational analysis. Nucleic Acids Res. 1989, 17, 3551–3561. [Google Scholar] [CrossRef]
- Garcia, J.A.; Harrich, D.; Pearson, L.; Mitsuyasu, R.; Gaynor, R.B. Functional domains required for tat-induced transcriptional activation of the HIV-1 long terminal repeat. EMBO J. 1988, 7, 3143–3147. [Google Scholar]
- Jeang, K.T.; Xiao, H.; Rich, E.A. Multifaceted activities of the HIV-1 transactivator of transcription, Tat. J. Biol. Chem. 1999, 274, 28837–28840. [Google Scholar]
- Dorn, P.; DaSilva, L.; Martarano, L.; Derse, D. Equine infectious anemia virus tat: Insights into the structure, function, and evolution of lentivirus trans-activator proteins. J. Virol. 1990, 64, 1616–1624. [Google Scholar]
- Noiman, S.; Yaniv, A.; Tsach, T.; Miki, T.; Tronick, S.R.; Gazit, A. The Tat protein of equine infectious anemia virus is encoded by at least three types of transcripts. Virology 1991, 184, 521–530. [Google Scholar] [CrossRef]
- Roy, S.; Katze, M.G.; Parkin, N.T.; Edery, I.; Hovanessian, A.G.; Sonenberg, N. Control of the interferon-induced 68-kilodalton protein kinase by the HIV-1 tat gene product. Science 1990, 247, 1216–1219. [Google Scholar]
- Churcher, M.J.; Lamont, C.; Hamy, F.; Dingwall, C.; Green, S.M.; Lowe, A.D.; Butler, J.G.; Gait, M.J.; Karn, J. High affinity binding of TAR RNA by the human immunodeficiency virus type-1 tat protein requires base-pairs in the RNA stem and amino acid residues flanking the basic region. J. Mol. Biol. 1993, 230, 90–110. [Google Scholar] [CrossRef]
- Dingwall, C.; Ernberg, I.; Gait, M.J.; Green, S.M.; Heaphy, S.; Karn, J.; Lowe, A.D.; Singh, M.; Skinner, M.A.; Valerio, R. Human immunodeficiency virus 1 tat protein binds trans-activation-responsive region (TAR) RNA in vitro. Proc. Natl. Acad. Sci. U. S. A. 1989, 86, 6925–6929. [Google Scholar]
- Weeks, K.M.; Crothers, D.M. RNA recognition by Tat-derived peptides: Interaction in the major groove? Cell 1991, 66, 577–588. [Google Scholar] [CrossRef]
- Chang, Y.N.; Jeang, K.T. The basic RNA-binding domain of HIV-2 Tat contributes to preferential trans-activation of a TAR2-containing LTR. Nucleic Acids Res. 1992, 20, 5465–5472. [Google Scholar] [CrossRef]
- de Mareuil, J.; Carre, M.; Barbier, P.; Campbell, G.R.; Lancelot, S.; Opi, S.; Esquieu, D.; Watkins, J.D.; Prevot, C.; Braguer, D.; et al. HIV-1 Tat protein enhances microtubule polymerization. Retrovirology 2005, 2, 5. [Google Scholar] [CrossRef] [Green Version]
- Fiorelli, V.; Barillari, G.; Toschi, E.; Sgadari, C.; Monini, P.; Sturzl, M.; Ensoli, B. IFN-gamma induces endothelial cells to proliferate and to invade the extracellular matrix in response to the HIV-1 Tat protein: Implications for AIDS-Kaposi's sarcoma pathogenesis. J. Immunol. 1999, 162, 1165–1170. [Google Scholar]
- Barillari, G.; Gendelman, R.; Gallo, R.C.; Ensoli, B. The Tat protein of human immunodeficiency virus type 1, a growth factor for AIDS Kaposi sarcoma and cytokine-activated vascular cells, induces adhesion of the same cell types by using integrin receptors recognizing the RGD amino acid sequence. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 7941–7945. [Google Scholar]
- Kaehlcke, K.; Dorr, A.; Hetzer-Egger, C.; Kiermer, V.; Henklein, P.; Schnoelzer, M.; Loret, E.; Cole, P.A.; Verdin, E.; Ott, M. Acetylation of Tat defines a cyclinT1-independent step in HIV transactivation. Mol. Cell 2003, 12, 167–176. [Google Scholar]
- Bres, V.; Tagami, H.; Peloponese, J.M.; Loret, E.; Jeang, K.T.; Nakatani, Y.; Emiliani, S.; Benkirane, M.; Kiernan, R.E. Differential acetylation of Tat coordinates its interaction with the co-activators cyclin T1 and PCAF. EMBO J. 2002, 21, 6811–6819. [Google Scholar] [CrossRef]
- Dorr, A.; Kiermer, V.; Pedal, A.; Rackwitz, H.R.; Henklein, P.; Schubert, U.; Zhou, M.M.; Verdin, E.; Ott, M. Transcriptional synergy between Tat and PCAF is dependent on the binding of acetylated Tat to the PCAF bromodomain. EMBO J. 2002, 21, 2715–2723. [Google Scholar]
- Pantano, S.; Marcello, A.; Ferrari, A.; Gaudiosi, D.; Sabo, A.; Pellegrini, V.; Beltram, F.; Giacca, M.; Carloni, P. Insights on HIV-1 Tat:P/CAF bromodomain molecular recognition from in vivo experiments and molecular dynamics simulations. Proteins 2006, 62, 1062–1073. [Google Scholar]
- de Vries, S.J.; van Dijk, A.D.; Krzeminski, M.; van Dijk, M.; Thureau, A.; Hsu, V.; Wassenaar, T.; Bonvin, A.M. HADDOCK versus HADDOCK: New features and performance of HADDOCK2.0 on the CAPRI targets. Proteins 2007, 69, 726–733. [Google Scholar] [CrossRef]
- Dominguez, C.; Boelens, R.; Bonvin, A.M. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar]
- Lowry, J.A.; Gamsjaeger, R.; Thong, S.Y.; Hung, W.; Kwan, A.H.; Broitman-Maduro, G.; Matthews, J.M.; Maduro, M.; Mackay, J.P. Structural analysis of MED-1 reveals unexpected diversity in the mechanism of DNA recognition by GATA-type zinc finger domains. J. Biol. Chem. 2009, 284, 5827–5835. [Google Scholar]
- Gelis, I.; Bonvin, A.M.; Keramisanou, D.; Koukaki, M.; Gouridis, G.; Karamanou, S.; Economou, A.; Kalodimos, C.G. Structural basis for signal-sequence recognition by the translocase motor SecA as determined by NMR. Cell 2007, 131, 756–769. [Google Scholar] [CrossRef]
- Wang, J.; Hu, W.; Cai, S.; Lee, B.; Song, J.; Chen, Y. The intrinsic affinity between E2 and the Cys domain of E1 in ubiquitin-like modifications. Mol. Cell. 2007, 27, 228–237. [Google Scholar] [CrossRef]
- Lowry, J.A.; Gamsjaeger, R.; Thong, S.Y.; Hung, W.; Kwan, A.H.; Broitman-Maduro, G.; Matthews, J.M.; Maduro, M.; Mackay, J.P. Structural analysis of MED-1 reveals unexpected diversity in the mechanism of DNA recognition by GATA-type zinc finger domains. J. Biol. Chem. 2009, 284, 5827–5835. [Google Scholar]
- Haynes, S.R.; Dollard, C.; Winston, F.; Beck, S.; Trowsdale, J.; Dawid, I.B. The bromodomain: A conserved sequence found in human, Drosophila and yeast proteins. Nucleic Acids Res. 1992, 20, 2603. [Google Scholar]
- Tamkun, J.W. The role of brahma and related proteins in transcription and development. Curr. Opin. Genet. Dev. 1995, 5, 473–477. [Google Scholar] [CrossRef]
- Tamkun, J.W.; Deuring, R.; Scott, M.P.; Kissinger, M.; Pattatucci, A.M.; Kaufman, T.C.; Kennison, J.A. brahma: A regulator of Drosophila homeotic genes structurally related to the yeast transcriptional activator SNF2/SWI2. Cell 1992, 68, 561–572. [Google Scholar] [CrossRef]
- Umehara, T.; Nakamura, Y.; Jang, M.K.; Nakano, K.; Tanaka, A.; Ozato, K.; Padmanabhan, B.; Yokoyama, S. Structural basis for acetylated histone H4 recognition by the human BRD2 bromodomain. J. Biol. Chem. 2010, 285, 7610–7618. [Google Scholar]
- Hewings, D.S.; Wang, M.; Philpott, M.; Fedorov, O.; Uttarkar, S.; Filippakopoulos, P.; Picaud, S.; Vuppusetty, C.; Marsden, B.; Knapp, S.; et al. 3,5-dimethylisoxazoles act as acetyl-lysine-mimetic bromodomain ligands. J. Med. Chem. 2011, 54, 6761–6770. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38, 27–28. [Google Scholar] [CrossRef]
- Case, D.A.; Darden, T.A.; Cheatham, T.E.; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Walker, R.C.; Zhang, W.; Merz, K.M.; et al. Amber 11; University of California: San Francisco, CA, USA, 2010. [Google Scholar]
- Machado, M.R.; Dans, P.D.; Pantano, S. Isoform-specific determinants in the HP1 binding to histone 3: Insights from molecular simulations. Amino Acids 2010, 38, 1571–1581. [Google Scholar] [CrossRef]
- De Mori, G.M.; Colombo, G.; Micheletti, C. Study of the Villin headpiece folding dynamics by combining coarse-grained Monte Carlo evolution and all-atom molecular dynamics. Proteins 2005, 58, 459–471. [Google Scholar]
- HADDOCK, version 2.1 program; free of charge for non commercial users. Available online: http://www.nmr.chem.uu.nl/haddock/download.html (accessed on 31 July 2012).
- van Dijk, A.D.; Bonvin, A.M. Solvated docking: Introducing water into the modelling of biomolecular complexes. Bioinformatics 2006, 22, 2340–2347. [Google Scholar] [CrossRef]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar] [CrossRef]
- Makov, G.; Payne, M.C. Periodic boundary conditions in ab initio calculations. Phys. Rev. B Condens Matter 1995, 51, 4014–4022. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N [center-dot] log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995, 8, 127–134. [Google Scholar]
- Zeng, L.; Li, J.; Muller, M.; Yan, S.; Mujtaba, S.; Pan, C.; Wang, Z.; Zhou, M.M. Selective small molecules blocking HIV-1 Tat and coactivator PCAF association. J. Am. Chem. Soc. 2005, 127, 2376–2377. [Google Scholar]
- Pan, C.; Mezei, M.; Mujtaba, S.; Muller, M.; Zeng, L.; Li, J.; Wang, Z.; Zhou, M.M. Structure-guided optimization of small molecules inhibiting human immunodeficiency virus 1 Tat association with the human coactivator p300/CREB binding protein-associated factor. J. Med. Chem. 2007, 50, 2285–2288. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar]
- Umehara, T.; Nakamura, Y.; Wakamori, M.; Ozato, K.; Yokoyama, S.; Padmanabhan, B. Structural implications for K5/K12-di-acetylated histone H4 recognition by the second bromodomain of BRD2. FEBS Lett. 2010, 584, 3901–3908. [Google Scholar]
- Mujtaba, S.; He, Y.; Zeng, L.; Yan, S.; Plotnikova, O.; Sachchidanand; Sanchez, R.; Zeleznik-Le, N.J.; Ronai, Z.; Zhou, M.M. Structural mechanism of the bromodomain of the coactivator CBP in p53 transcriptional activation. Mol. Cell 2004, 13, 251–263. [Google Scholar] [CrossRef]
- Chandy, M.; Gutierrez, J.L.; Prochasson, P.; Workman, J.L. SWI/SNF displaces SAGA-acetylated nucleosomes. Eukaryot. Cell 2006, 5, 1738–1747. [Google Scholar] [CrossRef]
- Singh, M.; D'Silva, L.; Holak, T.A. DNA-binding properties of the recombinant high-mobility-group-like AT-hook-containing region from human BRG1 protein. Biol. Chem. 2006, 387, 1469–1478. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, J.; Shen, W.; Wang, X.; Wu, J.; Shi, Y. Solution structure of the second bromodomain of Brd2 and its specific interaction with acetylated histone tails. BMC Struct. Biol. 2007, 7, 57. [Google Scholar] [CrossRef]
- Chung, C.W.; Coste, H.; White, J.H.; Mirguet, O.; Wilde, J.; Gosmini, R.L.; Delves, C.; Magny, S.M.; Woodward, R.; Hughes, S.A.; et al. Discovery and characterization of small molecule inhibitors of the BET family bromodomains. J. Med. Chem. 2011, 54, 3827–3838. [Google Scholar]
- Chung, C.W.; Witherington, J. Progress in the discovery of small-molecule inhibitors of bromodomain—Histone interactions. J. Biomol. Screen 2011, 16, 1170–1185. [Google Scholar] [CrossRef]
- Chung, C.W.; Dean, A.W.; Woolven, J.M.; Bamborough, P. Fragment-based discovery of bromodomain inhibitors part 1: Inhibitor binding modes and implications for lead discovery. J. Med. Chem. 2012, 55, 576–586. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Picaud, S.; Fedorov, O.; Keller, M.; Wrobel, M.; Morgenstern, O.; Bracher, F.; Knapp, S. Benzodiazepines and benzotriazepines as protein interaction inhibitors targeting bromodomains of the BET family. Bioorg. Med. Chem. 2012, 20, 1878–1886. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Quy, V.C.; Pantano, S.; Rossetti, G.; Giacca, M.; Carloni, P. HIV-1 Tat Binding to PCAF Bromodomain: Structural Determinants from Computational Methods. Biology 2012, 1, 277-296. https://doi.org/10.3390/biology1020277
Quy VC, Pantano S, Rossetti G, Giacca M, Carloni P. HIV-1 Tat Binding to PCAF Bromodomain: Structural Determinants from Computational Methods. Biology. 2012; 1(2):277-296. https://doi.org/10.3390/biology1020277
Chicago/Turabian StyleQuy, Vo Cam, Sergio Pantano, Giulia Rossetti, Mauro Giacca, and Paolo Carloni. 2012. "HIV-1 Tat Binding to PCAF Bromodomain: Structural Determinants from Computational Methods" Biology 1, no. 2: 277-296. https://doi.org/10.3390/biology1020277