Computer-Aided Approaches for Targeting HIVgp41

Abstract
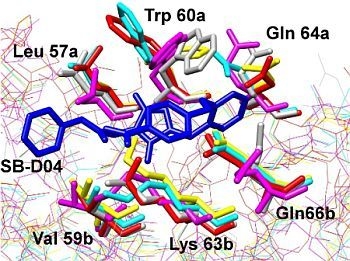
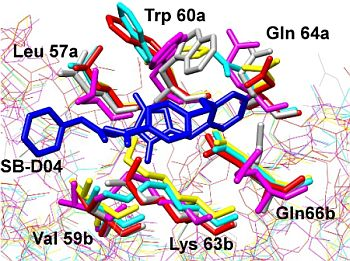
:
1. Introduction
2. Virus-Cell Fusion and Structural Biology of HIVgp41
2.1. HIV Envelope Proteins Originate from the env Gene
2.2. Virus-Cell Fusion is Initiated Through Receptor/Co-receptor Recognition
2.3. HIVgp41 as a Target for Fusion Inhibition

3. Experimental Models of the gp41 Ectodomain
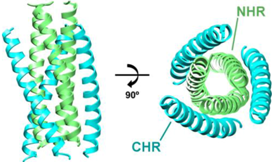
3.1. NHR/CHR Peptide Mixtures
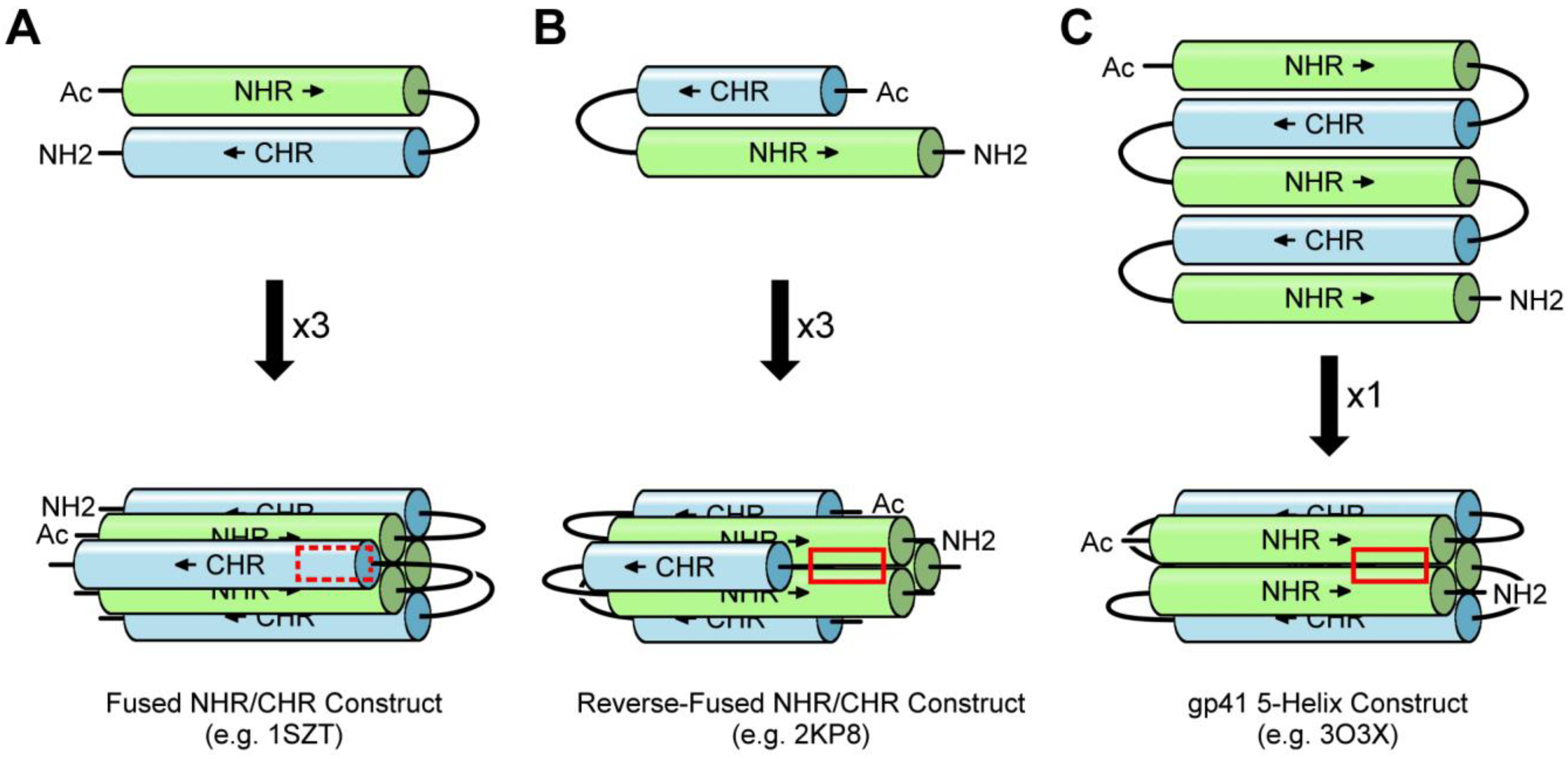
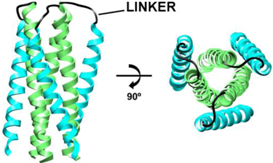
3.2. Fused NHR/CHR Constructs
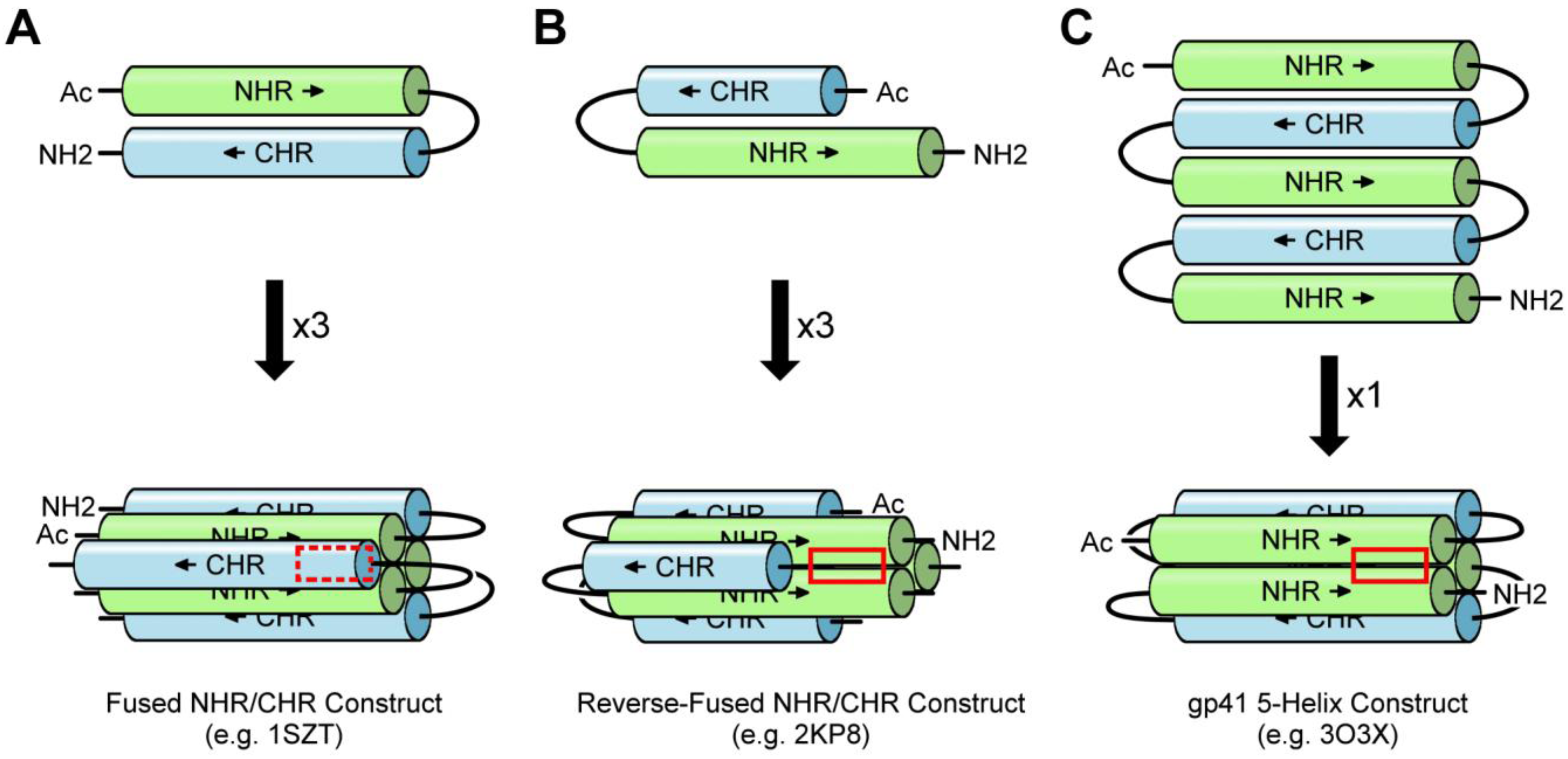
3.3. Stabilized NHR Constructs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
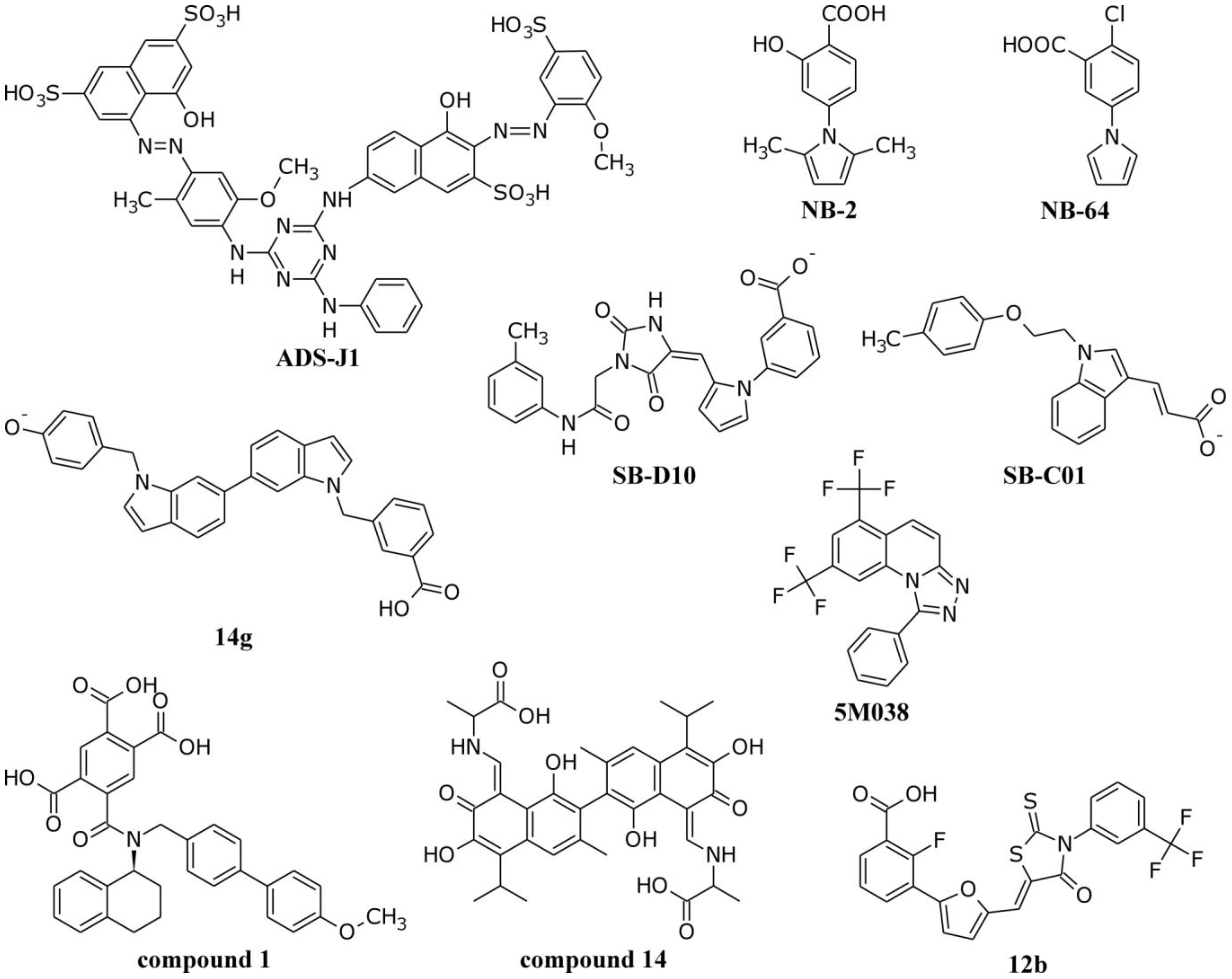
| NHR/CHR Peptide Mixtures | |
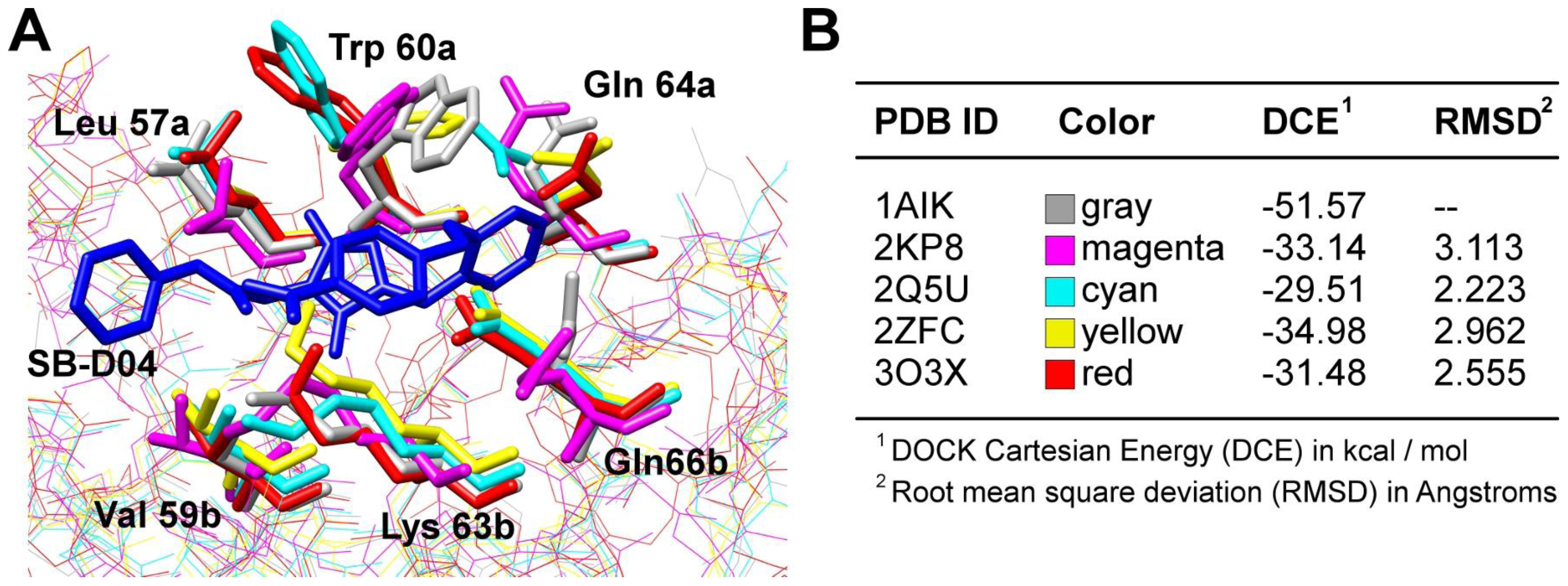
| Example Structure (PDB 1AIK): | Summary of PDB Structures:1 |
 | N36/C34: 1AIK [38]. |
| N36/C34-Mutants: 2ZZO [85 ]; 3AHA [86 ]; 2Z2T [N/A]. | |
| N36/Sifuvirtide: 3VIE [60 ]. | |
| T21/Cp621-652: 3VGX [87 ]. | |
| Fused NHR/CHR Constructs | |
| Example Structure (PDB 1SZT): | Summary of PDB Structures:1 |
 | Standard Fused, e.g., N34(L6)C28: 1SZT [39]; 1QR8, 1QR9 [88]; 1DLB [89]; 1DF4, 1DF5 [90]; 1F23 [94]; 1I5X, 1I5Y [91]; 1K33, 1K34 [92]; 2OT5, 3CP1, 3CYO [93]; 2X7R [95]; 3K9A [96]. |
| Reverse Fused: 2KP8 (NMR) [65 ]. | |
| 5-Helix: 3O3X, 3O3Z, 3O40, 3O43 [98 ]; 4DZU, 4DZV [99 ]; 3O42 [N/A]. | |
| Stabilized NHR Constructs | |
| Example Structure (PDB 1CE0): | Summary of PDB Structures:1 |
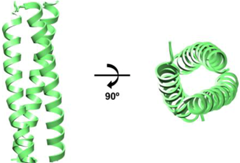 | Chimeras with GCN4 (pII41N): 1ENV [40]; 1FAV [73]. |
| Chimeras with GCN4 (IQN17): 1CZQ, 2Q3I; 2Q5U, 2Q7C [101 ]; 1GZL [102 ]; 2R3C, 2R5B, 2R5D [103 ]; 3L35, 3L36, 3L37 [104 ]. | |
| Heavily Mutated: 1CE0 [105 ]; 2ZFC [106 ]. | |
| α/β-Peptide Foldamers: 3F4Y, 3F4Z, 3F50, 3G7A [107 ]; 3O3Y [N/A]. | |
3.4. Antibody-Bound gp41-Derived Peptides
3.5. Apo gp41-Derived Peptides
| Antibody-bound gp41-derived Peptides | |||
| Antibody: | Target: | Summary of PDB Codes:1 | |
| 2F5 | MPER | 1TJG, 1TJH, 1TJI [108]; 2P8L, 2P8M, 2P8P, 3D0L, 3D0V, 3DRO, 3DRQ [109]; 1U8H, 1U8I, 1U8J, 1U8L, 1U8M, 1U8N, 1U8O, 1U8P, 1U8Q, 1U91, 1U92, 1U93, 1U95, 2F5B, 2PW1, 2PW2, 3IDG, 3IDI, 3IDJ, 3IDM, 3IDN [110]; 3DRT, 3EGS [111]; 3LEX, 3LEY [81]; 1U8K, 3MOA, 3MOB, 3MOD [N/A]. | |
| 4e10 | MPER | 1TZG [112]; 2FX7, 2FX8, 2FX9 [79]. | |
| 13H11 | MPER | 3MNW, 3MNZ, 3MO1 [N/A]. | |
| Z13e1 | MPER | 3FN0 [80]. | |
| Various | gp41 multimer | 2CMR [113]; 2XRA [114]; 3MA9, 3MAC [115]; 3P30 [116]. | |
| Apo gp41-derived Peptides | |||
| Peptide Origin: | Summary of PDB Codes:1 | ||
| FP | 1ERF (IR) [119]; 1P5A (IR) [120]; 2ARI (NMR) [117]; 2PJV (NMR) [121]; 2JNR (NMR) [122]. | ||
| Loop | 1IM7 (NMR), 1J8N (NMR), 1J8Z (NMR), 1J9V (NMR), 1JAA (NMR), 1JAR (NMR), 1JC8 (NMR), 1JCP (NMR), 1JD8 (NMR), 1JDK (NMR) [123]. | ||
| MPER | 1JAU (NMR), 1JAV (NMR) [124]; 1LB0 (NMR), 1LCX (NMR) [118]; 1MZI (NMR) [125]; 2PV6 (NMR) [126]; 3G9R [127]. | ||
| CTD | 3GWO, 3H00, 3H01 [128]. | ||
3.6. Electron Microscopy-Derived Models
4. Computational Modeling of gp41 and Fusion Inhibitors
4.1. Interactions of Small Molecule Inhibitors with gp41
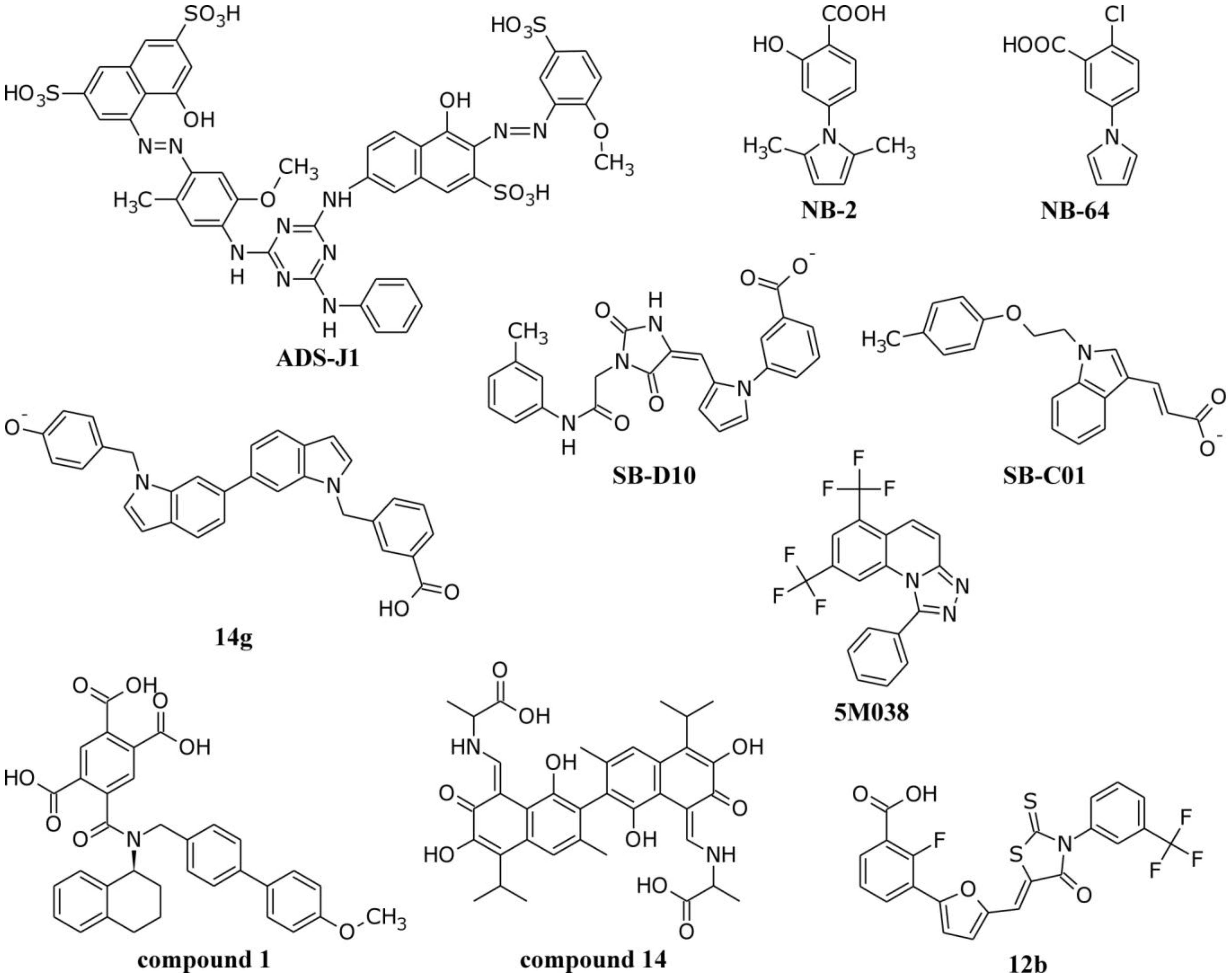

4.2. Molecular Dynamics Simulations of the gp41 NHR/CHR Core
4.3. Molecular Dynamics Simulations of the gp41 Fusion Peptide
4.4. Molecular Dynamics Simulations of the Transmembrane Domain
4.5. Molecular Dynamics Simulations of T20 and Other CHR-Derived Peptides
5. Future Directions and Concluding Remarks
Acknowledgments
References
- Barre-Sinoussi, F.; Chermann, J.C.; Rey, F.; Nugeyre, M.T.; Chamaret, S.; Gruest, J.; Dauguet, C.; Axler-Blin, C.; Vezinet-Brun, F.; Rouzioux, C.; et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 1983, 220, 868–871. [Google Scholar]
- Gallo, R.C.; Salahuddin, S.Z.; Popovic, M.; Shearer, G.M.; Kaplan, M.; Haynes, B.F.; Palker, T.J.; Redfield, R.; Oleske, J.; Safai, B.; et al. Frequent detection and isolation of cytopathic retroviruses (HTLV-III) from patients with AIDS and at risk for AIDS. Science 1984, 224, 500–503. [Google Scholar]
- Global HIV/AIDS Response: Epidemic Update and Health Sector Progress towards Universal Access: Progress Report 2011; World Health Organization: Geneva, Switzerland, 2011.
- Structure-Based Drug Design Fact Sheet. Available online: http://www.nigms.nih.gov/Education/structure_drugs.htm (accessed on 1 May 2012).
- Antiretroviral Drugs Used in the Treatment of HIV Infection. Available online: http://www.fda.gov/ForConsumers/byAudience/ForPatientAdvocates/HIVandAIDSActivities/ucm118915.htm (accessed on 1 May 2012).
- Johnson, V.A.; Calvez, V.; Gunthard, H.F.; Paredes, R.; Pillay, D.; Shafer, R.; Wensing, A.M.; Richman, D.D. 2011 update of the drug resistance mutations in HIV-1. Top. Antivir. Med. 2011, 19, 156–164. [Google Scholar]
- Shafer, R.W.; Schapiro, J.M. HIV-1 drug resistance mutations: An updated framework for the second decade of HAART. AIDS Rev. 2008, 10, 67–84. [Google Scholar]
- Wild, C.; Greenwell, T.; Matthews, T. A synthetic peptide from HIV-1 gp41 is a potent inhibitor of virus-mediated cell-cell fusion. AIDS Res. Hum. Retroviruses 1993, 9, 1051–1053. [Google Scholar] [CrossRef]
- Kilby, J.M.; Hopkins, S.; Venetta, T.M.; DiMassimo, B.; Cloud, G.A.; Lee, J.Y.; Alldredge, L.; Hunter, E.; Lambert, D.; Bolognesi, D.; et al. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat. Med. 1998, 4, 1302–1307. [Google Scholar]
- Eckert, D.M.; Kim, P.S. Design of potent inhibitors of HIV-1 entry from the gp41 N-peptide region. Proc. Natl. Acad. Sci. USA 2001, 98, 11187–11192. [Google Scholar] [CrossRef]
- Liu, S.; Lu, H.; Niu, J.; Xu, Y.; Wu, S.; Jiang, S. Different from the HIV fusion inhibitor C34, the anti-HIV drug fuzeon (T-20) inhibits HIV-1 entry by targeting multiple sites in gp41 and gp120. J. Biol. Chem. 2005, 280, 11259–11273. [Google Scholar]
- Baldwin, C.E.; Sanders, R.W.; Deng, Y.; Jurriaans, S.; Lange, J.M.; Lu, M.; Berkhout, B. Emergence of a drug-dependent human immunodeficiency virus type 1 variant during therapy with the T20 fusion inhibitor. J. Virol. 2004, 78, 12428–12437. [Google Scholar]
- Xu, L.; Pozniak, A.; Wildfire, A.; Stanfield-Oakley, S.A.; Mosier, S.M.; Ratcliffe, D.; Workman, J.; Joall, A.; Myers, R.; Smit, E.; et al. Emergence and evolution of enfuvirtide resistance following long-term therapy involves heptad repeat 2 mutations within gp41. Antimicrob. Agents Chemother. 2005, 49, 1113–1119. [Google Scholar]
- Mink, M.; Mosier, S.M.; Janumpalli, S.; Davison, D.; Jin, L.; Melby, T.; Sista, P.; Erickson, J.; Lambert, D.; Stanfield-Oakley, S.A.; et al. Impact of human immunodeficiency virus type 1 gp41 amino acid substitutions selected during enfuvirtide treatment on gp41 binding and antiviral potency of enfuvirtide in vitro. J. Virol. 2005, 79, 12447–12454. [Google Scholar]
- Jorgensen, W.L. The many roles of computation in drug discovery. Science 2004, 303, 1813–1818. [Google Scholar] [CrossRef]
- Jorgensen, W.L. Efficient drug lead discovery and optimization. Acc. Chem. Res. 2009, 42, 724–733. [Google Scholar] [CrossRef]
- Cai, L.; Jiang, S. Development of peptide and small-molecule HIV-1 fusion inhibitors that target gp41. ChemMedChem 2010, 5, 1813–1824. [Google Scholar] [CrossRef]
- Pan, C.; Liu, S.; Jiang, S. HIV1 gp41 fusion intermediate: A target for HIV therapeutics. J. Formos. Med. Assoc. 2010, 109, 94–105. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Shai, Y. Insights into the mechanism of HIV-1 envelope induced membrane fusion as revealed by its inhibitory peptides. Eur. Biophys. J. 2011, 40, 349–357. [Google Scholar] [CrossRef]
- Checkley, M.A.; Luttge, B.G.; Freed, E.O. HIV-1 envelope glycoprotein biosynthesis, trafficking, and incorporation. J. Mol. Biol. 2011, 410, 582–608. [Google Scholar] [CrossRef]
- Gochin, M.; Zhou, G. Amphipathic properties of HIV-1 gp41 fusion inhibitors. Curr. Top. Med. Chem. 2011, 11, 3022–3032. [Google Scholar] [CrossRef]
- Berkhout, B.; Eggink, D.; Sanders, R.W. Is there a future for antiviral fusion inhibitors? Curr. Opin. Virol. 2012, 2, 50–59. [Google Scholar] [CrossRef]
- Hallenberger, S.; Bosch, V.; Angliker, H.; Shaw, E.; Klenk, H.D.; Garten, W. Inhibition of furin-mediated cleavage activation of HIV-1 glycoprotein gp160. Nature 1992, 360, 358–361. [Google Scholar]
- Zanetti, G.; Briggs, J.A.G.; Grunewald, K.; Sattentau, Q.J.; Fuller, S.D. Cryo-electron tomographic structure of an immunodeficiency virus envelope complex in situ. PLoS Pathog. 2006, 2, e83. [Google Scholar] [CrossRef]
- Zhu, P.; Liu, J.; Bess, J., Jr.; Chertova, E.; Lifson, J.D.; Grise, H.; Ofek, G.A.; Taylor, K.A.; Roux, K.H. Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature 2006, 441, 847–852. [Google Scholar]
- Zhu, P.; Winkler, H.; Chertova, E.; Taylor, K.A.; Roux, K.H. Cryoelectron tomography of HIV-1 envelope spikes: Further evidence for tripod-like legs. PLoS Pathog. 2008, 4, e1000203. [Google Scholar] [CrossRef]
- Chan, D.C.; Kim, P.S. HIV entry and its inhibition. Cell 1998, 93, 681–684. [Google Scholar] [CrossRef]
- Salzwedel, K.; Smith, E.D.; Dey, B.; Berger, E.A. Sequential CD4-coreceptor interactions in human immunodeficiency virus type 1 Env function: Soluble CD4 activates Env for coreceptor-dependent fusion and reveals blocking activities of antibodies against cryptic conserved epitopes on gp120. J. Virol. 2000, 74, 326–333. [Google Scholar] [CrossRef]
- Moore, J.P.; Kitchen, S.G.; Pugach, P.; Zack, J.A. The CCR5 and CXCR4 coreceptors-central to understanding the transmission and pathogenesis of human immunodeficiency virus type 1 infection. AIDS Res. Hum. Retroviruses 2004, 20, 111–126. [Google Scholar] [CrossRef]
- Jones, P.L.S.J.; Korte, T.; Blumenthal, R. Conformational changes in cell surface HIV-1 envelope glycoproteins are triggered by cooperation between cell surface CD4 and co-receptors. J. Biol. Chem. 1998, 273, 404–409. [Google Scholar]
- Eckert, D.M.; Kim, P.S. Mechanisms of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 2001, 70, 777–810. [Google Scholar] [CrossRef]
- Furuta, R.A.; Wild, C.T.; Weng, Y.; Weiss, C.D. Capture of an early fusion-active conformation of HIV-1 gp41. Nat. Struct. Biol. 1998, 5, 276–279. [Google Scholar] [CrossRef]
- Bradshaw, J.P.; Darkes, M.J.M.; Harroun, T.A.; Katsaras, J.; Epand, R.M. Oblique membrane insertion of viral fusion peptide probed by neutron diffraction. Biochemistry 2000, 39, 6581–6585. [Google Scholar] [CrossRef]
- Schmick, S.D.; Weliky, D.P. Major antiparallel and minor parallel β sheet populations detected in the membrane-associated human immunodeficiency virus fusion peptide. Biochemistry 2010, 49, 10623–10635. [Google Scholar] [CrossRef]
- Yang, J.; Prorok, M.; Castellino, F.J.; Weliky, D.P. Oligomeric β-structure of the membrane-bound HIV-1 fusion peptide formed from soluble monomers. Biophys. J. 2004, 87, 1951–1963. [Google Scholar] [CrossRef]
- Sackett, K.; Shai, Y. The HIV fusion peptide adopts intermolecular parallel β-sheet structure in membranes when stabilized by the adjacent N-terminal heptad repeat: A 13C FTIR study. J. Mol. Biol. 2005, 350, 790–805. [Google Scholar] [CrossRef]
- Hughson, F.M. Enveloped viruses: A common mode of membrane fusion? Curr. Biol. 1997, 7, R565–R569. [Google Scholar] [CrossRef]
- Chan, D.C.; Fass, D.; Berger, J.M.; Kim, P.S. Core structure of gp41 from the HIV envelope glycoprotein. Cell 1997, 89, 263–273. [Google Scholar] [CrossRef]
- Tan, K.; Liu, J.-H.; Wang, J.-H.; Shen, S.; Lu, M. Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc. Natl. Acad. Sci. USA 1997, 94, 12303–12308. [Google Scholar] [CrossRef]
- Weissenhorn, W.; Dessen, A.; Harrison, S.C.; Skehel, J.J.; Wiley, D.C. Atomic structure of the ectodomain from HIV-1 gp41. Nature 1997, 387, 426–430. [Google Scholar]
- Melikyan, G.B.; Markosyan, R.M.; Hemmati, H.; Delmedico, M.K.; Lambert, D.M.; Cohen, F.S. Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J. Cell Biol. 2000, 151, 413–423. [Google Scholar] [CrossRef]
- Cohen, F.S.; Melikyan, G.B. The energetics of membrane fusion from binding, through hemifusion, pore formation, and pore enlargement. J. Membr. Biol. 2004, 199, 1–14. [Google Scholar] [CrossRef]
- Chernomordik, L.V.; Kozlov, M.M. Membrane hemifusion: Crossing a chasm in two leaps. Cell 2005, 123, 375–382. [Google Scholar] [CrossRef]
- Harrison, S.C. Viral membrane fusion. Nat. Struct. Mol. Biol. 2008, 15, 690–698. [Google Scholar] [CrossRef]
- Yang, X.; Kurteva, S.; Ren, X.; Lee, S.; Sodroski, J. Stoichiometry of envelope glycoprotein trimers in the entry of human immunodeficiency virus type 1. J. Virol. 2005, 79, 12132–12147. [Google Scholar]
- Herrera, C.; Klasse, P.J.; Kibler, C.W.; Michael, E.; Moore, J.P.; Beddows, S. Dominant-negative effect of hetero-oligomerization on the function of the human immunodeficiency virus type 1 envelope glycoprotein complex. Virology 2006, 351, 121–132. [Google Scholar] [CrossRef]
- Sougrat, R.; Bartesaghi, A.; Lifson, J.D.; Bennett, A.E.; Bess, J.W.; Zabransky, D.J.; Subramaniam, S. Electron tomography of the contact between T cells and SIV/HIV-1: Implications for viral entry. PLoS Pathog. 2007, 3, e63. [Google Scholar] [CrossRef]
- Nowak, S.A.; Chou, T. Mechanisms of receptor/coreceptor-mediated entry of enveloped viruses. Biophys. J. 2009, 96, 2624–2636. [Google Scholar] [CrossRef]
- Magnus, C.; Rusert, P.; Bonhoeffer, S.; Trkola, A.; Regoes, R.R. Estimating the stoichiometry of human immunodeficiency virus entry. J. Virol. 2009, 83, 1523–1531. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.; Boyle, T.; Lyerly, H.; Cullen, B. Identification of the envelope V3 loop as the primary determinant of cell tropism in HIV-1. Science 1991, 253, 71–74. [Google Scholar]
- Los Alamos National Laboratory, HIV Sequence Compendium 2011; Los Alamos National Laboratory: Los Alamos, NM, USA, 2011.
- Doms, R.W.; Moore, J.P. HIV-1 membrane fusion: Targets of opportunity. J. Cell Biol. 2000, 151, F9–F13. [Google Scholar] [CrossRef]
- Liu, S.; Jing, W.; Cheung, B.; Lu, H.; Sun, J.; Yan, X.; Niu, J.; Farmar, J.; Wu, S.; Jiang, S. HIV gp41 C-terminal heptad repeat contains multifunctional domains: Relation to mechanisms of action of anti-HIV peptides. J. Biol. Chem. 2007, 282, 9612–9620. [Google Scholar]
- Greenberg, M.L.; Cammack, N. Resistance to enfuvirtide, the first HIV fusion inhibitor. J. Antimicrob. Chemother. 2004, 54, 333–340. [Google Scholar] [CrossRef]
- Chan, D.C.; Chutkowski, C.T.; Kim, P.S. Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. Proc. Natl. Acad. Sci. USA 1998, 95, 15613–15617. [Google Scholar]
- Eggink, D.; Baldwin, C.E.; Deng, Y.; Langedijk, J.P.M.; Lu, M.; Sanders, R.W.; Berkhout, B. Selection of T1249-resistant human immunodeficiency virus type 1 variants. J. Virol. 2008, 82, 6678–6688. [Google Scholar] [CrossRef]
- Qi, Z.; Shi, W.; Xue, N.; Pan, C.; Jing, W.; Liu, K.; Jiang, S. Rationally designed anti-HIV peptides containing multifunctional domains as molecule probes for studying the mechanisms of action of the first and second generation HIV fusion inhibitors. J. Biol. Chem. 2008, 283, 30376–30384. [Google Scholar]
- He, Y.; Xiao, Y.; Song, H.; Liang, Q.; Ju, D.; Chen, X.; Lu, H.; Jing, W.; Jiang, S.; Zhang, L. Design and evaluation of sifuvirtide, a novel HIV-1 fusion inhibitor. J. Biol. Chem. 2008, 283, 11126–11134. [Google Scholar]
- Wang, R.-R.; Yang, L.-M.; Wang, Y.-H.; Pang, W.; Tam, S.-C.; Tien, P.; Zheng, Y.-T. Sifuvirtide, a potent HIV fusion inhibitor peptide. Biochem. Biophys. Res. Commun. 2009, 382, 540–544. [Google Scholar] [CrossRef]
- Yao, X.; Chong, H.; Zhang, C.; Waltersperger, S.; Wang, M.; Cui, S.; He, Y. Broad antiviral activity and crystal structure of HIV-1 fusion inhibitor sifuvirtide. J. Biol. Chem. 2012, 287, 6788–6796. [Google Scholar]
- Debnath, A.K.; Radigan, L.; Jiang, S. Structure-based identification of small molecule antiviral compounds targeted to the gp41 core structure of the human immunodeficiency virus type 1. J. Med. Chem. 1999, 42, 3203–3209. [Google Scholar] [CrossRef]
- Jiang, S.; Lu, H.; Liu, S.; Zhao, Q.; He, Y.; Debnath, A.K. N-substituted pyrrole derivatives as novel human immunodeficiency virus type 1 entry inhibitors that interfere with the gp41 six-helix bundle formation and block virus fusion. Antimicrob. Agents Chemother. 2004, 48, 4349–4359. [Google Scholar] [CrossRef]
- Frey, G.; Rits-Volloch, S.; Zhang, X.Q.; Schooley, R.T.; Chen, B.; Harrison, S.C. Small molecules that bind the inner core of gp41 and inhibit HIV envelope-mediated fusion. Proc. Natl. Acad. Sci. USA 2006, 103, 13938–13943. [Google Scholar]
- Katritzky, A.R.; Tala, S.R.; Lu, H.; Vakulenko, A.V.; Chen, Q.-Y.; Sivapackiam, J.; Pandya, K.; Jiang, S.; Debnath, A.K. Design, synthesis, and structure-activity relationship of a novel series of 2-aryl 5-(4-oxo-3-phenethyl-2-thioxothiazolidinylidenemethyl)furans as HIV-1 entry inhibitors. J. Med. Chem. 2009, 52, 7631–7639. [Google Scholar] [CrossRef]
- Stewart, K.D.; Huth, J.R.; Ng, T.I.; McDaniel, K.; Hutchinson, R.N.; Stoll, V.S.; Mendoza, R.R.; Matayoshi, E.D.; Carrick, R.; Mo, H.; et al. Non-peptide entry inhibitors of HIV-1 that target the gp41 coiled coil pocket. Bioorg. Med. Chem. Lett. 2010, 20, 612–617. [Google Scholar]
- Zhou, G.; Wu, D.; Hermel, E.; Balogh, E.; Gochin, M. Design, synthesis, and evaluation of indole compounds as novel inhibitors targeting gp41. Bioorg. Med. Chem. Lett. 2010, 20, 1500–1503. [Google Scholar] [CrossRef]
- Jiang, S.; Tala, S.R.; Lu, H.; Abo-Dya, N.E.; Avan, I.; Gyanda, K.; Lu, L.; Katritzky, A.R.; Debnath, A.K. Design, synthesis, and biological activity of novel 5-((arylfuran/1H-pyrrol-2-yl)methylene)-2-thioxo-3-(3-(trifluoromethyl)phenyl)thiazolidin-4-ones as HIV-1 fusion inhibitors targeting gp41. J. Med. Chem. 2011, 54, 572–579. [Google Scholar]
- He, X.-Y.; Zou, P.; Qiu, J.; Hou, L.; Jiang, S.; Liu, S.; Xie, L. Design, synthesis and biological evaluation of 3-substituted 2,5-dimethyl-N-(3-(1H-tetrazol-5-yl)phenyl)pyrroles as novel potential HIV-1 gp41 inhibitors. Bioorg. Med. Chem. 2011, 19, 6726–6734. [Google Scholar]
- Holden, P.M.; Kaur, H.; Goyal, R.; Gochin, M.; Rizzo, R.C. Footprint-based identification of viral entry inhibitors targeting HIVgp41. Bioorg. Med. Chem. Lett. 2012, 22, 3011–3016. [Google Scholar]
- Whitby, L.R.; Boyle, K.E.; Cai, L.; Yu, X.; Gochin, M.; Boger, D.L. Discovery of HIV fusion inhibitors targeting gp41 using a comprehensive α-helix mimetic library. Bioorg. Med. Chem. Lett. 2012, 22, 2861–2865. [Google Scholar]
- Yang, J.; Zhang, F.; Li, J.; Chen, G.; Wu, S.; Ouyang, W.; Pan, W.; Yu, R.; Yang, J.; Tien, P. Synthesis and antiviral activities of novel gossypol derivatives. Bioorg. Med. Chem. Lett. 2012, 22, 1415–1420. [Google Scholar]
- Yi, H.A.; Diaz-Aguilar, B.; Bridon, D.; Quraishi, O.; Jacobs, A. Permanent inhibition of viral entry by covalent entrapment of HIV gp41 on the virus surface. Biochemistry 2011, 50, 6966–6972. [Google Scholar]
- Zhou, G.; Ferrer, M.; Chopra, R.; Kapoor, T.M.; Strassmaier, T.; Weissenhorn, W.; Skehel, J.J.; Oprian, D.; Schreiber, S.L.; Harrison, S.C.; et al. The structure of an HIV-1 specific cell entry inhibitor in complex with the HIV-1 gp41 trimeric core. Bioorg. Med. Chem. 2000, 8, 2219–2227. [Google Scholar] [CrossRef]
- Hildinger, M.; Dittmar, M.T.; Schult-Dietrich, P.; Fehse, B.; Schnierle, B.S.; Thaler, S.; Stiegler, G.; Welker, R.; von Laer, D. Membrane-anchored peptide inhibits human immunodeficiency virus entry. J. Virol. 2001, 75, 3038–3042. [Google Scholar]
- Peisajovich, S.G.; Gallo, S.A.; Blumenthal, R.; Shai, Y. C-terminal octylation rescues an inactive T20 mutant: Implications for the mechanism of HIV/simian immunodeficiency virus-induced membrane fusion. J. Biol. Chem. 2003, 278, 21012–21017. [Google Scholar]
- Melikyan, G.B.; Egelhofer, M.; von Laer, D. Membrane-anchored inhibitory peptides capture human immunodeficiency virus type 1 gp41 conformations that engage the target membrane prior to fusion. J. Virol. 2006, 80, 3249–3258. [Google Scholar] [CrossRef]
- Zhang, H.; Schneider, S.E.; Bray, B.L.; Friedrich, P.E.; Tvermoes, N.A.; Mader, C.J.; Whight, S.R.; Niemi, T.E.; Silinski, P.; Picking, T.; et al. Process development of TRI-999, a fatty-acid-modified HIV fusion inhibitory peptide. Org. Process Res. Dev. 2008, 12, 101–110. [Google Scholar] [CrossRef]
- Ingallinella, P.; Bianchi, E.; Ladwa, N.A.; Wang, Y.-J.; Hrin, R.; Veneziano, M.; Bonelli, F.; Ketas, T.J.; Moore, J.P.; Miller, M.D.; et al. Addition of a cholesterol group to an HIV-1 peptide fusion inhibitor dramatically increases its antiviral potency. Proc. Natl. Acad. Sci. USA 2009, 106, 5801–5806. [Google Scholar]
- Cardoso, R.M.F.; Brunel, F.M.; Ferguson, S.; Zwick, M.; Burton, D.R.; Dawson, P.E.; Wilson, I.A. Structural basis of enhanced binding of extended and helically constrained peptide epitopes of the broadly neutralizing HIV-1 antibody 4E10. J. Mol. Biol. 2007, 365, 1533–1544. [Google Scholar] [CrossRef]
- Pejchal, R.; Gach, J.S.; Brunel, F.M.; Cardoso, R.M.; Stanfield, R.L.; Dawson, P.E.; Burton, D.R.; Zwick, M.B.; Wilson, I.A. A conformational switch in human immunodeficiency virus gp41 revealed by the structures of overlapping epitopes recognized by neutralizing antibodies. J. Virol. 2009, 83, 8451–8462. [Google Scholar] [CrossRef]
- Ofek, G.; Guenaga, F.J.; Schief, W.R.; Skinner, J.; Baker, D.; Wyatt, R.; Kwong, P.D. Elicitation of structure-specific antibodies by epitope scaffolds. Proc. Natl. Acad. Sci. USA 2010, 107, 17880–17887. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar]
- Protein Data Bank. Available online: http://www.pdb.org (accessed on 1 June 2012).
- Lu, M.; Blacklow, S.C.; Kim, P.S. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat. Struct. Biol. 1995, 2, 1075–1082. [Google Scholar] [CrossRef]
- Watabe, T.; Terakawa, Y.; Watanabe, K.; Ohno, H.; Nakano, H.; Nakatsu, T.; Kato, H.; Izumi, K.; Kodama, E.; Matsuoka, M.; et al. X-ray crystallographic study of an HIV-1 fusion inhibitor with the gp41 S138A substitution. J. Mol. Biol. 2009, 392, 657–665. [Google Scholar] [CrossRef]
- Izumi, K.; Nakamura, S.; Nakano, H.; Shimura, K.; Sakagami, Y.; Oishi, S.; Uchiyama, S.; Ohkubo, T.; Kobayashi, Y.; Fujii, N.; et al. Characterization of HIV-1 resistance to a fusion inhibitor, N36, derived from the gp41 amino-terminal heptad repeat. Antivir. Res. 2010, 87, 179–186. [Google Scholar]
- Structural insight of HIV-1 fusion inhibitor CP621-652 discovers the critical residues for viral entry and inhibition. J. Biol. Chem. 2012, in press.
- Ji, H.; Shu, W.; Burling, F.T.; Jiang, S.; Lu, M. Inhibition of human immunodeficiency virus type 1 infectivity by the gp41 core: Role of a conserved hydrophobic cavity in membrane fusion. J. Virol. 1999, 73, 8578–8586. [Google Scholar]
- Shu, W.; Liu, J.; Ji, H.; Radigen, L.; Jiang, S.; Lu, M. Helical interactions in the HIV-1 gp41 core reveal structural basis for the inhibitory activity of gp41 peptides. Biochemistry 2000, 39, 1634–1642. [Google Scholar]
- Shu, W.; Ji, H.; Lu, M. Interactions between HIV-1 gp41 core and detergents and their implications for membrane fusion. J. Biol. Chem. 2000, 275, 1839–1845. [Google Scholar]
- Lu, M.; Stoller, M.O.; Wang, S.; Liu, J.; Fagan, M.B.; Nunberg, J.H. Structural and functional analysis of interhelical interactions in the human immunodeficiency virus type 1 gp41 envelope glycoprotein by alanine-scanning mutagenesis. J. Virol. 2001, 75, 11146–11156. [Google Scholar]
- Wang, S.; York, J.; Shu, W.; Stoller, M.O.; Nunberg, J.H.; Lu, M. Interhelical interactions in the gp41 core: Implications for activation of HIV-1 membrane fusion. Biochemistry 2002, 41, 7283–7292. [Google Scholar]
- Bai, X.; Wilson, K.L.; Seedorff, J.E.; Ahrens, D.; Green, J.; Davison, D.K.; Jin, L.; Stanfield-Oakley, S.A.; Mosier, S.M.; Melby, T.E.; et al. Impact of the enfuvirtide resistance mutation N43D and the associated baseline polymorphism E137K on peptide sensitivity and six-helix bundle structure. Biochemistry 2008, 47, 6662–6670. [Google Scholar]
- Liu, J.; Shu, W.; Fagan, M.B.; Nunberg, J.H.; Lu, M. Structural and functional analysis of the HIV gp41 core containing an Ile573 to Thr substitution: Implications for membrane fusion. Biochemistry 2001, 40, 2797–2807. [Google Scholar]
- Buzon, V.; Natrajan, G.; Schibli, D.; Campelo, F.; Kozlov, M.M.; Weissenhorn, W. Crystal structure of HIV-1 gp41 including both fusion peptide and membrane proximal external regions. PLoS Pathog. 2010, 6, e1000880. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.-X.; Bohon, J.; Han, D.-P.; Habte, H.; Qin, Y.-L.; Cho, M.W.; Chance, M.R. Structural characterization of HIV gp41 with the membrane-proximal external region. J. Biol. Chem. 2010, 285, 24290–24298. [Google Scholar]
- Root, M.J.; Kay, M.S.; Kim, P.S. Protein design of an HIV-1 entry inhibitor. Science 2001, 291, 884–888. [Google Scholar]
- Johnson, L.M.; Horne, W.S.; Gellman, S.H. Broad distribution of energetically important contacts across an extended protein interface. J. Am. Chem. Soc. 2011, 133, 10038–10041. [Google Scholar]
- Johnson, L.M.; Mortenson, D.E.; Yun, H.G.; Horne, W.S.; Ketas, T.J.; Lu, M.; Moore, J.P.; Gellman, S.H. Enhancement of α-helix mimicry by an α/β-peptide foldamer via incorporation of a dense ionic side-chain array. J. Am. Chem. Soc. 2012, 134, 7317–7320. [Google Scholar]
- Harbury, P.B.; Kim, P.S.; Alber, T. Crystal structure of an isoleucine-zipper trimer. Nature 1994, 371, 80–83. [Google Scholar]
- Eckert, D.M.; Malashkevich, V.N.; Hong, L.H.; Carr, P.A.; Kim, P.S. Inhibiting HIV-1 entry: Discovery of D-peptide inhibitors that target the gp41 coiled-coil pocket. Cell 1999, 99, 103–115. [Google Scholar] [CrossRef]
- Sia, S.K.; Carr, P.A.; Cochran, A.G.; Malashkevich, V.N.; Kim, P.S. Short constrained peptides that inhibit HIV-1 entry. Proc. Natl. Acad. Sci. USA 2002, 99, 14664–14669. [Google Scholar]
- Welch, B.D.; VanDemark, A.P.; Heroux, A.; Hill, C.P.; Kay, M.S. Potent D-peptide inhibitors of HIV-1 entry. Proc. Natl. Acad. Sci. USA 2007, 104, 16828–16833. [Google Scholar]
- Welch, B.D.; Francis, J.N.; Redman, J.S.; Paul, S.; Weinstock, M.T.; Reeves, J.D.; Lie, Y.S.; Whitby, F.G.; Eckert, D.M.; Hill, C.P.; et al. Design of a potent D-peptide HIV-1 entry inhibitor with a strong barrier to resistance. J. Virol. 2010, 84, 11235–11244. [Google Scholar]
- Shu, W.; Ji, H.; Lu, M. Trimerization specificity in HIV-1 gp41: Analysis with a GCN4 leucine zipper model. Biochemistry 1999, 38, 5378–5385. [Google Scholar]
- Dwyer, J.J.; Wilson, K.L.; Martin, K.; Seedorff, J.E.; Hasan, A.; Medinas, R.J.; Davison, D.K.; Feese, M.D.; Richter, H.-T.; Kim, H.; et al. Design of an engineered N-terminal HIV-1 gp41 trimer with enhanced stability and potency. Protein Sci. 2008, 17, 633–643. [Google Scholar] [CrossRef]
- Horne, W.S.; Johnson, L.M.; Ketas, T.J.; Klasse, P.J.; Lu, M.; Moore, J.P.; Gellman, S.H. Structural and biological mimicry of protein surface recognition by α/β-peptide foldamers. Proc. Natl. Acad. Sci. USA 2009, 106, 14751–14756. [Google Scholar]
- Ofek, G.; Tang, M.; Sambor, A.; Katinger, H.; Mascola, J.R.; Wyatt, R.; Kwong, P.D. Structure and mechanistic analysis of the anti-human immunodeficiency virus type 1 antibody 2F5 in complex with its gp41 epitope. J. Virol. 2004, 78, 10724–10737. [Google Scholar]
- Julien, J.-P.; Bryson, S.; Nieva, J.L.; Pai, E.F. Structural details of HIV-1 recognition by the broadly neutralizing monoclonal antibody 2F5: Epitope conformation, antigen-recognition loop mobility, and anion-binding site. J. Mol. Biol. 2008, 384, 377–392. [Google Scholar] [CrossRef]
- Bryson, S.; Julien, J.-P.; Hynes, R.C.; Pai, E.F. Crystallographic definition of the epitope promiscuity of the broadly neutralizing anti-human immunodeficiency virus type 1 antibody 2F5: Vaccine design implications. J. Virol. 2009, 83, 11862–11875. [Google Scholar] [CrossRef]
- de la Arada, I.; Julien, J.-P.; de la Torre, B.G.; Huarte, N.; Andreu, D.; Pai, E.F.; Arrondo, J.L.R.; Nieva, J.L. Structural constraints imposed by the conserved fusion peptide on the HIV-1 gp41 epitope recognized by the broadly neutralizing antibody 2F5. J. Phys. Chem. B 2009, 113, 13626–13637. [Google Scholar]
- Cardoso, R.M.F.; Zwick, M.B.; Stanfield, R.L.; Kunert, R.; Binley, J.M.; Katinger, H.; Burton, D.R.; Wilson, I.A. Broadly neutralizing anti-HIV antibody 4E10 recognizes a helical conformation of a highly conserved fusion-associated motif in gp41. Immunity 2005, 22, 163–173. [Google Scholar] [CrossRef]
- Luftig, M.A.; Mattu, M.; Di, G.P.; Geleziunas, R.; Hrin, R.; Barbato, G.; Bianchi, E.; Miller, M.D.; Pessi, A.; Carfi, A. Structural basis for HIV-1 neutralization by a gp41 fusion intermediate-directed antibody. Nat. Struct. Mol. Biol. 2006, 13, 740–747. [Google Scholar]
- Sabin, C.; Corti, D.; Buzon, V.; Seaman, M.S.; Hulsik, D.L.; Hinz, A.; Vanzetta, F.; Agatic, G.; Silacci, C.; Mainetti, L.; et al. Crystal structure and size-dependent neutralization properties of HK20, a human monoclonal antibody binding to the highly conserved heptad repeat 1 of gp41. PLoS Pathog. 2010, 6, e1001195. [Google Scholar] [CrossRef] [Green Version]
- Gustchina, E.; Li, M.; Louis, J.M.; Anderson, D.E.; Lloyd, J.; Frisch, C.; Bewley, C.A.; Gustchina, A.; Wlodawer, A.; Clore, G.M. Structural basis of HIV-1 neutralization by affinity matured Fabs directed against the internal trimeric coiled-coil of gp41. PLoS Pathog. 2010, 6, e1001182. [Google Scholar] [CrossRef]
- Frey, G.; Chen, J.; Rits-Volloch, S.; Freeman, M.M.; Zolla-Pazner, S.; Chen, B. Distinct conformational states of HIV-1 gp41 are recognized by neutralizing and non-neutralizing antibodies. Nat. Struct. Mol. Biol. 2010, 17, 1486–1491. [Google Scholar]
- Jaroniec, C.P.; Kaufman, J.D.; Stahl, S.J.; Viard, M.; Blumenthal, R.; Wingfield, P.T.; Bax, A. Structure and dynamics of micelle-associated human immunodeficiency virus gp41 fusion domain. Biochemistry 2005, 44, 16167–16180. [Google Scholar] [CrossRef]
- Biron, Z.; Khare, S.; Samson, A.O.; Hayek, Y.; Naider, F.; Anglister, J. A monomeric 310-helix is formed in water by a 13-residue peptide representing the neutralizing determinant of HIV-1 on gp41. Biochemistry 2002, 41, 12687–12696. [Google Scholar] [CrossRef]
- Gordon, L.M.; Mobley, P.W.; Pilpa, R.; Sherman, M.A.; Waring, A.J. Conformational mapping of the N-terminal peptide of HIV-1 gp41 in membrane environments using 13C-enhanced Fourier transform infrared spectroscopy. Biochim. Biophys. Acta 2002, 1559, 96–120. [Google Scholar]
- Gordon, L.M.; Mobley, P.W.; Lee, W.; Eskandari, S.; Kaznessis, Y.N.; Sherman, M.A.; Waring, A.J. Conformational mapping of the N-terminal peptide of HIV-1 gp41 in lipid detergent and aqueous environments using 13C-enhanced Fourier transform infrared spectroscopy. Protein Sci. 2004, 13, 1012–1030. [Google Scholar] [CrossRef]
- Li, Y.; Tamm, L.K. Structure and plasticity of the human immunodeficiency virus gp41 fusion domain in lipid micelles and bilayers. Biophys. J. 2007, 93, 876–885. [Google Scholar] [CrossRef]
- Munch, J.; Standker, L.; Adermann, K.; Schulz, A.; Schindler, M.; Chinnadurai, R.; Pohlmann, S.; Chaipan, C.; Biet, T.; Peters, T.; et al. Discovery and optimization of a natural HIV-1 entry inhibitor targeting the gp41 fusion peptide. Cell 2007, 129, 263–275. [Google Scholar] [CrossRef]
- Du, A.P.C.; Limal, D.; Semetey, V.; Dali, H.; Jolivet, M.; Desgranges, C.; Cung, M.T.; Briand, J.-P.; Petit, M.-C.; Muller, S. Structural and immunological characterisation of heteroclitic peptide analogues corresponding to the 600–612 region of the HIV envelope gp41 glycoprotein. J. Mol. Biol. 2002, 323, 503–521. [Google Scholar] [CrossRef]
- Schibli, D.J.; Montelaro, R.C.; Vogel, H.J. The membrane-proximal tryptophan-rich region of the HIV glycoprotein, gp41, forms a well-defined helix in dodecylphosphocholine micelles. Biochemistry 2001, 40, 9570–9578. [Google Scholar] [CrossRef]
- Barbato, G.; Bianchi, E.; Ingallinella, P.; Hurni, W.H.; Miller, M.D.; Ciliberto, G.; Cortese, R.; Bazzo, R.; Shiver, J.W.; Pessi, A. Structural analysis of the epitope of the anti-HIV antibody 2F5 sheds light into its mechanism of neutralization and HIV fusion. J. Mol. Biol. 2003, 330, 1101–1115. [Google Scholar] [CrossRef]
- Sun, Z.-Y.J.; Oh, K.J.; Kim, M.; Yu, J.; Brusic, V.; Song, L.; Qiao, Z.; Wang, J.-H.; Wagner, G.; Reinherz, E.L. HIV-1 broadly neutralizing antibody extracts its epitope from a kinked gp41 ectodomain region on the viral membrane. Immunity 2008, 28, 52–63. [Google Scholar] [CrossRef]
- Liu, J.; Deng, Y.; Dey, A.K.; Moore, J.P.; Lu, M. Structure of the HIV-1 gp41 membrane-proximal ectodomain region in a putative prefusion conformation. Biochemistry 2009, 48, 2915–2923. [Google Scholar]
- Liu, J.; Deng, Y.; Li, Q.; Dey, A.K.; Moore, J.P.; Lu, M. Role of a putative gp41 dimerization domain in human immunodeficiency virus type 1 membrane fusion. J. Virol. 2010, 84, 201–209. [Google Scholar] [CrossRef]
- Roux, K.H.; Taylor, K.A. AIDS virus envelope spike structure. Curr. Opin. Struct. Biol. 2007, 17, 244–252. [Google Scholar] [CrossRef]
- Liu, J.; Bartesaghi, A.; Borgnia, M.J.; Sapiro, G.; Subramaniam, S. Molecular architecture of native HIV-1 gp120 trimers. Nature 2008, 455, 109–113. [Google Scholar]
- Pancera, M.; Majeed, S.; Ban, Y.-E.A.; Chen, L.; Huang, C.-C.; Kong, L.; Kwon, Y.D.; Stuckey, J.; Zhou, T.; Robinson, J.E.; et al. Structure of HIV-1 gp120 with gp41-interactive region reveals layered envelope architecture and basis of conformational mobility. Proc. Natl. Acad. Sci. USA 2010, 107, 1166–1171. [Google Scholar]
- Hu, G.; Liu, J.; Taylor, K.A.; Roux, K.H. Structural comparison of HIV-1 envelope spikes with and without the V1/V2 loop. J. Virol. 2011, 85, 2741–2750. [Google Scholar] [CrossRef]
- Subramaniam, S. The SIV surface spike imaged by electron tomography: One leg or three? PLoS Pathog. 2006, 2, e91. [Google Scholar] [CrossRef]
- Jiang, S.; Debnath, A.K. Development of HIV entry inhibitors targeted to the coiled-coil regions of gp41. Biochem. Biophys. Res. Commun. 2000, 269, 641–646. [Google Scholar] [CrossRef]
- DesJarlais, R.L.; Sheridan, R.P.; Seibel, G.L.; Dixon, J.S.; Kuntz, I.D.; Venkataraghavan, R. Using shape complementarity as an initial screen in designing ligands for a receptor binding site of known three-dimensional structure. J. Med. Chem. 1988, 31, 722–729. [Google Scholar] [CrossRef]
- Shoichet, B.K.; Bodian, D.L.; Kuntz, I.D. Molecular docking using shape descriptors. J. Comput. Chem. 1992, 13, 380–397. [Google Scholar] [CrossRef]
- Manetti, F.; Tintori, C.; Armand-Ugon, M.; Clotet-Codina, I.; Massa, S.; Ragno, R.; Este, J.A.; Botta, M. A combination of molecular dynamics and docking calculations to explore the binding mode of ADS-J1, a polyanionic compound endowed with anti-HIV-1 activity. J. Chem. Inf. Model. 2006, 46, 1344–1351. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar]
- Wang, H.; Qi, Z.; Guo, A.; Mao, Q.; Lu, H.; An, X.; Xia, C.; Li, X.; Debnath, A.K.; Wu, S.; et al. ADS-J1 inhibits human immunodeficiency virus type 1 entry by interacting with the gp41 pocket region and blocking fusion-active gp41 core formation. Antimicrob. Agents Chemother. 2009, 53, 4987–4998. [Google Scholar]
- He, Y.; Liu, S.; Li, J.; Lu, H.; Qi, Z.; Liu, Z.; Debnath, A.K.; Jiang, S. Conserved salt bridge between the N- and C-terminal heptad repeat regions of the human immunodeficiency virus type 1 gp41 core structure is critical for virus entry and inhibition. J. Virol. 2008, 82, 11129–11139. [Google Scholar]
- Liu, K.; Lu, H.; Hou, L.; Qi, Z.; Teixeira, C.; Barbault, F.; Fan, B.-T.; Liu, S.; Jiang, S.; Xie, L. Design, synthesis, and biological evaluation of N-carboxyphenylpyrrole derivatives as potent HIV fusion inhibitors targeting gp41. J. Med. Chem. 2008, 51, 7843–7854. [Google Scholar]
- Teixeira, C.; Barbault, F.; Rebehmed, J.; Liu, K.; Xie, L.; Lu, H.; Jiang, S.; Fan, B.; Maurel, F. Molecular modeling studies of N-substituted pyrrole derivatives-potential HIV-1 gp41 inhibitors. Bioorg. Med. Chem. 2008, 16, 3039–3048. [Google Scholar]
- Irwin, J.J.; Shoichet, B.K. ZINC—A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef]
- Lang, P.T.; Brozell, S.R.; Mukherjee, S.; Pettersen, E.F.; Meng, E.C.; Thomas, V.; Rizzo, R.C.; Case, D.A.; James, T.L.; Kuntz, I.D. DOCK 6: Combining techniques to model RNA-small molecule complexes. RNA 2009, 15, 1219–1230. [Google Scholar] [CrossRef]
- Moustakas, D.T.; Lang, P.T.; Pegg, S.; Pettersen, E.; Kuntz, I.D.; Brooijmans, N.; Rizzo, R.C. Development and validation of a modular, extensible docking program: DOCK 5. J. Comput.-Aided Mol. Des. 2006, 20, 601–619. [Google Scholar] [CrossRef]
- Tan, J.J.; Chen, W.Z.; Wang, C.X. Investigating interactions between HIV-1 gp41 and inhibitors by molecular dynamics simulation and MM-PBSA/GBSA calculations. J. Mol. Struct. 2006, 766, 77–82. [Google Scholar]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Dubay, J.W.; Roberts, S.J.; Brody, B.; Hunter, E. Mutations in the leucine zipper of the human immunodeficiency virus type 1 transmembrane glycoprotein affect fusion and infectivity. J. Virol. 1992, 66, 4748–4756. [Google Scholar]
- Bao, J.; Zhang, D.W.; Zhang, J.Z.H.; Huang, P.L.; Huang, P.L.; Lee-Huang, S. Computational study of bindings of olive leaf extract (OLE) to HIV-1 fusion protein gp41. FEBS Lett. 2007, 581, 2737–2742. [Google Scholar]
- Song, K.; Bao, J.; Sun, Y.; Zhang, J.Z.H. Computational characterization of binding of small molecule inhibitors to HIV-1 gp41. Chin. J. Chem. 2011, 29, 1307–1311. [Google Scholar] [CrossRef]
- Zhou, G.; Wu, D.; Snyder, B.; Ptak, R.G.; Kaur, H.; Gochin, M. Development of indole compounds as small molecule fusion inhibitors targeting HIV-1 glycoprotein-41. J. Med. Chem. 2011, 54, 7220–7231. [Google Scholar] [CrossRef]
- Tan, J.J.; Zhang, B.; Cong, X.J.; Yang, L.F.; Liu, B.; Kong, R.; Kui, Z.Y.; Wang, C.X.; Hu, L.M. Computer-aided design, synthesis, and biological activity evaluation of potent fusion inhibitors targeting HIV-1 gp41. Med. Chem. 2011, 7, 309–316. [Google Scholar] [CrossRef]
- Zhao, Q.; Ernst, J.T.; Hamilton, A.D.; Debnath, A.K.; Jiang, S. XTT formazan widely used to detect cell viability inhibits HIV type 1 infection in vitro by targeting gp41. AIDS Res. Hum. Retroviruses 2002, 18, 989–997. [Google Scholar] [CrossRef]
- Neurath, A.R.; Strick, N.; Jiang, S.; Li, Y.-Y.; Debnath, A.K. Anti-HIV-1 activity of cellulose acetate phthalate: Synergy with soluble CD4 and induction of “dead-end” gp41 six-helix bundles. BMC Infect. Dis. 2002, 2, 6. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Lu, H.; Zhao, Q.; He, Y.; Niu, J.; Debnath, A.K.; Wu, S.; Jiang, S. Theaflavin derivatives in black tea and catechin derivatives in green tea inhibit HIV-1 entry by targeting gp41. Biochim. Biophys. Acta 2005, 1723, 270–281. [Google Scholar]
- Protein Data Bank code 3P7K. Available online: http://www.pdb.org (accessed on 1 June 2012).
- Gochin, M.; Zhou, G.-Y.; Phillips, A.H. Paramagnetic relaxation assisted docking of a small indole compound in the HIV-1 gp41 hydrophobic pocket. ACS Chem. Biol. 2011, 6, 267–274. [Google Scholar] [CrossRef]
- Siebert, X.; Hummer, G. Hydrophobicity maps of the N-peptide coiled coil of HIV-1 gp41. Biochemistry 2002, 41, 2956–2961. [Google Scholar] [CrossRef]
- Pohorille, A.; Pratt, L.R. Cavities in molecular liquids and the theory of hydrophobic solubilities. J. Am. Chem. Soc. 1990, 112, 5066–5074. [Google Scholar] [CrossRef]
- Strockbine, B.; Rizzo, R.C. Binding of antifusion peptides with HIVgp41 from molecular dynamics simulations: Quantitative correlation with experiment. Proteins: Struct. Funct. Bioinf. 2007, 67, 630–642. [Google Scholar] [CrossRef]
- Tan, J.J.; Kong, R.; Wang, C.X.; Chen, W.Z. Molecular dynamics simulation on the complexes of N-terminal region of HIV-1 gp41 and its C-peptide inhibitors. J. Mol. Struct. 2004, 682, 9–15. [Google Scholar]
- McGillick, B.E.; Balius, T.E.; Mukherjee, S.; Rizzo, R.C. Origins of resistance to the HIVgp41 viral entry inhibitor T20. Biochemistry 2010, 49, 3575–3592. [Google Scholar]
- Caffrey, M.; Cai, M.; Kaufman, J.; Stahl, S.J.; Wingfield, P.T.; Gronenborn, A.M.; Clore, G.M. Determination of the secondary structure and global topology of the 44 kDa ectodomain of gp41 of the simian immunodeficiency virus by multidimensional nuclear magnetic resonance spectroscopy. J. Mol. Biol. 1997, 271, 819–826. [Google Scholar] [CrossRef]
- Caffrey, M.; Cai, M.; Kaufman, J.; Stahl, S.J.; Wingfield, P.T.; Covell, D.G.; Gronenborn, A.M.; Clore, M.G. Three-dimensional solution structure of the 44 kDa ectodomain of SIV gp41. EMBO J. 1998, 17, 4572–4584. [Google Scholar]
- Caffrey, M. Model for the structure of the HIV gp41 ectodomain: Insight into the intermolecular interactions of the gp41 loop. Biochim. Biophys. Acta 2001, 1536, 116–122. [Google Scholar]
- Qiu, S.; Yi, H.; Hu, J.; Cao, Z.; Wu, Y.; Li, W. The binding mode of fusion inhibitor T20 onto HIV-1 gp41 and relevant T20-resistant mechanisms explored by computational study. Curr. HIV Res. 2012, 10, 182–194. [Google Scholar] [CrossRef]
- Qiang, W.; Bodner, M.L.; Weliky, D.P. Solid-state NMR spectroscopy of human immunodeficiency virus fusion peptides associated with host-cell-like membranes: 2D correlation spectra and distance measurements support a fully extended conformation and models for specific antiparallel strand registries. J. Am. Chem. Soc. 2008, 130, 5459–5471. [Google Scholar] [CrossRef]
- Qiang, W.; Weliky, D.P. HIV fusion peptide and its cross-linked oligomers: Efficient syntheses, significance of the trimer in fusion activity, correlation of β strand conformation with membrane cholesterol, and proximity to lipid headgroups. Biochemistry 2009, 48, 289–301. [Google Scholar]
- Reichert, J.; Grasnick, D.; Afonin, S.; Buerck, J.; Wadhwani, P.; Ulrich, A.S. A critical evaluation of the conformational requirements of fusogenic peptides in membranes. Eur. Biophys. J. 2007, 36, 405–413. [Google Scholar] [CrossRef]
- Sackett, K.; Nethercott, M.J.; Epand, R.F.; Epand, R.M.; Kindra, D.R.; Shai, Y.; Weliky, D.P. Comparative analysis of membrane-associated fusion peptide secondary structure and lipid mixing function of HIV gp41 constructs that model the early pre-hairpin intermediate and final hairpin conformations. J. Mol. Biol. 2010, 397, 301–315. [Google Scholar] [CrossRef]
- Kamath, S.; Wong, T.C. Membrane structure of the human immunodeficiency virus gp41 fusion domain by molecular dynamics simulation. Biophys. J. 2002, 83, 135–143. [Google Scholar] [CrossRef]
- Wong, T.C. Membrane structure of the human immunodeficiency virus gp41 fusion peptide by molecular dynamics simulation II. The glycine mutants. Biochim. Biophys. Acta 2003, 1609, 45–54. [Google Scholar]
- Huang, Q.; Chen, C.-L.; Herrmann, A. Bilayer conformation of fusion peptide of influenza virus hemagglutinin: A molecular dynamics simulation study. Biophys. J. 2004, 87, 14–22. [Google Scholar] [CrossRef]
- Grasnick, D.; Sternberg, U.; Strandberg, E.; Wadhwani, P.; Ulrich, A.S. Irregular structure of the HIV fusion peptide in membranes demonstrated by solid-state NMR and MD simulations. Eur. Biophys. J. 2011, 40, 529–543. [Google Scholar] [CrossRef]
- Venken, T.; Krnavek, D.; Muench, J.; Kirchhoff, F.; Henklein, P.; De, M.M.; Voet, A. An optimized MM/PBSA virtual screening approach applied to an HIV-1 gp41 fusion peptide inhibitor. Proteins: Struct. Funct. Bioinf. 2011, 79, 3221–3235. [Google Scholar] [CrossRef]
- Kim, J.H.; Hartley, T.L.; Curran, A.R.; Engelman, D.M. Molecular dynamics studies of the transmembrane domain of gp41 from HIV-1. Biochim. Biophys. Acta 2009, 1788, 1804–1812. [Google Scholar]
- Martins do Canto, A.M.T.; Carvalho, A.J.P.; Ramalho, J.P.P.; Loura, L.M.S. T-20 and T-1249 HIV fusion inhibitors’ structure and conformation in solution: A molecular dynamics study. J. Pept. Sci. 2008, 14, 442–447. [Google Scholar] [CrossRef]
- Martins do Canto, A.M.T.; Carvalho, A.J.P.; Ramalho, J.P.P.; Loura, L.M.S. Molecular dynamics simulations of T-20 HIV fusion inhibitor interacting with model membranes. Biophys. Chem. 2011, 159, 275–286. [Google Scholar] [CrossRef]
- Martins do Canto, A.M.T.; Palace, C.A.J.; Prates, R.J.P.; Loura, L.M.S. Structure and conformation of HIV fusion inhibitor peptide T-1249 in presence of model membranes: A molecular dynamics study. J. Mol. Struct. 2010, 946, 119–124. [Google Scholar]
- Singh, P.; Sharma, P.; Bisetty, K.; Corcho, F.J.; Perez, J.J. Comparative structural studies of T-20 analogues using molecular dynamics. Comput. Theor. Chem. 2011, 974, 122–132. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Allen, W.J.; Rizzo, R.C. Computer-Aided Approaches for Targeting HIVgp41. Biology 2012, 1, 311-338. https://doi.org/10.3390/biology1020311
Allen WJ, Rizzo RC. Computer-Aided Approaches for Targeting HIVgp41. Biology. 2012; 1(2):311-338. https://doi.org/10.3390/biology1020311
Chicago/Turabian StyleAllen, William J., and Robert C. Rizzo. 2012. "Computer-Aided Approaches for Targeting HIVgp41" Biology 1, no. 2: 311-338. https://doi.org/10.3390/biology1020311
APA StyleAllen, W. J., & Rizzo, R. C. (2012). Computer-Aided Approaches for Targeting HIVgp41. Biology, 1(2), 311-338. https://doi.org/10.3390/biology1020311