

Comparison of Alternative Splicing Landscapes Revealed by Long-Read Sequencing in Hepatocyte-Derived HepG2 and Huh7 Cultured Cells and Human Liver Tissue
 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Liver Tissue
2.2. RNA Isolation, Library Preparation, and Long-Read Sequencing
2.3. Data Analysis
3. Results
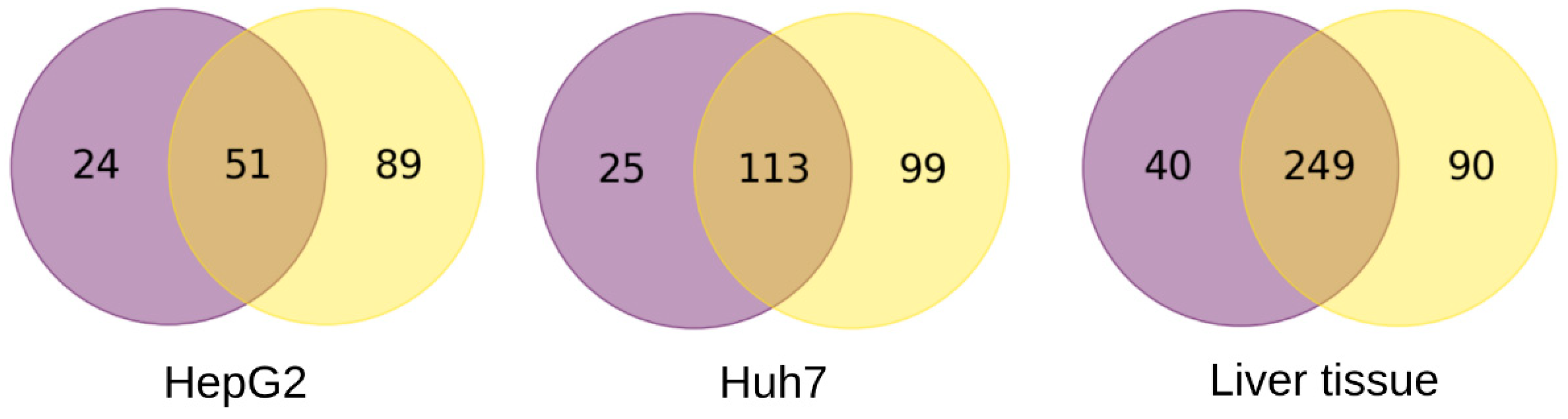
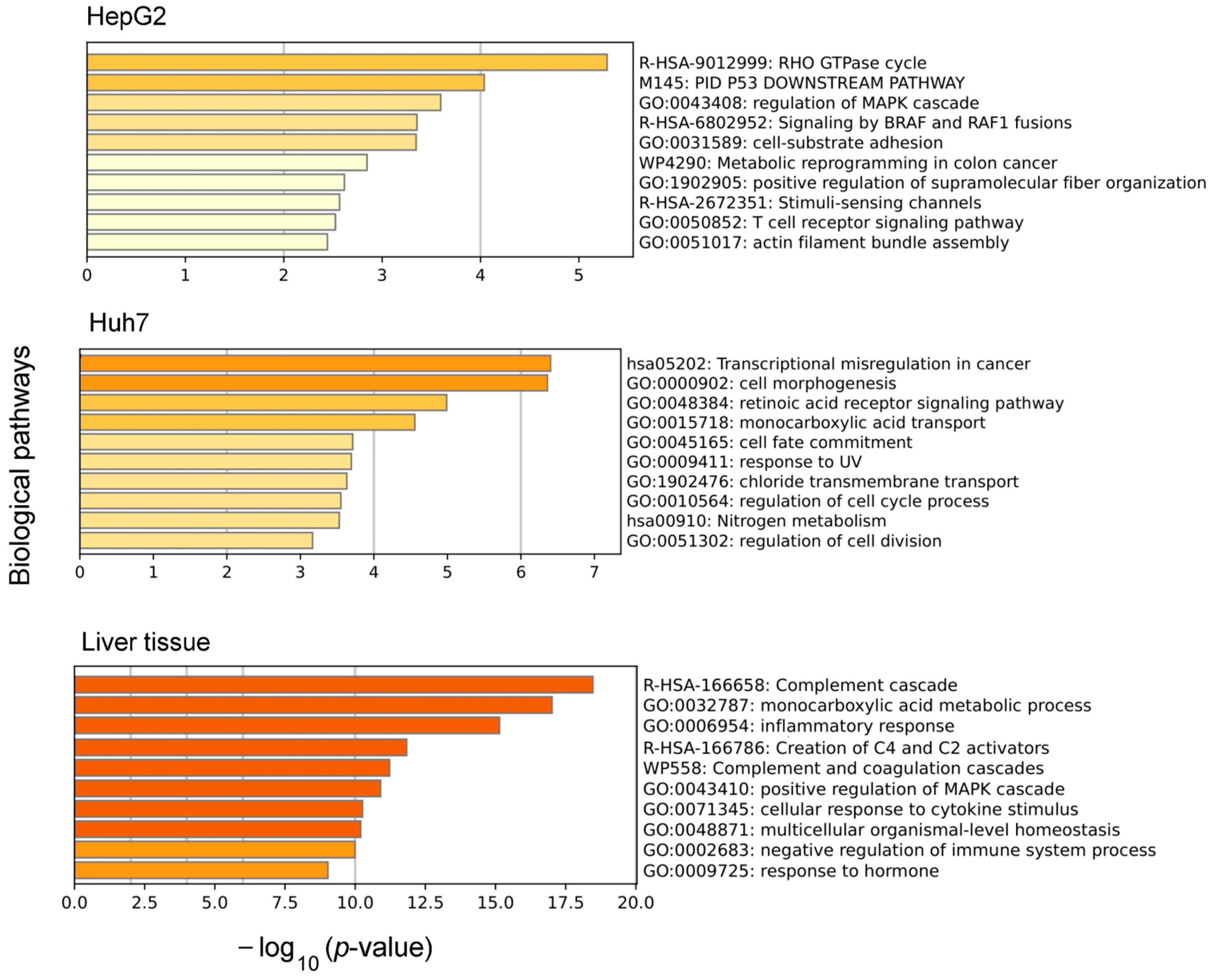
3.1. Biological Pathways Influenced by Differences in Alternative Splicing in Liver Tissue and Hepatocyte-Derived Cell Lines
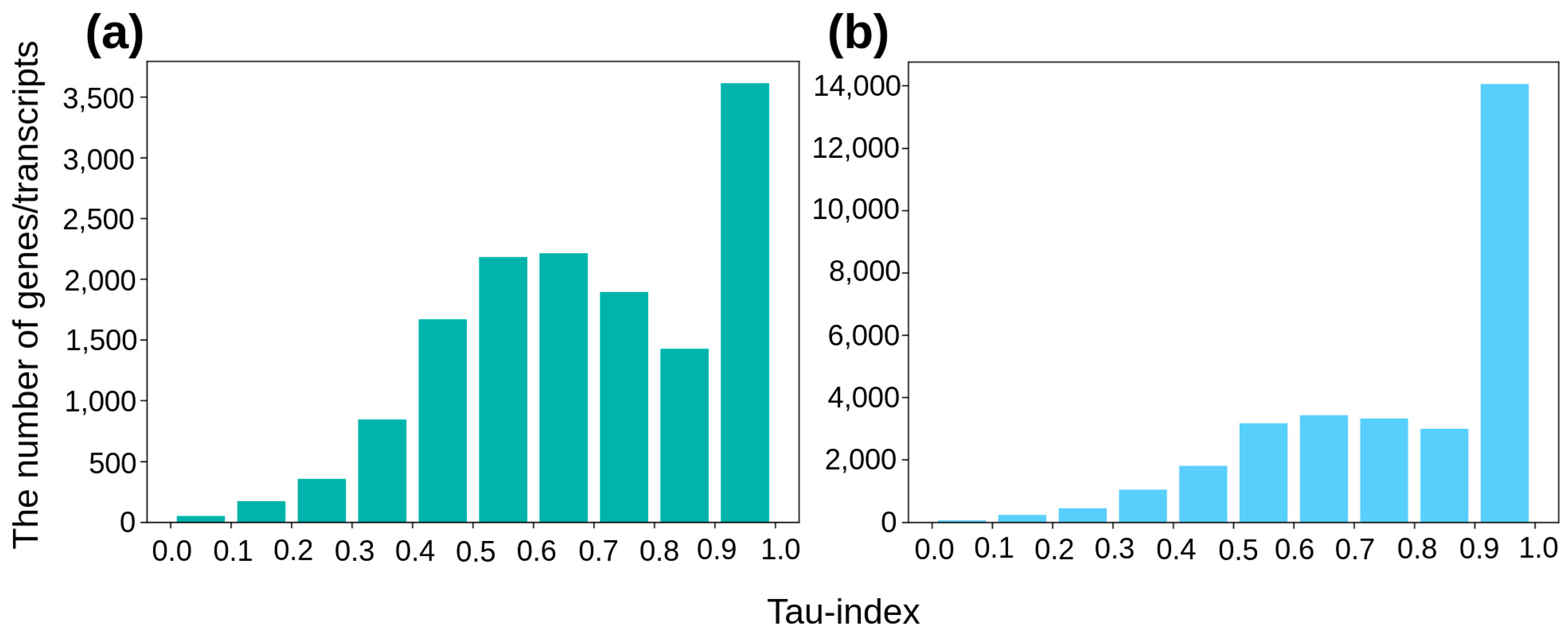
3.2. Genes with a Phenotype-Specific Splice Variant or Integral Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nilsen, T.W.; Graveley, B.R. Expansion of the Eukaryotic Proteome by Alternative Splicing. Nature 2010, 463, 457–463. [Google Scholar] [CrossRef]
- Jiang, W.; Chen, L. Alternative Splicing: Human Disease and Quantitative Analysis from High-Throughput Sequencing. Comput. Struct. Biotechnol. J. 2021, 19, 183–195. [Google Scholar] [CrossRef]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA Sequencing: The Teenage Years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential Analyses for RNA-Seq: Transcript-Level Estimates Improve Gene-Level Inferences. F1000Research 2016, 4, 1521. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, B.; Lin, L.-L.; Zhao, S. Evaluation and Comparison of Computational Tools for RNA-Seq Isoform Quantification. BMC Genom. 2017, 18, 583. [Google Scholar] [CrossRef]
- Anders, S.; Reyes, A.; Huber, W. Detecting Differential Usage of Exons from RNA-Seq Data. Genome Res. 2012, 22, 2008–2017. [Google Scholar] [CrossRef]
- Schafer, S.; Miao, K.; Benson, C.C.; Heinig, M.; Cook, S.A.; Hubner, N. Alternative Splicing Signatures in RNA-seq Data: Percent Spliced in (PSI). Curr. Protoc. Hum. Genet. 2015, 87, 11–16. [Google Scholar] [CrossRef]
- Halperin, R.F.; Hegde, A.; Lang, J.D.; Raupach, E.A.; C4RCD Research Group; Narayanan, V.; Huentelman, M.; Belnap, N.; Aziz, A.-M.; Ramsey, K.; et al. Improved Methods for RNAseq-Based Alternative Splicing Analysis. Sci. Rep. 2021, 11, 10740. [Google Scholar] [CrossRef]
- Benegas, G.; Fischer, J.; Song, Y.S. Robust and Annotation-Free Analysis of Alternative Splicing across Diverse Cell Types in Mice. eLife 2022, 11, e73520. [Google Scholar] [CrossRef]
- Leshkowitz, D.; Kedmi, M.; Fried, Y.; Pilzer, D.; Keren-Shaul, H.; Ainbinder, E.; Dassa, B. Exploring Differential Exon Usage via Short- and Long-Read RNA Sequencing Strategies. Open Biol. 2022, 12, 220206. [Google Scholar] [CrossRef]
- Wang, L.; Shi, L.; Liang, Y.; Ng, J.K.-W.; Yin, C.H.; Wang, L.; Hou, J.; Wang, Y.; Fung, C.S.-H.; Chiu, P.K.-F.; et al. Dissecting the Effects of METTL3 on Alternative Splicing in Prostate Cancer. Front. Oncol. 2023, 13, 1227016. [Google Scholar] [CrossRef]
- Hughes, A.E.O.; Montgomery, M.C.; Liu, C.; Weimer, E.T. Allele-Specific Quantification of Human Leukocyte Antigen Transcript Isoforms by Nanopore Sequencing. Front. Immunol. 2023, 14, 1199618. [Google Scholar] [CrossRef]
- Aguzzoli Heberle, B.; Brandon, J.A.; Page, M.L.; Nations, K.A.; Dikobe, K.I.; White, B.J.; Gordon, L.A.; Fox, G.A.; Wadsworth, M.E.; Doyle, P.H.; et al. Using Deep Long-Read RNAseq in Alzheimer’s Disease Brain to Assess Clinical Relevance of RNA Isoform Diversity. bioRxiv 2023, bioRxiv:2023:2023-08. [Google Scholar] [CrossRef]
- Yao, T.; Zhang, Z.; Li, Q.; Huang, R.; Hong, Y.; Li, C.; Zhang, F.; Huang, Y.; Fang, Y.; Cao, Q.; et al. Long-Read Sequencing Reveals Alternative Splicing-Driven, Shared Immunogenic Neoepitopes Regardless of SF3B1 Status in Uveal Melanoma. Cancer Immunol. Res. 2023, OF1–OF17. [Google Scholar] [CrossRef]
- Halstead, M.M.; Islas-Trejo, A.; Goszczynski, D.E.; Medrano, J.F.; Zhou, H.; Ross, P.J. Large-Scale Multiplexing Permits Full-Length Transcriptome Annotation of 32 Bovine Tissues From a Single Nanopore Flow Cell. Front. Genet. 2021, 12, 664260. [Google Scholar] [CrossRef]
- Sarygina, E.; Kozlova, A.; Deinichenko, K.; Radko, S.; Ptitsyn, K.; Khmeleva, S.; Kurbatov, L.K.; Spirin, P.; Prassolov, V.S.; Ilgisonis, E.; et al. Principal Component Analysis of Alternative Splicing Profiles Revealed by Long-Read ONT Sequencing in Human Liver Tissue and Hepatocyte-Derived HepG2 and Huh7 Cell Lines. Int. J. Mol. Sci. 2023, 24, 15502. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Yanai, I.; Benjamin, H.; Shmoish, M.; Chalifa-Caspi, V.; Shklar, M.; Ophir, R.; Bar-Even, A.; Horn-Saban, S.; Safran, M.; Domany, E.; et al. Genome-Wide Midrange Transcription Profiles Reveal Expression Level Relationships in Human Tissue Specification. Bioinformatics 2005, 21, 650–659. [Google Scholar] [CrossRef]
- Oku, Y.; Madia, F.; Lau, P.; Paparella, M.; McGovern, T.; Luijten, M.; Jacobs, M.N. Analyses of Transcriptomics Cell Signalling for Pre-Screening Applications in the Integrated Approach for Testing and Assessment of Non-Genotoxic Carcinogens. Int. J. Mol. Sci. 2022, 23, 12718. [Google Scholar] [CrossRef]
- Bortolomeazzi, M.; Keddar, M.R.; Ciccarelli, F.D.; Benedetti, L. Identification of Non-Cancer Cells from Cancer Transcriptomic Data. Biochim. Biophys. Acta BBA - Gene Regul. Mech. 2020, 1863, 194445. [Google Scholar] [CrossRef]
- Liu, W.; Zhu, P.; Li, M.; Li, Z.; Yu, Y.; Liu, G.; Du, J.; Wang, X.; Yang, J.; Tian, R.; et al. Large-scale across Species Transcriptomic Analysis Identifies Genetic Selection Signatures Associated with Longevity in Mammals. EMBO J. 2023, 42, e112740. [Google Scholar] [CrossRef]
- Fu, W.; Wang, R.; Nanaei, H.A.; Wang, J.; Hu, D.; Jiang, Y. RGD v2.0: A Major Update of the Ruminant Functional and Evolutionary Genomics Database. Nucleic Acids Res. 2022, 50, D1091–D1099. [Google Scholar] [CrossRef] [PubMed]
- Larouche, J.-D.; Trofimov, A.; Hesnard, L.; Ehx, G.; Zhao, Q.; Vincent, K.; Durette, C.; Gendron, P.; Laverdure, J.-P.; Bonneil, É.; et al. Widespread and Tissue-Specific Expression of Endogenous Retroelements in Human Somatic Tissues. Genome Med. 2020, 12, 40. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Schrooders, Y.; Hauser, D.; Van Herwijnen, M.; Albrecht, W.; Ter Braak, B.; Brecklinghaus, T.; Castell, J.V.; Elenschneider, L.; Escher, S.; et al. Comparing in Vitro Human Liver Models to in Vivo Human Liver Using RNA-Seq. Arch. Toxicol. 2021, 95, 573–589. [Google Scholar] [CrossRef] [PubMed]
- Arzumanian, V.A.; Kiseleva, O.I.; Poverennaya, E.V. The Curious Case of the HepG2 Cell Line: 40 Years of Expertise. Int. J. Mol. Sci. 2021, 22, 13135. [Google Scholar] [CrossRef]
- Choi, S.; Sainz, B.; Corcoran, P.; Uprichard, S.; Jeong, H. Characterization of Increased Drug Metabolism Activity in Dimethyl Sulfoxide (DMSO)-Treated Huh7 Hepatoma Cells. Xenobiotica 2009, 39, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Krelle, A.C.; Okoli, A.S.; Mendz, G.L. Huh-7 Human Liver Cancer Cells: A Model System to Understand Hepatocellular Carcinoma and Therapy. J. Cancer Ther. 2013, 04, 606–631. [Google Scholar] [CrossRef]
- Miranda, C.A.; Beretta, E.M.; Ferreira, L.A.; Da Silva, E.S.; Coimbra, B.Z.; Pereira, P.T.; Miranda, R.G.; Dorta, D.J.; Rodrigues, F.T.V.; Mingatto, F.E. Role of Biotransformation in the Diazinon-Induced Toxicity in HepG2 Cells and Antioxidant Protection by Tetrahydrocurcumin. Toxicol. Rep. 2023, 10, 32–39. [Google Scholar] [CrossRef]
- Ćwiklińska-Jurkowska, M.; Wiese-Szadkowska, M.; Janciauskiene, S.; Paprocka, R. Disparities in Cisplatin-Induced Cytotoxicity—A Meta-Analysis of Selected Cancer Cell Lines. Molecules 2023, 28, 5761. [Google Scholar] [CrossRef]
- Yin, L.; Liu, P.; Jin, Y.; Ning, Z.; Yang, Y.; Gao, H. Ferroptosis-Related Small-Molecule Compounds in Cancer Therapy: Strategies and Applications. Eur. J. Med. Chem. 2022, 244, 114861. [Google Scholar] [CrossRef]
- Alexia, C.; Fallot, G.; Lasfer, M.; Schweizer-Groyer, G.; Groyer, A. An Evaluation of the Role of Insulin-like Growth Factors (IGF) and of Type-I IGF Receptor Signalling in Hepatocarcinogenesis and in the Resistance of Hepatocarcinoma Cells against Drug-Induced Apoptosis. Biochem. Pharmacol. 2004, 68, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Cocciadiferro, L.; Miceli, V.; Granata, O.M.; Carruba, G. Merlin, the Product of NF2 Gene, Is Associated with Aromatase Expression and Estrogen Formation in Human Liver Tissues and Liver Cancer Cells. J. Steroid Biochem. Mol. Biol. 2017, 172, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Deynichenko, K.A.; Ptitsyn, K.G.; Radko, S.P.; Kurbatov, L.K.; Vakhrushev, I.V.; Buromski, I.V.; Markin, S.S.; Archakov, A.I.; Lisitsa, A.V.; Ponomarenko, E.A. Splice Variants of mRNA of Cytochrome P450 Genes: Analysis by the Nanopore Sequencing Method in Human Liver Tissue and HepG2 Cell Line. Biochem. Mosc. Suppl. Ser. B Biomed. Chem. 2022, 16, 318–327. [Google Scholar] [CrossRef]
- Li, X.; Yu, K.; Li, F.; Lu, W.; Wang, Y.; Zhang, W.; Bai, Y. Novel Method of Full-Length RNA-Seq That Expands the Identification of Non-Polyadenylated RNAs Using Nanopore Sequencing. Anal. Chem. 2022, 94, 12342–12351. [Google Scholar] [CrossRef]
- Shapovalova, V.V.; Radko, S.P.; Ptitsyn, K.G.; Krasnov, G.S.; Nakhod, K.V.; Konash, O.S.; Vinogradina, M.A.; Ponomarenko, E.A.; Druzhilovskiy, D.S.; Lisitsa, A.V. Processing Oxford Nanopore Long Reads Using Amazon Web Services. Biomed. Chem. Res. Methods 2020, 3, e00131. [Google Scholar] [CrossRef]
- Lanfear, R.; Schalamun, M.; Kainer, D.; Wang, W.; Schwessinger, B. MinIONQC: Fast and Simple Quality Control for MinION Sequencing Data. Bioinformatics 2019, 35, 523–525. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Martens, M.; Ammar, A.; Riutta, A.; Waagmeester, A.; Slenter, D.N.; Hanspers, K.; A Miller, R.; Digles, D.; Lopes, E.N.; Ehrhart, F.; et al. WikiPathways: Connecting Communities. Nucleic Acids Res. 2021, 49, D613–D621. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for Taxonomy-Based Analysis of Pathways and Genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef] [PubMed]
- harmonizome: A Collection of Processed Datasets Gathered to Serve and Mine Knowledge about Genes and Proteins|Database|Oxford Academic. Available online: https://academic.oup.com/database/article/doi/10.1093/database/baw100/2630482 (accessed on 27 October 2023).
- Su, T.; Hollas, M.A.R.; Fellers, R.T.; Kelleher, N.L. Identification of Splice Variants and Isoforms in Transcriptomics and Proteomics. Annu. Rev. Biomed. Data Sci. 2023, 6, 357–376. [Google Scholar] [CrossRef]
- Mancini, E.; Rabinovich, A.; Iserte, J.; Yanovsky, M.; Chernomoretz, A. ASpli: Integrative Analysis of Splicing Landscapes through RNA-Seq Assays. Bioinformatics 2021, 37, 2609–2616. [Google Scholar] [CrossRef] [PubMed]
- Costa-Silva, J.; Domingues, D.S.; Menotti, D.; Hungria, M.; Lopes, F.M. Temporal Progress of Gene Expression Analysis with RNA-Seq Data: A Review on the Relationship between Computational Methods. Comput. Struct. Biotechnol. J. 2023, 21, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Vasko, R. Peroxisomes and Kidney Injury. Antioxid. Redox Signal. 2016, 25, 217–231. [Google Scholar] [CrossRef]
- Malik, A.; Thanekar, U.; Amarachintha, S.; Mourya, R.; Nalluri, S.; Bondoc, A.; Shivakumar, P. “Complimenting the Complement”: Mechanistic Insights and Opportunities for Therapeutics in Hepatocellular Carcinoma. Front. Oncol. 2021, 10, 627701. [Google Scholar] [CrossRef]
- Trefts, E.; Gannon, M.; Wasserman, D.H. The Liver. Curr. Biol. 2017, 27, R1147–R1151. [Google Scholar] [CrossRef]
- Nwosu, Z.C.; Battello, N.; Rothley, M.; Piorońska, W.; Sitek, B.; Ebert, M.P.; Hofmann, U.; Sleeman, J.; Wölfl, S.; Meyer, C.; et al. Liver Cancer Cell Lines Distinctly Mimic the Metabolic Gene Expression Pattern of the Corresponding Human Tumours. J. Exp. Clin. Cancer Res. 2018, 37, 211. [Google Scholar] [CrossRef]
- Wang, X.; Cairns, M.J. SeqGSEA: A Bioconductor Package for Gene Set Enrichment Analysis of RNA-Seq Data Integrating Differential Expression and Splicing. Bioinformatics 2014, 30, 1777–1779. [Google Scholar] [CrossRef]
- Choi, J.M.; Oh, S.J.; Lee, S.Y.; Im, J.H.; Oh, J.M.; Ryu, C.S.; Kwak, H.C.; Lee, J.-Y.; Kang, K.W.; Kim, S.K. HepG2 Cells as an in Vitro Model for Evaluation of Cytochrome P450 Induction by Xenobiotics. Arch. Pharm. Res. 2015, 38, 691–704. [Google Scholar] [CrossRef]
- High Expression of RPL27A Predicts Poor Prognosis in Patients with Hepatocellular Carcinoma|World Journal of Surgical Oncology|Full Text. Available online: https://wjso.biomedcentral.com/articles/10.1186/s12957-023-03102-w (accessed on 27 October 2023).
- Sciarrillo, R.; Wojtuszkiewicz, A.; Assaraf, Y.G.; Jansen, G.; Kaspers, G.J.L.; Giovannetti, E.; Cloos, J. The Role of Alternative Splicing in Cancer: From Oncogenesis to Drug Resistance. Drug Resist. Updates 2020, 53, 100728. [Google Scholar] [CrossRef]
- Kahles, A.; Lehmann, K.-V.; Toussaint, N.C.; Hüser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; Rätsch, G.; et al. Comprehensive Analysis of Alternative Splicing Across Tumors from 8705 Patients. Cancer Cell 2018, 34, 211–224. [Google Scholar] [CrossRef]
- Climente-González, H.; Porta-Pardo, E.; Godzik, A.; Eyras, E. The Functional Impact of Alternative Splicing in Cancer. Cell Rep. 2017, 20, 2215–2226. [Google Scholar] [CrossRef]
- Badve, S.; Logdberg, L.; Lal, A.; De Davila, M.T.G.; Greco, M.A.; Mitsudo, S.; Saxena, R. Small Cells in Hepatoblastoma Lack “Oval” Cell Phenotype. Mod. Pathol. 2003, 16, 930–936. [Google Scholar] [CrossRef]
- Zhuang, H.; Peng, Y.; Chen, T.; Jiang, Y.; Luo, Y.; Zhang, Q.; Yang, Z. The Comparison of Grey-Scale Ultrasonic and Clinical Features of Hepatoblastoma and Hepatocellular Carcinoma in Children: A Retrospective Study for Ten Years. BMC Gastroenterol. 2011, 11, 78. [Google Scholar] [CrossRef]
- Haga, R.B.; Ridley, A.J. Rho GTPases: Regulation and Roles in Cancer Cell Biology. Small GTPases 2016, 7, 207–221. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phenotype | Transcript (Splice Variant) Name | Protein UniProt ID | Current Proteoform Status |
|---|---|---|---|
| Liver tissue | SLC17A1-201 | Q14916-1 | canonic |
| Liver tissue | SLC17A1-204 | Q14916-1 | canonic |
| Liver tissue | ALDOB-203 | P05062 | canonic |
| Liver tissue | ALDOB-207 | A0A3B3IS80 | predicted |
| Liver tissue | APOC3-202 | B0YIW2 | predicted |
| Liver tissue | APOC3-205 | B0YIW2 | predicted |
| Huh7 cells | UROS-211 | A0A087WZB7 | predicted |
| Huh7 cells | UROS-203 | P10746 | canonic |
| Huh7 cells | GJC1-204 | Q5H9P2 | predicted |
| Huh7 cells | GJC1-206 | P36383 | canonic |
| HepG2 cells | ASPHD1-203 | I3L2A5 | predicted |
| HepG2 cells | ASPHD1-201 | Q5U4P2 | canonic |
| HepG2 cells | NEDD4L-205 | Q96PU5 | canonic |
| HepG2 cells | NEDD4L-225 | K7EKL1 | predicted |
| HepG2 cells | SLC13A3-201 | Q8WWT9 | canonic |
| HepG2 cells | SLC13A3-205 | C9J4A3 | predicted |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kozlova, A.; Sarygina, E.; Deinichenko, K.; Radko, S.; Ptitsyn, K.; Khmeleva, S.; Kurbatov, L.; Spirin, P.; Prassolov, V.; Ilgisonis, E.; et al. Comparison of Alternative Splicing Landscapes Revealed by Long-Read Sequencing in Hepatocyte-Derived HepG2 and Huh7 Cultured Cells and Human Liver Tissue. Biology 2023, 12, 1494. https://doi.org/10.3390/biology12121494
Kozlova A, Sarygina E, Deinichenko K, Radko S, Ptitsyn K, Khmeleva S, Kurbatov L, Spirin P, Prassolov V, Ilgisonis E, et al. Comparison of Alternative Splicing Landscapes Revealed by Long-Read Sequencing in Hepatocyte-Derived HepG2 and Huh7 Cultured Cells and Human Liver Tissue. Biology. 2023; 12(12):1494. https://doi.org/10.3390/biology12121494
Chicago/Turabian StyleKozlova, Anna, Elizaveta Sarygina, Kseniia Deinichenko, Sergey Radko, Konstantin Ptitsyn, Svetlana Khmeleva, Leonid Kurbatov, Pavel Spirin, Vladimir Prassolov, Ekaterina Ilgisonis, and et al. 2023. "Comparison of Alternative Splicing Landscapes Revealed by Long-Read Sequencing in Hepatocyte-Derived HepG2 and Huh7 Cultured Cells and Human Liver Tissue" Biology 12, no. 12: 1494. https://doi.org/10.3390/biology12121494