


Cellular Stress: Modulator of Regulated Cell Death

Department of Microbiology and Immunology, University of Texas Medical Branch, Galveston, TX 77555, USA
*
Author to whom correspondence should be addressed.
Biology 2023, 12(9), 1172; https://doi.org/10.3390/biology12091172
Submission received: 28 July 2023
/
Revised: 22 August 2023
/
Accepted: 22 August 2023
/
Published: 25 August 2023
(This article belongs to the Special Issue Cell Self-Destruction (Programmed Cell Death), Immunonutrition and Metabolism)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Stress and regulated cell death signaling are activated simultaneously during disease conditions. However, the crosstalk between the two signaling pathways is poorly understood. There has been a renewed focus on understanding this crosstalk. This review summarizes the current state of knowledge about the crosstalk between stress and regulated cell death.
Abstract
Cellular stress response activates a complex program of an adaptive response called integrated stress response (ISR) that can allow a cell to survive in the presence of stressors. ISR reprograms gene expression to increase the transcription and translation of stress response genes while repressing the translation of most proteins to reduce the metabolic burden. In some cases, ISR activation can lead to the assembly of a cytoplasmic membraneless compartment called stress granules (SGs). ISR and SGs can inhibit apoptosis, pyroptosis, and necroptosis, suggesting that they guard against uncontrolled regulated cell death (RCD) to promote organismal homeostasis. However, ISR and SGs also allow cancer cells to survive in stressful environments, including hypoxia and during chemotherapy. Therefore, there is a great need to understand the molecular mechanism of the crosstalk between ISR and RCD. This is an active area of research and is expected to be relevant to a range of human diseases. In this review, we provided an overview of the interplay between different cellular stress responses and RCD pathways and their modulation in health and disease.
Keywords:
apoptosis; GCN2; HRI; integrated stress response; necroptosis; programmed cell death; PERK; PKR; pyroptosis; stress granules1. Introduction
On the first of July 1858, at the meeting of the Linnean Society in London, Charles Darwin’s theory of evolution by natural selection was presented along with Alfred Russel Wallace’s manuscript entitled “Evolution through natural selection of the fittest individuals” [1]. Organisms come across changing external or internal environments, and only the fittest survive. The original concept of the survival of the fittest can be adopted at a cellular level. Cells have sophisticasted physiological mechanisms to sense environmental stress and initiate an adaptive mode; if successful, they survive, otherwise, death is inevitable.
Cells are exposed to various external stressors such as temperature fluctuations, infections, nutrient deprivations, radiation, chemicals, and toxins. In addition, changes in redox states, ionic imbalances, accumulation of unfolded proteins, oncogene activation, etc., contribute to intrinsic cellular stress [2,3,4,5,6,7]. Various signaling pathways that are activated during stress, such as the heat shock response (HSR), unfolded protein response (UPR), DNA damage response (DDR), and response to oxidative stress, efficiently revert the stress-induced alterations to restore cellular homeostasis and prevent cell death.
Cell fate decisions under stress are context dependent and depend on the nature and duration of the trigger [8]. Cells survive when pro-survival pathways are turned ON. Cells entering a pro-death mode activate one of the regulated cell death (RCD) pathways. We focus on apoptosis, necroptosis, and pyroptosis because they have been studied in greater detail than other RCD pathways [9,10,11]. In the current review, we are highlighting cellular stress response mediated translation arrest by various stress kinases in tumor microenvironments (TME) and viral infections. We also discuss how the stress response is being modulated in tumors and infected cells. Finally, we highlight the role of the cellular stress response, especially stress granules (SG), in modulating cell survival, signaling, and RCD.
2. Cellular Stress Response and Translation
Cellular homeostasis is maintained by a balance between cell survival and cell death. The rate of physiological cell turnover, in which replicative aged terminally differentiated cells are being continuously replaced (except neurons), varies with cell types [6,12,13,14]. Cells experience replicative stress during the regenerative process, eventually leading to cellular senescence and death of dysfunctional cells [15,16]. In contrast, cells exposed to a wide range of exogenous and endogenous stressors respond to resolve them before initiating a thermodynamically costly RCD pathway. Eukaryotes have a common adaptive signaling pathway named the integrated stress response (ISR) to deal with cellular stress. The ISR pathway reprograms cellular translational machinery to promote the survival of stressed cells [17,18,19].
The complex pathway of translation consisting of four steps, namely, initiation, elongation, termination and ribosome recycling, has been extensively reviewed by others [20,21]. Initiation of translation involves the formation of the 43S pre-initiation complex (PIC) in parallel to the formation of the eukaryotic initiation factor 4F (eIF4F) and the cap binding complex (eIF4F-CBC). Regulation of protein synthesis at the translation initiation step is a major component of translational control. Owing to the fact that translation initiation involves the formation of two independent complexes (PIC and eIF4F-CBC), its regulation can be eIF2α dependent or eIF2α independent [22]. Both of these complexes can be targeted by ISR to repress translation.
Diverse stress insults activate the distinct ISR kinases [23,24,25,26], namely, General control nonderepressible 2 (GCN2), double-stranded RNA dependent serine/threonine protein kinase R (PKR), PKR-like endoplasmic reticulum kinase (PERK) and heme regulated inhibitor (HRI) kinase that phosphorylates eIF2α leading to translation arrest (Figure 1A).
Amino acid deprivations or defects in tRNA synthetase activity increases cellular levels of uncharged tRNAs. Recognition of uncharged tRNAs by the tRNA synthetase domain of GCN2 activates its kinase activity [27]. Double-stranded RNA generated during viral replication activates PKR [24,28]. Physiological or pathological conditions that increase the accumulation of mistranslated or unfolded proteins lead to ER stress-mediated activation of PERK [25,29]. Heme deficiency [26,30], oxidative stress [26,31] and heat shock [32,33,34] activate the HRI kinase. The ISR kinases are regulated by distinct stressors; however, their catalytic kinase domain phosphorylates eIF2α, allowing stress signals to converge on translation initiation inhibition, which is a critical step in translational control. The affinity of eIF2B for phosphorylated eIF2α-GDP is 150-fold higher than that for eIF2α-GDP [35], which allows inhibition of translation initiation.
Figure 1.
Regulation of stress kinases: (A) Heme deficiency, oxidative stress, heat shock, starvation, viral infection, and ER stressors activate HRI, GCN2, PKR and PERK kinases, respectively. Kinase activation phosphorylates eIF2α leading to attenuation of cap dependent translation [23,24,25,26]. (B) Cellular stress leads to an increase in local concentration of mRNAs and ribonucleoproteins. RNA binding proteins such as T-cell intracellular antigen 1 (TIA1), GTPase-activating protein-(SH3 domain)-binding protein1 (G3BP1) and poly(A) binding proteins (PABPs) act as nucleation factors for stress granule (SG) assembly [36,37,38]. SGs can sequester proteins involved in apoptosis [4,10] and pyroptosis [9] to inhibit them. ISR can inhibit necroptosis by reducing RIPK3 mediated phosphorylation of MLKL through an unknown mechanism. ER stress activates stress kinase PERK [11] and inhibits phosphorylation of RIPK1 at S166, which inhibits downstream signal transduction to RIPK3 and MLKL, restricting necroptosis [11].
Figure 1.
Regulation of stress kinases: (A) Heme deficiency, oxidative stress, heat shock, starvation, viral infection, and ER stressors activate HRI, GCN2, PKR and PERK kinases, respectively. Kinase activation phosphorylates eIF2α leading to attenuation of cap dependent translation [23,24,25,26]. (B) Cellular stress leads to an increase in local concentration of mRNAs and ribonucleoproteins. RNA binding proteins such as T-cell intracellular antigen 1 (TIA1), GTPase-activating protein-(SH3 domain)-binding protein1 (G3BP1) and poly(A) binding proteins (PABPs) act as nucleation factors for stress granule (SG) assembly [36,37,38]. SGs can sequester proteins involved in apoptosis [4,10] and pyroptosis [9] to inhibit them. ISR can inhibit necroptosis by reducing RIPK3 mediated phosphorylation of MLKL through an unknown mechanism. ER stress activates stress kinase PERK [11] and inhibits phosphorylation of RIPK1 at S166, which inhibits downstream signal transduction to RIPK3 and MLKL, restricting necroptosis [11].

Stress-mediated translation arrest increases the local concentration of mRNA and 40S ribosome-containing ribonucleoproteins (mRNPs) and subsequent sequestering of mRNPs in membraneless cytoplasmic foci called SGs. RNA binding proteins (RBPs) such as T-cell intracellular antigen-1 (TIA1), TIA1-related protein (TIAL1), poly(A) binding proteins (PABPs), GTPase-activating protein-(SH3 domain)-binding protein 1 (G3BP1) and several DEAD-box proteins promote SG assembly [36,37,38,39]. In addition to mRNPs, SGs also sequester central components of the RCD pathways that modulate apoptosis [4,10], pyroptosis [9], and necroptosis [40] mediated cell death (Figure 1B).
3. Regulated Cell Death
Cell death is a normal physiological process. However, during infections and several pathological conditions, pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) elicit robust immune responses, which can be detrimental to the host. Therefore, cells have developed a regulated death system to modulate the immune response. Thus, cell death can be immunologically silent, such as apoptosis or immunogenic, and pyroptosis or necroptosis (Figure 2). Several RCD pathways have been described that differ in their mechanisms of activation and effect on host homeostasis. In this review, we will focus on apoptosis, necroptosis, pyroptosis, and ferroptosis because their regulation by ISR and SGs has been studied the most.
3.1. Apoptosis
Apoptosis plays a crucial role in physiological cell turnover. Various pathological conditions or insults may lead to apoptotic cell death. The trigger for apoptosis can be extrinsic or intrinsic (Figure 2A).
Binding of death ligands such as tumor necrosis factor (TNF), fas ligand (FASL), or TNF-related apoptosis-inducing ligand (TRAIL) to their cognate cell surface death receptors, TNF receptor 1 (TNFR1), FAS, or TRAIL receptor 1/2 (TRAILR1/2), respectively, leads to receptor oligomerization, which initiates recruitment of adaptor proteins TRADD (TNFR1/2-associated death domain protein), and FADD (Fas-associated death domain protein). This is followed by FADD-mediated recruitment of the initiator caspase-8 (CASP8), forming a signaling platform called DISC (Death-inducing signaling complex). The formation of DISC initiates autocatalytic proteolysis and activation of CASP8, triggering the execution phase of apoptosis [41,42].
Mitochondrial outer membrane permeabilization (MOMP) initiates intrinsic apoptosis by releasing the pro-apoptotic protein cytochrome c from the mitochondrial intermembrane space into the cytoplasm [77]. Cytochrome c binds to APAF1, which recruits the initiator caspase-9 (CASP9) to form a complex called apoptosome. Autocatalytic proteolysis and activation of CASP9 in the apoptosome initiate the execution phase of apoptosis [41,42].
Cleaved initiator caspases (CASP8 and CASP9) proteolytically activate executioner caspases, caspase-3 (CASP3) and caspase-7 (CASP7). Active executioner caspases cleave their target proteins, including cytoskeletal components and are responsible for the morphological and biochemical alterations that ultimately result in apoptosis [43].
3.2. Pyroptosis
Pyroptosis is an inflammasome-dependent inflammatory cell death pathway mediated by the pore-forming protein gasdermin d (GSDMD). Inflammasome sensors such as Nucleotide oligomerization domain (NOD) like receptor (NLR) pyrin (PYD) domain-containing protein 1 (NLRP1) [44], NLRP3 [45,46,47,48,49,50,51], NLR family caspase activation and recruitment domain (CARD) containing protein 4 (NLRC4) [52,53,54], absent in melanoma 2 (AIM2) [57,58,59,60], and PYRIN [55,56] sense different PAMPs and DAMPs (Figure 2B). Activated inflammasome sensors oligomerize, forming a signaling platform to which apoptosis-associated speck-like protein containing a CARD (ASC, also known as PYCARD) is recruited, which in turn recruits caspase-1 (CASP1) where it gets activated by proximity-induced autoproteolysis [61,62,63,64]. NLRP1 and NLRC4 sensors themselves contain a CARD domain and are capable of recruiting CASP1 independent of ASC [78,79]. Active CASP1 cleaves GSDMD [70,71], interleukin-1β (IL-1β) [65,66,67] and IL-18 [68,69] among others. The N-terminal fragment of GSDMD (GSDMD-N) translocates to the plasma membrane to form pores that result in the osmotic lysis of cells. GSDMD-N pores also facilitate the release of several cytosolic inflammatory mediators, including active IL-1β and IL-18, in the absence of cell lysis [70,72].
3.3. Necroptosis
Necroptosis is a regulated form of cell death originally found to be triggered by the activation of TNFR, leading to mixed lineage kinase domain-like pseudokinase (MLKL) activation downstream of RIPK3 (receptor-interacting protein kinase 3) (Figure 2C). One of the most studied necroptosis occurs in cells when CASP8 is inhibited in the presence of TNFR1 activation [42,73]. TNFR1 signaling (in the absence of CASP8) recruits RIPK1, which interacts with RIPK3 through the RHIM (RIP homotypic interaction motif) domain. RIPK1 and RIPK3 form a complex called necrosome. MLKL is recruited and phosphorylated in the necrosome. The phosphorylated MLKL is trafficked to the plasma membrane, where MLKL oligomers execute cell death through the disruption of plasma membranes [11,42,74,75,76]. Depending on the stimuli, other RHIM domains containing proteins such as Z-DNA binding protein 1 (ZBP1) and Toll–IL-1 receptor domain-containing adaptor-inducing IFN-β (TRIF) can interact with RIPK1 [42].
4. Stress Kinases in Regulation of Apoptosis
4.1. GCN2 in Regulation of Apoptosis
Human GCN2 is a 1649 amino acid long protein that forms a homodimer (Figure 3) and has five conserved domains, namely, an N-terminal RWD domain (RING finger-containing protein, WD repeat-containing protein and yeast DEAD-like helicase), a pseudokinase domain, a catalytically active kinase domain (KD), a histidyl-tRNA synthetase like (HisRS) domain and a C-terminal domain (CTD) [80,81]. At a basal metabolic state, the interaction between CTD and KD auto-inhibits the kinase activity of GCN2. Additionally, CTD-CTD and CTD-HisRS interactions stabilize the inactive dimeric architecture of the GCN2 [80,81]. Starvation, in particular amino acid deprivation, increases the amount of deacylated tRNA in cells. In addition, glucose deprivation shunts the amino acids to the cytosolic vacuoles [82,83], increasing the uncharged tRNA pool. The binding of uncharged tRNA to the HisRS domain of GCN2 [80,81,84] weakens the autoinhibitory interactions followed by structural rearrangement that activates the kinase activity of GCN2 [84], leading to eIF2α phosphorylation and translation inhibition.
Cellular metabolism in normal cells maintains physiological homeostasis during cell growth and proliferation. In contrast, uncontrolled cell proliferation during malignancies leads to nutrient deficiencies and metabolic stress. However, metabolic reprogramming occurs in cancer cells [88,89] to provide nutrients for the growth of tumor cells (Figure 4A). As such, GCN2 is highly expressed in papillary renal cell carcinoma [90], head and neck small cell carcinoma [91], prostate cancer [92], cisplatin-resistant human gastric cancer cells [93], hematological malignancies such as multiple myeloma, acute myeloid leukemia, and acute lymphoid leukemia [94,95], hepatocellular carcinoma [96] and pancreatic cancer cells [95]. Inhibition of GCN2 leads to an increase in apoptosis of tumor cells [95,96], suggesting that the stress kinase GCN2 helps malignant cells to cope with nutrient limitation favoring tumor growth.
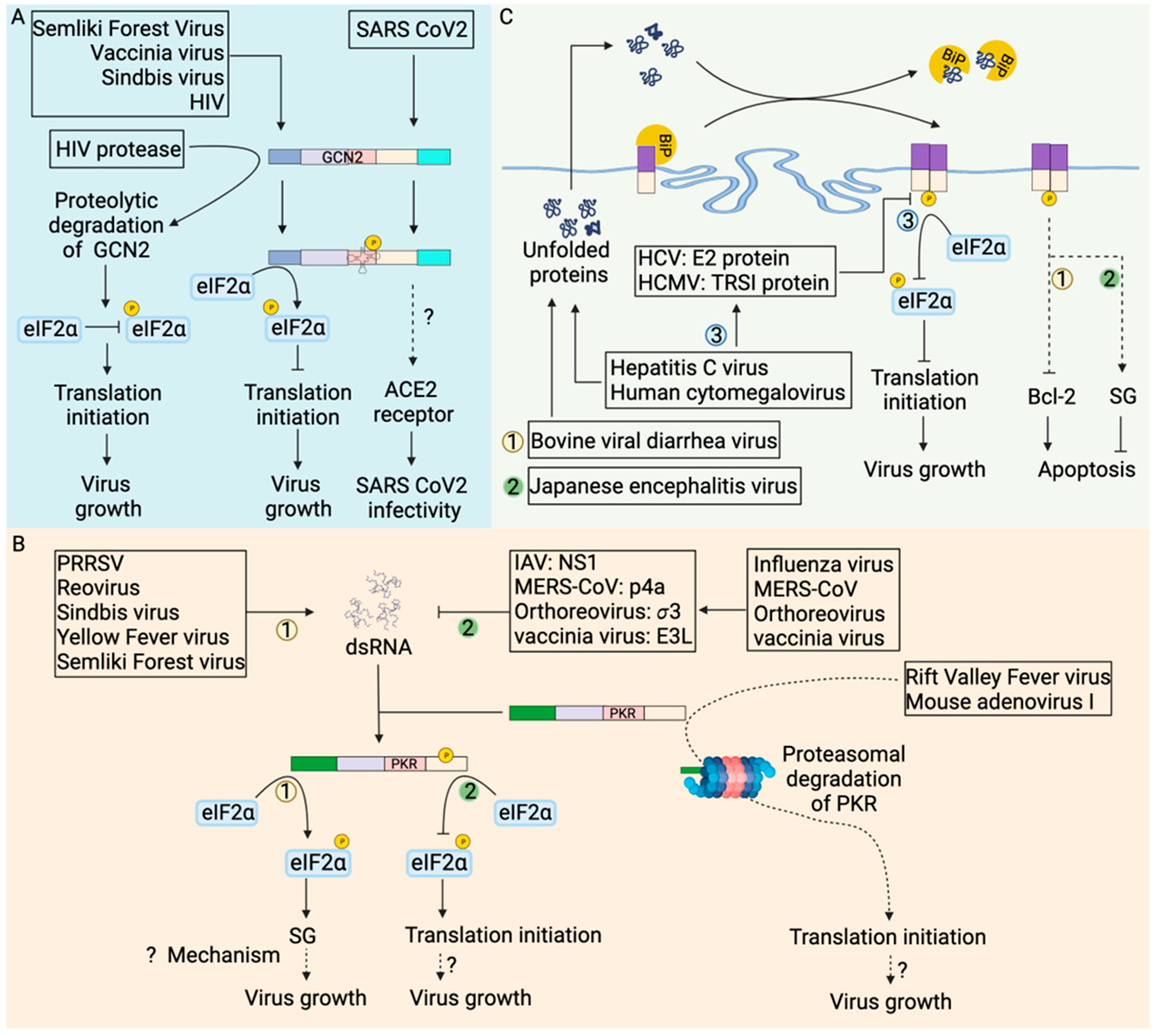
Viruses also manipulate the host-cell metabolic pathway [118] to provide biomolecules, in particular amino acids, for the production of new virions. However, the role of GCN2 stress kinase (Figure 5A) in regulating viral infection dynamics is contextual. Angiotensin-converting enzyme 2 (ACE2), the receptor for severe acute respiratory coronavirus 2 (SARS-CoV-2), is also involved in the intestinal uptake of amino acids [119]. GCN2 increased expression of SARS-CoV-2 receptor in amino acid starved human colonic epithelial cells [120] aggravates the disease. Semliki Forest virus and vaccinia virus infections increase the kinase activity of GCN2 and subsequent attenuation of viral replication [121]. Genomic RNA of Sindbis virus (SV) [121] or human immunodeficiency virus-1 (HIV-1) [122] activate GCN2 kinase activity which blocks translation, including viral protein synthesis hindering virus replication. It is unclear if SG assembly plays a role in viral replication inhibition in these cases.
Although activation of GCN2 kinase activity and subsequent phosphorylation of eIF2α can lead to SG assembly, the formation of SGs in tumor cells has been challenging to detect. One possible reason might be that the downregulation of SG components in these cells inhibits SG assembly. Another reason could be that the dynamic nature of SG assembly:disassembly processes make it hard to capture them in vivo. The formation of SGs in tumor cells treated with chemotherapeutic agents [110,134] has provided indirect evidence for SG formation. Additionally, viral proteases, such as HIV-1 protease, cleave GCN2 and abrogate translation inhibition [122] to increase viral protein synthesis and viral replication. Taken together, modulation of SG formation might be an alternative approach to alter cancer progression and virus infection outcomes.
4.2. PKR in Regulation of Apoptosis
Protein Kinase R (PKR) is a 551 amino acid long monomeric protein consisting of a regulatory N-terminal double-stranded RNA (dsRNA) binding domain and catalytic C-terminal kinase domain joined by a 20 amino acid linker region (Figure 3). The two tandem copies of dsRNA binding motifs, dsRBM1 and dsRBM2, in the N-terminal domain, bind dsRNA in a sequence-independent manner. The binding of dsRNA to the regulatory domain of PKR induces dimerization of the kinase domain and phosphorylation-mediated activation of PKR [28,85]. Activated PKR phosphorylates eIF2α to inhibit translation initiation and induce SG assembly.
PKR has been implicated in the regulation of various cellular processes in tumors (Figure 4B). dsRNA from diverse cellular sources, including those liberated from dying cells in the TME, are subsequently internalized by the tumor cells and are recognized by RNA sensors [135,136], including PKR. Studies have shown a dual role of PKR in tumors, tumor-suppressive or tumorigenic. Initial studies suggested an anti-proliferative role of PKR pertaining to the inhibition of translation initiation [137]. NIH 3T3 cells expressing PKR exhibited a delay in malignant transformation in nude mice [103]. Similarly, PKR downregulated HCT116 human colon cancer cells grew rapidly in nude mice [104], suggesting a tumor-suppressive role of PKR. PKR overexpressing cells undergo Fas-associated death domain (FADD) mediated activation of caspase 8 [105,106,107], leading to apoptosis. Additionally, 3T3 L1 cells expressing catalytically inactive PKR were resistant to apoptosis and underwent malignant transformation [105].
In contrast to the tumor-suppressing role of PKR discussed above, transcriptomics analysis showed overexpression of PKR in several cancer types [28]. Overexpression of PKR or increased PKR activity is seen in pancreatic cancer [28], melanoma [97], colon cancer [97], breast cancer [98] and hepatocellular carcinoma [99], suggesting its role in tumorigenesis. Furthermore, it has been suggested that increased PKR activity promotes tumor progression and invasion [97,100,101,102]. Interestingly, tumors consist of a heterogeneous population of cancer cells [138], and there is spatial heterogeneity of PKR expression, at least in head and neck squamous cell carcinoma [139]. Intriguingly, cancer cells with high PKR expression were present in the core of tumors, while those with low levels were at the periphery [139]. The significance of this observation remains to be unraveled.
The contradicting role of PKR in apoptosis could be due to the differential role of PKR in normal and cancer cells. Although the knockdown of PKR leads to apoptosis of both normal and cancer cells, there was a greater increase in tumor cell apoptosis, suggesting a strong anti-cancer therapeutic potential of PKR inhibition and emphasizing the role of PKR in tumor cell survival [140]. In addition, PKR has been shown to finely tune the balance between the pro-survival and pro-death signal by activating both nuclear factor kappa-B (NF-κB) and eIF2α in a sequential manner [141]. In NIH3T3 cells expressing PKR (tet-off system), NF-κB is activated at the earliest time point, promoting cell survival independent of PKR kinase activity. On the other hand, catalytically active PKR phosphorylates eIF2α at a later time point and subsequently promotes apoptosis [141]. Although sustained elevation of NF-κB activity, a key player in tumorigenesis [142], might explain the survival of cancer cells. The mystery that remains unresolved is “how kinase activity of PKR is being differentially modulated in the tumor cells?”.
PKR is activated by dsRNA, a byproduct of RNA virus replication and some DNA viruses (Figure 5B). Activated PKR phosphorylates eIF2α to attenuate translation leading to formation of stress granules and subsequently triggering apoptosis. However, many viruses avoid the formation of stress granules in host cells confirming that viruses can manipulate PKR-mediated cellular stress responses for their benefit. The Middle East Respiratory Syndrome Coronavirus (MERS-CoV) accessory protein p4a [127], mammalian orthoreovirus σ3 protein [128], vaccinia virus E3L protein [129] and influenza A virus nonstructural protein 1 (NS1) [126] bind and sequester dsRNA to prevent PKR activation in infected cells. Viral proteins, for instance, influenza NS1 [143] and vaccinia virus E3L [129] protein, can directly bind with PKR to prevent PKR-mediated eIF2α phosphorylation. In addition, viral proteins can degrade PKR [123,124,125] by mechanisms that are incompletely understood for efficient viral replication. During rift valley fever virus [123,124] and mouse adenovirus type 1 infection [125], PKR is degraded in the proteasome. Overall, viruses evolved mechanisms to inhibit PKR activation avoiding SG formation for efficient virus replication.
Nevertheless, many viruses activate PKR and form SGs. Reovirus [128,144], sindbis virus [145], semliki forest virus [146], yellow fever virus [147] and porcine reproductive and respiratory syndrome virus (PRRSV) [148] infections lead to PKR-mediated formation of SGs in infected cells. PKR-mediated activation of cellular stress response facilitated reovirus replication by relocating viral transcripts to unknown sites where limited cellular translational machinery is accumulated [144]. In contrast, PPRSV replicase protein nsp1β localizes and interacts with PKR within SG in infected cells [148], utilizing SGs for viral benefit. The status of cell death in virus-infected cells that form SGs has not been explored except for Sindbis virus-infected cells where PKR activation leads to inhibition of translation of anti-apoptotic B-cell lymphoma 2 (bcl-2) protein and apoptosis of the infected cells [145].
4.3. PERK in Regulation of Apoptosis
PKR-like endoplasmic reticulum kinase (PERK) is an 1116 amino acid long transmembrane protein consisting of an N-terminal endoplasmic reticulum (ER) lumenal domain and a C-terminal cytosolic domain (Figure 3). The regulatory lumenal domain helps in dimerization, while the cytosolic domain has kinase activity and consists of autophosphorylation sites [86]. Immunoglobulin binding protein (BiP) maintains PERK as inactive monomers [109]. However, ER stress causes BiP to dissociate from PERK, facilitating its dimerization and activation of its kinase domain [109]. Activated PERK phosphorylates eIF2α leading to translational arrest [149]. Perturbation of ER homeostasis has been identified in several pathological conditions, including cancer [150], viral infections [151] or neurodegeneration [152].
Cancer cells experience adverse physiological conditions such as hypoxia and nutrient deprivation in the TME. The cumulative effect is the accumulation of unfolded or misfolded proteins in the ER of tumor cells leading to ER stress [108]. Cells respond to deregulated ER homeostasis through the UPR signal, PERK being one of the sensors of ER stress (Figure 4C) [153]. PERK plays a vital role in oncogenesis and tumor progression. Differential expression of PERK is found in several tumors and cancers [153]; neuronal cancer, liver cancer, lung cancer, breast cancer, gastric carcinoma and cholangiocarcinoma have high expression, while thyroid cancer, lymphoma, sarcoma and colorectal cancer have low expression [153] suggesting variation in ER stress among different cancer types. How different levels of ER stress fine-tune the progression of various tumor types is an exciting area for future research. However, another possibility for differential expression exists in the spatial heterogeneity in PERK expression arising from heterogeneous tumor cell populations.
Although tumor cells have sustained expression of PERK, no histological evidence of them forming SGs in an eIF2α kinase-dependent manner exists. Human cervical cancer HeLa cells form SG under hypoxia [111]; this might provide intriguing evidence to implicate SG formation in cancer cells within TME. SG formation helps the cancer cells to survive the extreme physiological conditions of TME and avoid RCD [10,154]. In line with this, treatment with various anti-cancer drugs induces PERK-dependent formation of SGs in tumor cells [110,111,112,113]. Downregulation of PERK sensitizes tumor cells to chemotherapy leading to apoptosis [110,112,113]. Taken together, PERK expression in tumor cells supports oncogenesis.
Viruses are obligate intracellular pathogens and depend on host cell machinery for viral protein synthesis and replication. Virus infection involves the synthesis of viral proteins, which are routed to the ER for proper protein folding and post-translational modifications. In addition, accumulation of unfolded or misfolded viral proteins can occur in the ER and induce ER stress (Figure 5C). Under ER stress, BiP is released from PERK, leading to PERK homodimerization and autophosphorylation-mediated activation of PERK. Activation of PERK blocks translation as a measure to cope with stress. Sustained ER stress can lead to RCD.
Activation of ER stress has been detected in a range of viral infections. Flaviviruses and coronaviruses exploit ER as a site of viral protein synthesis, replication and assembly [155]. PERK autophosphorylation was observed in Madin-Darby Bovine Kidney (MDBK) cells infected with the bovine viral diarrhea virus (BVDV) [132]. Sustained ER stress downregulated anti-apoptotic protein Bcl-2 leading to apoptosis of BVDV-infected cells [132]. Japanese encephalitis virus (JEV), another ER tropic virus, involves the budding of progeny virions from the ER membrane of infected BHK-21 cells and initiates ER stress. However, PERK activation was not detected by mobility shift assay [156]. In contrast, PERK activation is observed by immunoblot in both neuronal cells and BHK-21 cells infected with JEV [133], suggesting variation in PERK activation can be cell type dependent or technique dependent In addition, PERK activation promoted SG formation in JEV-infected neuronal cells, which subsequently repress apoptosis [133].
Transmissible gastrointestinal virus (TGEV) infection of the IPEC-J2 jejunum epithelium induced ER stress. Activated PERK suppressed TGEV replication [157] through phospho-eIF2α mediated translation inhibition. Similar to what has been observed with TGEV, viruses such as human cytomegalovirus (HCMV) [158] and hepatitis C virus (HCV) [130] infections also activate PERK. However, HCV E2 protein binds with PERK to inhibit PERK activity and prevent eIF2α phosphorylation [130]. Similarly, HCMV immediate early protein TRS1 prevents eIF2α phosphorylation [131]. HCMV and HCV, thus, maintain protein translation to benefit virus replication. In summary, PERK is a critical regulator of cellular stress response to ER stress and plays a multifaceted role in tumorigenesis and host response to viral infections.
4.4. HRI in Regulation of Apoptosis
Heme-regulated inhibitor (HRI) kinase is a 630 amino acid long protein consisting of five domains, an N-terminal domain, a C-terminal domain, and a central kinase insertion (KI) domain flanked by two kinase domain (Kinase I and Kinase II) [86,87] (Figure 3). The heme binding site is located on the N-terminal and the central KI domain of HRI; however, the binding site on the KI domain performs the regulatory role [87]. Heme binding at the N-terminal heme binding site dimerizes the HRI. The HRI dimer is stable and inactive during heme sufficiency. However, heme deficiency HRI is activated by a mechanism of transactivation that subsequently leads to the phosphorylation of eIF2α to inhibit translation [30,159,160].
HRI being a sensor of heme, modulates erythropoiesis. During heme sufficiency, HRI is inhibited, and protein synthesis (the globin chains of hemoglobin) is activated. In contrast, heme deficiency activates HRI-mediated phosphorylation of eIF2α, which inhibits translation, in particular globin synthesis [31]. Strikingly, HRI senses heme availability to balance heme and the globin chain synthesis by increasing apoptosis of late erythroid precursors during iron deficiency anemia [159].
The expression of HRI is not limited to red blood cells and has been found in macrophages and the liver [86,161], suggesting the role of HRI beyond red blood cells (Figure 4D). Compounds such as N,N′-diarylureas that activate HRI inhibited the proliferation of tumor cells in an eIF2α phosphorylation-dependent manner [114,115,116]. HRI expression has been found to be high in tissue samples from lungs, ovary, breast and gastric cancers compared to normal tissue and is associated with poor survival [117]. Targeting HRI to trigger apoptosis might be a new strategy for anti-cancer therapy. However, following anti-cancer therapy, drug-tolerant persister cells are generated, which are dependent on sublethal cytochrome c (released due to incomplete MOMPs) mediated activation of HRI [117], suggesting translational reprogramming by HRI can promote tumorigenesis.
5. Emerging Concept on Crosstalk between Cellular Stress and Other Forms of Cell Death
The eIF2α kinases have long been known for their ability to modulate cellular homeostasis during stress, often activating apoptosis if the stress persists beyond a threshold. Apoptosis is not the only RCD program that can be triggered; cells can die by pyroptosis, necroptosis or other more recently discovered RCD pathways. RCD is a very active area of research. This has led to the discovery of several RCD pathways in the last decade [162]. Since then, other RCD pathways have been proposed, including PANoptosis, cuproptosis, oxeiptosis and ferroptosis [163,164,165,166]. The crosstalk between stress and most of these RCD pathways is poorly understood (Figure 6). In this review, we focus on apoptosis, necroptosis, pyroptosis, and ferroptosis and discuss their modulation by ISR since we have a better, albeit still incomplete, understanding of them.
5.1. Stress and Pyroptosis
The stress kinase PKR plays a crucial role in the activation of inflammasomes [169,177,178,179,180]. PKR physically interacted with NLRP1, NLRP3, NLRC4 and AIM2 inflammasome and induced activation of caspase 1 and IL-1β cleavage [169]. Inhibition of PKR activity suppressed inflammasome activation and IL-1β release [169,177,178,179,180]. In contrast, another study could not establish the role of PKR in inflammasome activation [181]. However, multiple lines of evidence suggested crosstalk between stress kinase PKR and pyroptosis. The induction of ER stress has been found to activate NLRP3 inflammasome activation and pyroptosis during fungal infection [168] and infection with Mycobacterium tuberculosis [167] (Figure 6A).
In contrast, activation of the GCN2/eIF2α pathway has been shown to suppress inflammasome activation, resulting in reduced pyroptosis during intestinal inflammation [170]. Additionally, induction of SGs has been shown to inhibit NLRP3 inflammasome [9] wherein SGs sequestered DDX3X activating a pro-survival mode; in contrast, the association of DDX3X with NLRP3 induces pyroptosis [9,182]. In summary, stress signaling has a pleiotropic effect on pyroptosis (Figure 6A). The molecular mechanisms of this pleiotropic effect are poorly understood and are an active area of research due to their immense therapeutic potential.
5.2. Stress and Necroptosis
RNA viruses and some DNA viruses replication generate dsRNA, which can adopt a right-handed conformation (A-RNA) or a left-handed conformation (Z-RNA). PKR and melanoma differentiation-associated protein 5 (MDA5) senses A-RNA [183], while Z-DNA binding protein 1 (ZBP1) senses Z-RNA [184]. Adenosine deaminase acting on RNA 1 (ADAR1) performs adenosine-to-inosine base editing of endogenous dsRNA [185] to prevent PKR [186] or MDA5 overactivation [187] and spontaneous activation of ZBP1 [184]. Accumulation of Z-RNA during infection with viruses such as influenza, SARS-CoV2, murine cytomegalovirus and vaccinia triggers ZBP1-RIPK3-mediated necroptosis [188,189,190,191,192]. In addition, ZBP1 is a key regulator of tumor cell necroptosis [193,194].
ADAR1, as well as ZBP1, the key molecules of the necroptotic pathway, localizes to arsenite-induced SGs (Figure 6B) in HeLa cells and L929 cells, respectively [40,195]. In the SG, ZBP1 forms ZBP1-RIPK3 necrosomes triggering necroptosis [40]. In contrast, various stressors (thapsigargin, brefeldin A, tunicamycin, MG132) inhibited necroptosis (Figure 6B) in bone marrow-derived macrophages [11]. The contrasting result might be due to differences in stressors used and the cell type under study.
Nutrient deprivation (glucose or serum) induces ZBP1-mediated necroptosis of breast cancer cells [194], suggesting ZBP1 expression is increased during starvation. However, tumor cells execute metabolic reprogramming to provide nutrients for proliferating cancer cells and possibly downregulate ZBP1 to prevent necroptosis. In agreement, increased ZBP1 expression correlated with breast cancer aggressiveness [196].
Taken together, these data support the notion that there is a crosstalk between necroptosis and SG. However, our understanding of the mechanism of this crosstalk remains in its infancy. Future research on this topic will greatly aid in the development of therapeutics against a range of infectious and inflammatory diseases.
5.3. Stress and Ferroptosis
Ferroptosis is an iron-dependent caspase-independent form of regulated cell death activated by oxidative stress [166]. The peculiar feature of ferroptosis is the peroxidation of membrane phospholipids leading to membrane rupture culminating in cell death [197]. Ferroptosis is inhibited by reduced glutathione (GSH) and glutathione peroxidase 4 (GPX4) [198,199]. The oxidative imbalance that leads to ferroptosis can activate ISR in several ways. However, the relationship between ISR and ferroptosis remains underexplored.
Multiple lines of evidence suggest crosstalk between ISR and ferroptosis (Figure 6C). Deficiency of selenium sensitizes cells to ferroptosis [171,172]. tRNA specific to selenium participates in the synthesis of GPX4, a selenoprotein that regulates ferroptosis [200]. In selenium deficiency, accumulation of selenium-specific uncharged tRNA possibly activates the stress kinase GCN2 suggesting a crosstalk between stress kinase and ferroptosis. During metabolic or oxidative stress, a cytosolic Nicotinamide Adenine Dinucleotide Phosphate (NADPH) phosphatase called MESH1 (Metazoan SpoT Homologue 1) is induced, depleting NADPH and resulting in ferroptosis [173,174]. In addition, MESH1 depletion-induced ER stress leading to activation of PERK and subsequent phosphorylation of eIF2α [174] suggests a crosstalk between ISR and ferroptosis; however, the mechanism and significance of this potential crosstalk remain unexplored. Iron accumulation (as in hemochromatosis) also sensitizes cells to ferroptosis by enhancing lipid peroxidation [175,176,201]. In contrast, as discussed earlier, heme (a form of iron) sufficiency inhibits activation of the stress kinase HRI. This represents another potential mechanism for crosstalk between ISR and ferroptosis. Lower amounts of GSH sensitize cells to ferroptosis. However, an increased amount of GSH has been found in West Nile Virus (WNV) infected cells that protects cells from oxidative damage [202]. In addition, increased amounts of GSH are correlated with the absence of stress granules in WNV-infected cells [202]. These observations suggest a possible crosstalk between ISR and ferroptosis during viral infection as well. Taken together, several avenues for crosstalk between ISR and ferroptosis are present but remain unexplored. These avenues represent opportunities for exciting new discoveries in the future.
6. Conclusions
Various pathogens and cancer cells activate stress kinases leading to the formation of SGs as an attempt to maintain cellular homeostasis. In addition, stress granules sequester components of apoptosis (executioner caspases 3/7) [203], pyroptosis [9] and necroptosis [40,204] to favor pro-survival pathways. Sustained exposure to stress triggers one or the other forms of the RCD pathway. The crosstalk between different cell death types is emerging. Therefore, it is intriguing to know how stress kinases bridge with RCDs and how they can fine-tune different forms of cell death. Understanding the complex interaction between cellular stress and RCD will help to identify novel therapeutic targets.
Author Contributions
P.P.L. and P.S. wrote the manuscript, with P.P.L. writing the first draft. P.S. provided supervision. All authors have read and agreed to the published version of the manuscript.
Funding
P.P.L. and P.S. are funded by a lab startup grant provided by the University of Texas Medical Branch to P.S.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank members of Samir lab for their critical feedback.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fields, S.; Johnston, M. The Law of Evolution: Darwin, Wallace, and the Survival of the Fittest. In Genetic Twists of Fate; MIT Press: Cambridge, MA, USA, 2010. [Google Scholar]
- Nover, L.; Scharf, K.D.; Neumann, D. Formation of Cytoplasmic Heat Shock Granules in Tomato Cell Cultures and Leaves. Mol. Cell. Biol. 1983, 3, 1648–1655. [Google Scholar] [CrossRef] [PubMed]
- Salloum-Asfar, S.; Engelke, R.; Mousa, H.; Goswami, N.; Thompson, I.R.; Palangi, F.; Kamal, K.; Al-Noubi, M.N.; Schmidt, F.; Abdulla, S.A.; et al. Hyperosmotic Stress Induces a Specific Pattern for Stress Granule Formation in Human-Induced Pluripotent Stem Cells. Stem Cells Int. 2021, 2021, e8274936. [Google Scholar] [CrossRef] [PubMed]
- Thedieck, K.; Holzwarth, B.; Prentzell, M.T.; Boehlke, C.; Kläsener, K.; Ruf, S.; Sonntag, A.G.; Maerz, L.; Grellscheid, S.-N.; Kremmer, E.; et al. Inhibition of MTORC1 by Astrin and Stress Granules Prevents Apoptosis in Cancer Cells. Cell 2013, 154, 859–874. [Google Scholar] [CrossRef]
- Moeller, B.J.; Cao, Y.; Li, C.Y.; Dewhirst, M.W. Radiation Activates HIF-1 to Regulate Vascular Radiosensitivity in Tumors: Role of Reoxygenation, Free Radicals, and Stress Granules. Cancer Cell 2004, 5, 429–441. [Google Scholar] [CrossRef]
- Magrassi, L.; Leto, K.; Rossi, F. Lifespan of Neurons Is Uncoupled from Organismal Lifespan. Proc. Natl. Acad. Sci. USA 2013, 110, 4374–4379. [Google Scholar] [CrossRef] [PubMed]
- Frydrýšková, K.; Mašek, T.; Pospíšek, M. Changing Faces of Stress: Impact of Heat and Arsenite Treatment on the Composition of Stress Granules. Wiley Interdiscip. Rev. RNA 2020, 11, e1956. [Google Scholar] [CrossRef] [PubMed]
- Reineke, L.C.; Neilson, J.R. Differences between Acute and Chronic Stress Granules, and How These Differences May Impact Function in Human Disease. Biochem. Pharmacol. 2019, 162, 123–131. [Google Scholar] [CrossRef]
- Samir, P.; Kesavardhana, S.; Patmore, D.M.; Gingras, S.; Malireddi, R.K.S.; Karki, R.; Guy, C.S.; Briard, B.; Place, D.E.; Bhattacharya, A.; et al. DDX3X Acts as a Live-or-Die Checkpoint in Stressed Cells by Regulating NLRP3 Inflammasome. Nature 2019, 573, 590–594. [Google Scholar] [CrossRef]
- Arimoto, K.; Fukuda, H.; Imajoh-Ohmi, S.; Saito, H.; Takekawa, M. Formation of Stress Granules Inhibits Apoptosis by Suppressing Stress-Responsive MAPK Pathways. Nat. Cell Biol. 2008, 10, 1324–1332. [Google Scholar] [CrossRef]
- Place, D.E.; Samir, P.; Malireddi, R.S.; Kanneganti, T.-D. Integrated Stress Response Restricts Macrophage Necroptosis. Life Sci. Alliance 2022, 5, 1. [Google Scholar] [CrossRef]
- Pellettieri, J.; Sánchez Alvarado, A. Cell Turnover and Adult Tissue Homeostasis: From Humans to Planarians. Annu. Rev. Genet. 2007, 41, 83–105. [Google Scholar] [CrossRef] [PubMed]
- Seim, I.; Ma, S.; Gladyshev, V.N. Gene Expression Signatures of Human Cell and Tissue Longevity. npj Aging Mech. Dis. 2016, 2, 16014. [Google Scholar] [CrossRef]
- Sender, R.; Milo, R. The Distribution of Cellular Turnover in the Human Body. Nat. Med. 2021, 27, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Mulia, J.C.; Schwerer, H.; Besnard, E.; Desprat, R.; Trevilla-Garcia, C.; Sima, J.; Bensadoun, P.; Zouaoui, A.; Gilbert, D.M.; Lemaitre, J.-M. Cellular Senescence Induces Replication Stress with Almost No Affect on DNA Replication Timing. Cell Cycle 2018, 17, 1667–1681. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; d’Adda di Fagagna, F. Cellular Senescence: When Bad Things Happen to Good Cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Advani, V.M.; Ivanov, P. Translational Control under Stress: Reshaping the Translatome. BioEssays 2019, 41, 1900009. [Google Scholar] [CrossRef]
- Liu, B.; Qian, S.-B. Translational Reprogramming in Cellular Stress Response. Wiley Interdiscip. Rev. RNA 2014, 5, 301–305. [Google Scholar] [CrossRef]
- Costa-Mattioli, M.; Walter, P. The Integrated Stress Response: From Mechanism to Disease. Science 2020, 368, eaat5314. [Google Scholar] [CrossRef]
- Jackson, R.J.; Hellen, C.U.T.; Pestova, T.V. The Mechanism of Eukaryotic Translation Initiation and Principles of Its Regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127. [Google Scholar] [CrossRef]
- Aitken, C.E.; Lorsch, J.R. A Mechanistic Overview of Translation Initiation in Eukaryotes. Nat. Struct. Mol. Biol. 2012, 19, 568–576. [Google Scholar] [CrossRef]
- Panas, M.D.; Ivanov, P.; Anderson, P. Mechanistic Insights into Mammalian Stress Granule Dynamics. J. Cell Biol. 2016, 215, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Wek, S.A.; Zhu, S.; Wek, R.C. The Histidyl-TRNA Synthetase-Related Sequence in the EIF-2α Protein Kinase GCN2 Interacts with TRNA and Is Required for Activation in Response to Starvation for Different Amino Acids. Mol. Cell. Biol. 1995, 15, 4497–4506. [Google Scholar] [CrossRef]
- Srivastava, S.P.; Kumar, K.U.; Kaufman, R.J. Phosphorylation of Eukaryotic Translation Initiation Factor 2 Mediates Apoptosis in Response to Activation of the Double-Stranded RNA-Dependent Protein Kinase. J. Biol. Chem. 1998, 273, 2416–2423. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk Is Essential for Translational Regulation and Cell Survival during the Unfolded Protein Response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef] [PubMed]
- McEwen, E.; Kedersha, N.; Song, B.; Scheuner, D.; Gilks, N.; Han, A.; Chen, J.-J.; Anderson, P.; Kaufman, R.J. Heme-Regulated Inhibitor Kinase-Mediated Phosphorylation of Eukaryotic Translation Initiation Factor 2 Inhibits Translation, Induces Stress Granule Formation, and Mediates Survival upon Arsenite Exposure. J. Biol. Chem. 2005, 280, 16925–16933. [Google Scholar] [CrossRef]
- Dever, T.E.; Hinnebusch, A.G. GCN2 Whets the Appetite for Amino Acids. Mol. Cell 2005, 18, 141–142. [Google Scholar] [CrossRef]
- Gal-Ben-Ari, S.; Barrera, I.; Ehrlich, M.; Rosenblum, K. PKR: A Kinase to Remember. Front. Mol. Neurosci. 2019, 11, 480. [Google Scholar] [CrossRef]
- Hetz, C. The Unfolded Protein Response: Controlling Cell Fate Decisions under ER Stress and Beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Chen, J.-J.; London, I.M. Regulation of Protein Synthesis by Heme-Regulated EIF-2α Kinase. Trends Biochem. Sci. 1995, 20, 105–108. [Google Scholar] [CrossRef]
- Chen, J.-J.; Zhang, S. Heme-Regulated EIF2α Kinase in Erythropoiesis and Hemoglobinopathies. Blood 2019, 134, 1697–1707. [Google Scholar] [CrossRef]
- Lu, L.; Han, A.-P.; Chen, J.-J. Translation Initiation Control by Heme-Regulated Eukaryotic Initiation Factor 2α Kinase in Erythroid Cells under Cytoplasmic Stresses. Mol. Cell. Biol. 2001, 21, 7971–7980. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Pal, J.K.; Thulasiraman, V.; Hahn, H.P.; Chen, J.J.; Matts, R.L. The Role of the 90-KDa Heat-Shock Protein and Its Associated Cohorts in Stabilizing the Heme-Regulated EIF-2alpha Kinase in Reticulocyte Lysates during Heat Stress. Eur. J. Biochem. 1997, 246, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Berwal, S.K.; Bhatia, V.; Bendre, A.; Suresh, C.G.; Chatterjee, S.; Pal, J.K. Activation of HRI Is Mediated by Hsp90 during Stress through Modulation of the HRI-Hsp90 Complex. Int. J. Biol. Macromol. 2018, 118, 1604–1613. [Google Scholar] [CrossRef] [PubMed]
- Rowlands, A.G.; Panniers, R.; Henshaw, E.C. The Catalytic Mechanism of Guanine Nucleotide Exchange Factor Action and Competitive Inhibition by Phosphorylated Eukaryotic Initiation Factor 2. J. Biol. Chem. 1988, 263, 5526–5533. [Google Scholar] [CrossRef] [PubMed]
- Riggs, C.L.; Kedersha, N.; Ivanov, P.; Anderson, P. Mammalian Stress Granules and P Bodies at a Glance. J. Cell Sci. 2020, 133, jcs242487. [Google Scholar] [CrossRef]
- Kedersha, N.L.; Gupta, M.; Li, W.; Miller, I.; Anderson, P. RNA-Binding Proteins TIA-1 and TIAR Link the Phosphorylation of EIF-2 Alpha to the Assembly of Mammalian Stress Granules. J. Cell Biol. 1999, 147, 1431–1442. [Google Scholar] [CrossRef]
- Hondele, M.; Sachdev, R.; Heinrich, S.; Wang, J.; Vallotton, P.; Fontoura, B.M.A.; Weis, K. DEAD-Box ATPases Are Global Regulators of Phase-Separated Organelles. Nature 2019, 573, 144–148. [Google Scholar] [CrossRef]
- Samir, P.; Kanneganti, T.-D. DEAD/H-Box Helicases in Immunity, Inflammation, Cell Differentiation, and Cell Death and Disease. Cells 2022, 11, 1608. [Google Scholar] [CrossRef]
- Szczerba, M.; Johnson, B.; Acciai, F.; Gogerty, C.; McCaughan, M.; Williams, J.; Kibler, K.V.; Jacobs, B.L. Canonical Cellular Stress Granules Are Required for Arsenite-Induced Necroptosis Mediated by Z-DNA–Binding Protein 1. Sci. Signal. 2023, 16, eabq0837. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, Pyroptosis and Apoptosis: An Intricate Game of Cell Death. Cell. Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef] [PubMed]
- Slee, E.A.; Adrain, C.; Martin, S.J. Executioner Caspase-3, -6, and -7 Perform Distinct, Non-Redundant Roles during the Demolition Phase of Apoptosis. J. Biol. Chem. 2001, 276, 7320–7326. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of ProIL-Beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Agostini, L.; Martinon, F.; Burns, K.; McDermott, M.F.; Hawkins, P.N.; Tschopp, J. NALP3 Forms an IL-1beta-Processing Inflammasome with Increased Activity in Muckle-Wells Autoinflammatory Disorder. Immunity 2004, 20, 319–325. [Google Scholar] [CrossRef]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin Activates the Inflammasome in Response to Toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Martinon, F.; Agostini, L.; Meylan, E.; Tschopp, J. Identification of Bacterial Muramyl Dipeptide as Activator of the NALP3/Cryopyrin Inflammasome. Curr. Biol. 2004, 14, 1929–1934. [Google Scholar] [CrossRef]
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-Associated Uric Acid Crystals Activate the NALP3 Inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica Crystals and Aluminum Salts Activate the NALP3 Inflammasome through Phagosomal Destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef]
- Kanneganti, T.-D.; Body-Malapel, M.; Amer, A.; Park, J.-H.; Whitfield, J.; Franchi, L.; Taraporewala, Z.F.; Miller, D.; Patton, J.T.; Inohara, N.; et al. Critical Role for Cryopyrin/Nalp3 in Activation of Caspase-1 in Response to Viral Infection and Double-Stranded RNA. J. Biol. Chem. 2006, 281, 36560–36568. [Google Scholar] [CrossRef]
- Kanneganti, T.-D.; Özören, N.; Body-Malapel, M.; Amer, A.; Park, J.-H.; Franchi, L.; Whitfield, J.; Barchet, W.; Colonna, M.; Vandenabeele, P.; et al. Bacterial RNA and Small Antiviral Compounds Activate Caspase-1 through Cryopyrin/Nalp3. Nature 2006, 440, 233–236. [Google Scholar] [CrossRef]
- Mariathasan, S.; Newton, K.; Monack, D.M.; Vucic, D.; French, D.M.; Lee, W.P.; Roose-Girma, M.; Erickson, S.; Dixit, V.M. Differential Activation of the Inflammasome by Caspase-1 Adaptors ASC and Ipaf. Nature 2004, 430, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Miao, E.A.; Alpuche-Aranda, C.M.; Dors, M.; Clark, A.E.; Bader, M.W.; Miller, S.I.; Aderem, A. Cytoplasmic Flagellin Activates Caspase-1 and Secretion of Interleukin 1beta via Ipaf. Nat. Immunol. 2006, 7, 569–575. [Google Scholar] [CrossRef]
- Miao, E.A.; Mao, D.P.; Yudkovsky, N.; Bonneau, R.; Lorang, C.G.; Warren, S.E.; Leaf, I.A.; Aderem, A. Innate Immune Detection of the Type III Secretion Apparatus through the NLRC4 Inflammasome. Proc. Natl. Acad. Sci. USA 2010, 107, 3076–3080. [Google Scholar] [CrossRef] [PubMed]
- Chae, J.J.; Cho, Y.-H.; Lee, G.-S.; Cheng, J.; Liu, P.P.; Feigenbaum, L.; Katz, S.I.; Kastner, D.L. Gain-of-Function Pyrin Mutations Induce NLRP3 Protein-Independent Interleukin-1β Activation and Severe Autoinflammation in Mice. Immunity 2011, 34, 755–768. [Google Scholar] [CrossRef]
- Xu, H.; Yang, J.; Gao, W.; Li, L.; Li, P.; Zhang, L.; Gong, Y.-N.; Peng, X.; Xi, J.J.; Chen, S.; et al. Innate Immune Sensing of Bacterial Modifications of Rho GTPases by the Pyrin Inflammasome. Nature 2014, 513, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Alnemri, T.; Yu, J.-W.; Datta, P.; Wu, J.; Alnemri, E.S. AIM2 Activates the Inflammasome and Cell Death in Response to Cytoplasmic DNA. Nature 2009, 458, 509–513. [Google Scholar] [CrossRef]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 Recognizes Cytosolic DsDNA and Forms a Caspase-1-Activating Inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef]
- Bürckstümmer, T.; Baumann, C.; Blüml, S.; Dixit, E.; Dürnberger, G.; Jahn, H.; Planyavsky, M.; Bilban, M.; Colinge, J.; Bennett, K.L.; et al. An Orthogonal Proteomic-Genomic Screen Identifies AIM2 as a Cytoplasmic DNA Sensor for the Inflammasome. Nat. Immunol. 2009, 10, 266–272. [Google Scholar] [CrossRef]
- Roberts, T.L.; Idris, A.; Dunn, J.A.; Kelly, G.M.; Burnton, C.M.; Hodgson, S.; Hardy, L.L.; Garceau, V.; Sweet, M.J.; Ross, I.L.; et al. HIN-200 Proteins Regulate Caspase Activation in Response to Foreign Cytoplasmic DNA. Science 2009, 323, 1057–1060. [Google Scholar] [CrossRef]
- Cai, X.; Chen, J.; Xu, H.; Liu, S.; Jiang, Q.-X.; Halfmann, R.; Chen, Z.J. Prion-like Polymerization Underlies Signal Transduction in Antiviral Immune Defense and Inflammasome Activation. Cell 2014, 156, 1207–1222. [Google Scholar] [CrossRef]
- Fernandes-Alnemri, T.; Wu, J.; Yu, J.-W.; Datta, P.; Miller, B.; Jankowski, W.; Rosenberg, S.; Zhang, J.; Alnemri, E.S. The Pyroptosome: A Supramolecular Assembly of ASC Dimers Mediating Inflammatory Cell Death via Caspase-1 Activation. Cell Death Differ. 2007, 14, 1590–1604. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.; Magupalli, V.G.; Ruan, J.; Yin, Q.; Atianand, M.K.; Vos, M.R.; Schröder, G.F.; Fitzgerald, K.A.; Wu, H.; Egelman, E.H. Unified Polymerization Mechanism for the Assembly of ASC-Dependent Inflammasomes. Cell 2014, 156, 1193–1206. [Google Scholar] [CrossRef] [PubMed]
- Franklin, B.S.; Bossaller, L.; De Nardo, D.; Ratter, J.M.; Stutz, A.; Engels, G.; Brenker, C.; Nordhoff, M.; Mirandola, S.R.; Al-Amoudi, A.; et al. The Adaptor ASC Has Extracellular and “prionoid” Activities That Propagate Inflammation. Nat. Immunol. 2014, 15, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Thornberry, N.A.; Bull, H.G.; Calaycay, J.R.; Chapman, K.T.; Howard, A.D.; Kostura, M.J.; Miller, D.K.; Molineaux, S.M.; Weidner, J.R.; Aunins, J. A Novel Heterodimeric Cysteine Protease Is Required for Interleukin-1 Beta Processing in Monocytes. Nature 1992, 356, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Howard, A.D.; Kostura, M.J.; Thornberry, N.; Ding, G.J.; Limjuco, G.; Weidner, J.; Salley, J.P.; Hogquist, K.A.; Chaplin, D.D.; Mumford, R.A. IL-1-Converting Enzyme Requires Aspartic Acid Residues for Processing of the IL-1 Beta Precursor at Two Distinct Sites and Does Not Cleave 31-KDa IL-1 Alpha. J. Immunol. 1991, 147, 2964–2969. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Allen, H.; Banerjee, S.; Franklin, S.; Herzog, L.; Johnston, C.; McDowell, J.; Paskind, M.; Rodman, L.; Salfeld, J. Mice Deficient in IL-1 Beta-Converting Enzyme Are Defective in Production of Mature IL-1 Beta and Resistant to Endotoxic Shock. Cell 1995, 80, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Ghayur, T.; Banerjee, S.; Hugunin, M.; Butler, D.; Herzog, L.; Carter, A.; Quintal, L.; Sekut, L.; Talanian, R.; Paskind, M.; et al. Caspase-1 Processes IFN-γ-Inducing Factor and Regulates LPS-Induced IFN-γ Production. Nature 1997, 386, 619–623. [Google Scholar] [CrossRef]
- Gu, Y.; Kuida, K.; Tsutsui, H.; Ku, G.; Hsiao, K.; Fleming, M.A.; Hayashi, N.; Higashino, K.; Okamura, H.; Nakanishi, K.; et al. Activation of Interferon-γ Inducing Factor Mediated by Interleukin-1β Converting Enzyme. Science 1997, 275, 206–209. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 Cleaves Gasdermin D for Non-Canonical Inflammasome Signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Sborgi, L.; Rühl, S.; Mulvihill, E.; Pipercevic, J.; Heilig, R.; Stahlberg, H.; Farady, C.J.; Müller, D.J.; Broz, P.; Hiller, S. GSDMD Membrane Pore Formation Constitutes the Mechanism of Pyroptotic Cell Death. EMBO J. 2016, 35, 1766–1778. [Google Scholar] [CrossRef]
- Yu, Z.; Jiang, N.; Su, W.; Zhuo, Y. Necroptosis: A Novel Pathway in Neuroinflammation. Front. Pharmacol. 2021, 12, 701564. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Jitkaew, S.; Cai, Z.; Choksi, S.; Li, Q.; Luo, J.; Liu, Z.-G. Mixed Lineage Kinase Domain-like Is a Key Receptor Interacting Protein 3 Downstream Component of TNF-Induced Necrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 5322–5327. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed Lineage Kinase Domain-like Protein Mediates Necrosis Signaling Downstream of RIP3 Kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.-F.; Wang, F.-S.; Wang, X. Mixed Lineage Kinase Domain-like Protein MLKL Causes Necrotic Membrane Disruption upon Phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Saelens, X.; Festjens, N.; Walle, L.V.; van Gurp, M.; van Loo, G.; Vandenabeele, P. Toxic Proteins Released from Mitochondria in Cell Death. Oncogene 2004, 23, 2861–2874. [Google Scholar] [CrossRef]
- Van Opdenbosch, N.; Gurung, P.; Vande Walle, L.; Fossoul, A.; Kanneganti, T.-D.; Lamkanfi, M. Activation of the NLRP1b Inflammasome Independently of ASC-Mediated Caspase-1 Autoproteolysis and Speck Formation. Nat. Commun. 2014, 5, 3209. [Google Scholar] [CrossRef]
- Case, C.L.; Shin, S.; Roy, C.R. Asc and Ipaf Inflammasomes Direct Distinct Pathways for Caspase-1 Activation in Response to Legionella Pneumophila. Infect. Immun. 2009, 77, 1981–1991. [Google Scholar] [CrossRef]
- Masson, G.R. Towards a Model of GCN2 Activation. Biochem. Soc. Trans. 2019, 47, 1481–1488. [Google Scholar] [CrossRef]
- Lageix, S.; Zhang, J.; Rothenburg, S.; Hinnebusch, A.G. Interaction between the TRNA-Binding and C-Terminal Domains of Yeast Gcn2 Regulates Kinase Activity In Vivo. PLoS Genet. 2015, 11, e1004991. [Google Scholar] [CrossRef]
- Yang, R.; Wek, S.A.; Wek, R.C. Glucose Limitation Induces GCN4 Translation by Activation of Gcn2 Protein Kinase. Mol. Cell. Biol. 2000, 20, 2706–2717. [Google Scholar] [CrossRef] [PubMed]
- Messenguy, F.; Colin, D.; ten Have, J.P. Regulation of Compartmentation of Amino Acid Pools in Saccharomyces Cerevisiae and Its Effects on Metabolic Control. Eur. J. Biochem. 1980, 108, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Qiu, H.; Garcia-Barrio, M.; Anderson, J.; Hinnebusch, A.G. Uncharged TRNA Activates GCN2 by Displacing the Protein Kinase Moiety from a Bipartite TRNA-Binding Domain. Mol. Cell 2000, 6, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Husain, B.; Mukerji, I.; Cole, J.L. Analysis of High Affinity Binding of PKR to DsRNA. Biochemistry 2012, 51, 8764. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, N.; Gorman, A.M.; Gupta, S.; Samali, A. The EIF2α Kinases: Their Structures and Functions. Cell. Mol. Life Sci. 2013, 70, 3493–3511. [Google Scholar] [CrossRef] [PubMed]
- Rafie-Kolpin, M.; Chefalo, P.J.; Hussain, Z.; Hahn, J.; Uma, S.; Matts, R.L.; Chen, J.-J. Two Heme-Binding Domains of Heme-Regulated Eukaryotic Initiation Factor-2α Kinase: N Terminus and Kinase Insertion. J. Biol. Chem. 2000, 275, 5171–5178. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic Reprogramming and Cancer Progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Yoshida, G.J. Metabolic Reprogramming: The Emerging Concept and Associated Therapeutic Strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111. [Google Scholar] [CrossRef]
- Ge, L.; Chen, W.; Cao, W.; Liu, G.; Zhang, Q.; Zhuang, J.; Zhang, M.; Yang, J.; Guo, S.; Zhao, X.; et al. GCN2 Is a Potential Prognostic Biomarker for Human Papillary Renal Cell Carcinoma. Cancer Biomark. 2018, 22, 395–403. [Google Scholar] [CrossRef]
- Lee, J.; Keam, B.; Kim, S.; Heo, J.-N.; Joung, E.; Kim, M.; Kim, T.M.; Kim, D.-W.; Heo, D.S. The Antitumor Activity of a Novel GCN2 Inhibitor in Head and Neck Squamous Cell Carcinoma Cell Lines. Transl. Oncol. 2022, 27, 101592. [Google Scholar] [CrossRef]
- Cordova, R.A.; Misra, J.; Amin, P.H.; Klunk, A.J.; Damayanti, N.P.; Carlson, K.R.; Elmendorf, A.J.; Kim, H.-G.; Mirek, E.T.; Elzey, B.D.; et al. GCN2 EIF2 Kinase Promotes Prostate Cancer by Maintaining Amino Acid Homeostasis. eLife 2022, 11, e81083. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-F.; Chen, M.-S.; Chou, Y.-C.; Ueng, Y.-F.; Yin, P.-H.; Yeh, T.-S.; Lee, H.-C. Mitochondrial Dysfunction Enhances Cisplatin Resistance in Human Gastric Cancer Cells via the ROS-Activated GCN2-EIF2α-ATF4-XCT Pathway. Oncotarget 2016, 7, 74132–74151. [Google Scholar] [CrossRef]
- Croucher, D.C.; Richards, L.M.; Tsofack, S.P.; Waller, D.; Li, Z.; Wei, E.N.; Huang, X.F.; Chesi, M.; Bergsagel, P.L.; Sebag, M.; et al. Longitudinal Single-Cell Analysis of a Myeloma Mouse Model Identifies Subclonal Molecular Programs Associated with Progression. Nat. Commun. 2021, 12, 6322. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Nambu, T.; Ebara, S.; Hasegawa, Y.; Toyoshima, K.; Tsuchiya, Y.; Tomita, D.; Fujimoto, J.; Kurasawa, O.; Takahara, C.; et al. Inhibition of GCN2 Sensitizes ASNS-Low Cancer Cells to Asparaginase by Disrupting the Amino Acid Response. Proc. Natl. Acad. Sci. USA 2018, 115, E7776–E7785. [Google Scholar] [CrossRef] [PubMed]
- Missiaen, R.; Anderson, N.M.; Kim, L.C.; Nance, B.; Burrows, M.; Skuli, N.; Carens, M.; Riscal, R.; Steensels, A.; Li, F.; et al. GCN2 Inhibition Sensitizes Arginine-Deprived Hepatocellular Carcinoma Cells to Senolytic Treatment. Cell Metab. 2022, 34, 1151–1167.e7. [Google Scholar] [CrossRef]
- Kim, S.H.; Gunnery, S.; Choe, J.K.; Mathews, M.B. Neoplastic Progression in Melanoma and Colon Cancer Is Associated with Increased Expression and Activity of the Interferon-Inducible Protein Kinase, PKR. Oncogene 2002, 21, 8741–8748. [Google Scholar] [CrossRef]
- Kim, S.H.; Forman, A.P.; Mathews, M.B.; Gunnery, S. Human Breast Cancer Cells Contain Elevated Levels and Activity of the Protein Kinase, PKR. Oncogene 2000, 19, 3086–3094. [Google Scholar] [CrossRef]
- Shimada, A.; Shiota, G.; Miyata, H.; Kamahora, T.; Kawasaki, H.; Shiraki, K.; Hino, S.; Terada, T. Aberrant Expression of Double-Stranded RNA-Dependent Protein Kinase in Hepatocytes of Chronic Hepatitis and Differentiated Hepatocellular Carcinoma. Cancer Res. 1998, 58, 4434–4438. [Google Scholar]
- Falletta, P.; Sanchez-del-Campo, L.; Chauhan, J.; Effern, M.; Kenyon, A.; Kershaw, C.J.; Siddaway, R.; Lisle, R.; Freter, R.; Daniels, M.J.; et al. Translation Reprogramming Is an Evolutionarily Conserved Driver of Phenotypic Plasticity and Therapeutic Resistance in Melanoma. Genes Dev. 2017, 31, 18–33. [Google Scholar] [CrossRef]
- Delgado André, N.; De Lucca, F.L. Knockdown of PKR Expression by RNAi Reduces Pulmonary Metastatic Potential of B16-F10 Melanoma Cells in Mice: Possible Role of NF-ΚB. Cancer Lett. 2007, 258, 118–125. [Google Scholar] [CrossRef]
- André, N.D.; Silva, V.A.O.; Watanabe, M.A.E.; De Lucca, F.L. Intratumoral Injection of PKR ShRNA Expressing Plasmid Inhibits B16-F10 Melanoma Growth. Oncol. Rep. 2014, 32, 2267–2273, Erratum in Oncol. Rep. 2018, 40, 3113. [Google Scholar] [CrossRef] [PubMed]
- Meurs, E.F.; Galabru, J.; Barber, G.N.; Katze, M.G.; Hovanessian, A.G. Tumor Suppressor Function of the Interferon-Induced Double-Stranded RNA-Activated Protein Kinase. Proc. Natl. Acad. Sci. USA 1993, 90, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Yoon, C.-H.; Lee, E.-S.; Lim, D.-S.; Bae, Y.-S. PKR, a P53 Target Gene, Plays a Crucial Role in the Tumor-Suppressor Function of P53. Proc. Natl. Acad. Sci. USA 2009, 106, 7852–7857. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, S.; Kim, C.N.; Yeh, W.-C.; Mak, T.W.; Bhalla, K.; Barber, G.N. Activation of the DsRNA-Dependent Protein Kinase, PKR, Induces Apoptosis through FADD-Mediated Death Signaling. EMBO J. 1998, 17, 6888–6902. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.; Esteban, M. Induction of Apoptosis by the DsRNA-Dependent Protein Kinase (PKR): Mechanism of Action. Apoptosis 2000, 5, 107–114. [Google Scholar] [CrossRef]
- Gil, J.; García, M.A.; Esteban, M. Caspase 9 Activation by the DsRNA-Dependent Protein Kinase, PKR: Molecular Mechanism and Relevance. FEBS Lett. 2002, 529, 249–255. [Google Scholar] [CrossRef]
- Lee, A.S.; Hendershot, L.M. ER Stress and Cancer. Cancer Biol. Ther. 2006, 5, 721–722. [Google Scholar] [CrossRef]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic Interaction of BiP and ER Stress Transducers in the Unfolded-Protein Response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Adjibade, P.; St-Sauveur, V.G.; Huberdeau, M.Q.; Fournier, M.-J.; Savard, A.; Coudert, L.; Khandjian, E.W.; Mazroui, R. Sorafenib, a Multikinase Inhibitor, Induces Formation of Stress Granules in Hepatocarcinoma Cells. Oncotarget 2015, 6, 43927–43943. [Google Scholar] [CrossRef]
- Timalsina, S.; Arimoto-Matsuzaki, K.; Kitamura, M.; Xu, X.; Wenzhe, Q.; Ishigami-Yuasa, M.; Kagechika, H.; Hata, Y. Chemical Compounds That Suppress Hypoxia-Induced Stress Granule Formation Enhance Cancer Drug Sensitivity of Human Cervical Cancer HeLa Cells. J. Biochem. 2018, 164, 381–391. [Google Scholar] [CrossRef]
- Martins, I.; Kepp, O.; Schlemmer, F.; Adjemian, S.; Tailler, M.; Shen, S.; Michaud, M.; Menger, L.; Gdoura, A.; Tajeddine, N.; et al. Restoration of the Immunogenicity of Cisplatin-Induced Cancer Cell Death by Endoplasmic Reticulum Stress. Oncogene 2011, 30, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Adjibade, P.; Simoneau, B.; Ledoux, N.; Gauthier, W.-N.; Nkurunziza, M.; Khandjian, E.W.; Mazroui, R. Treatment of Cancer Cells with Lapatinib Negatively Regulates General Translation and Induces Stress Granules Formation. PLoS ONE 2020, 15, e0231894. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Ozel, D.; Qiao, Y.; Harbinski, F.; Chen, L.; Denoyelle, S.; He, X.; Zvereva, N.; Supko, J.G.; Chorev, M.; et al. Chemical Genetics Identify EIF2α Kinase Heme-Regulated Inhibitor as an Anticancer Target. Nat. Chem. Biol. 2011, 7, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Yefidoff-Freedman, R.; Fan, J.; Yan, L.; Zhang, Q.; dos Santos, G.R.R.; Rana, S.; Contreras, J.I.; Sahoo, R.; Wan, D.; Young, J.; et al. Development of 1-((1,4-Trans)-4-Aryloxycyclohexyl)-3-Arylurea Activators of Heme-Regulated Inhibitor as Selective Activators of the Eukaryotic Initiation Factor 2 Alpha (EIF2α) Phosphorylation Arm of the Integrated Endoplasmic Reticulum Stress Response. J. Med. Chem. 2017, 60, 5392–5406. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Takrouri, K.; Hee-Hwang, S.; Rana, S.; Yefidoff-Freedman, R.; Halperin, J.; Natarajan, A.; Morisseau, C.; Hammock, B.; Chorev, M.; et al. Explorations of Substituted Urea Functionality for the Discovery of New Activators of the Heme-Regulated Inhibitor Kinase. J. Med. Chem. 2013, 56, 9457–9470. [Google Scholar] [CrossRef]
- Kalkavan, H.; Chen, M.J.; Crawford, J.C.; Quarato, G.; Fitzgerald, P.; Tait, S.W.G.; Goding, C.R.; Green, D.R. Sublethal Cytochrome c Release Generates Drug-Tolerant Persister Cells. Cell 2022, 185, 3356–3374.e22. [Google Scholar] [CrossRef]
- Allen, C.N.S.; Arjona, S.P.; Santerre, M.; Sawaya, B.E. Hallmarks of Metabolic Reprogramming and Their Role in Viral Pathogenesis. Viruses 2022, 14, 602. [Google Scholar] [CrossRef]
- Hashimoto, T.; Perlot, T.; Rehman, A.; Trichereau, J.; Ishiguro, H.; Paolino, M.; Sigl, V.; Hanada, T.; Hanada, R.; Lipinski, S.; et al. ACE2 Links Amino Acid Malnutrition to Microbial Ecology and Intestinal Inflammation. Nature 2012, 487, 477–481. [Google Scholar] [CrossRef]
- Hu, X.; Niu, Y.; Luo, P.; Xiao, F.; Yuan, F.; Yin, H.; Chen, S.; Guo, F. Amino Acid Sensor GCN2 Promotes SARS-CoV-2 Receptor ACE2 Expression in Response to Amino Acid Deprivation. Commun. Biol. 2022, 5, 651. [Google Scholar] [CrossRef]
- Berlanga, J.J.; Ventoso, I.; Harding, H.P.; Deng, J.; Ron, D.; Sonenberg, N.; Carrasco, L.; Haro, C. de Antiviral Effect of the Mammalian Translation Initiation Factor 2α Kinase GCN2 against RNA Viruses. EMBO J. 2006, 25, 1730–1740. [Google Scholar] [CrossRef]
- del Pino, J.; Jiménez, J.L.; Ventoso, I.; Castelló, A.; Muñoz-Fernández, M.Á.; de Haro, C.; Berlanga, J.J. GCN2 Has Inhibitory Effect on Human Immunodeficiency Virus-1 Protein Synthesis and Is Cleaved upon Viral Infection. PLoS ONE 2012, 7, e47272. [Google Scholar] [CrossRef] [PubMed]
- Habjan, M.; Pichlmair, A.; Elliott, R.M.; Överby, A.K.; Glatter, T.; Gstaiger, M.; Superti-Furga, G.; Unger, H.; Weber, F. NSs Protein of Rift Valley Fever Virus Induces the Specific Degradation of the Double-Stranded RNA-Dependent Protein Kinase. J. Virol. 2009, 83, 4365–4375. [Google Scholar] [CrossRef] [PubMed]
- Mudhasani, R.; Tran, J.P.; Retterer, C.; Kota, K.P.; Whitehouse, C.A.; Bavari, S. Protein Kinase R Degradation Is Essential for Rift Valley Fever Virus Infection and Is Regulated by SKP1-CUL1-F-Box (SCF)FBXW11-NSs E3 Ligase. PLoS Pathog. 2016, 12, e1005437. [Google Scholar] [CrossRef] [PubMed]
- Goodman, D.E.; Pretto, C.D.; Krepostman, T.A.; Carnahan, K.E.; Spindler, K.R. Enhanced Replication of Mouse Adenovirus Type 1 Following Virus-Induced Degradation of Protein Kinase R (PKR). mBio 2019, 10, e00668-19. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wambach, M.; Katze, M.G.; Krug, R.M. Binding of the Influenza Virus NS1 Protein to Double-Stranded RNA Inhibits the Activation of the Protein Kinase That Phosphorylates the EIF-2 Translation Initiation Factor. Virology 1995, 214, 222–228. [Google Scholar] [CrossRef]
- Rabouw, H.H.; Langereis, M.A.; Knaap, R.C.M.; Dalebout, T.J.; Canton, J.; Sola, I.; Enjuanes, L.; Bredenbeek, P.J.; Kikkert, M.; de Groot, R.J.; et al. Middle East Respiratory Coronavirus Accessory Protein 4a Inhibits PKR-Mediated Antiviral Stress Responses. PLoS Pathog. 2016, 12, e1005982. [Google Scholar] [CrossRef]
- Qin, Q.; Hastings, C.; Miller, C.L. Mammalian Orthoreovirus Particles Induce and Are Recruited into Stress Granules at Early Times Postinfection. J. Virol. 2009, 83, 11090–11101. [Google Scholar] [CrossRef]
- Romano, P.R.; Zhang, F.; Tan, S.-L.; Garcia-Barrio, M.T.; Katze, M.G.; Dever, T.E.; Hinnebusch, A.G. Inhibition of Double-Stranded RNA-Dependent Protein Kinase PKR by Vaccinia Virus E3: Role of Complex Formation and the E3 N-Terminal Domain. Mol. Cell. Biol. 1998, 18, 7304–7316. [Google Scholar] [CrossRef]
- Pavio, N.; Romano, P.R.; Graczyk, T.M.; Feinstone, S.M.; Taylor, D.R. Protein Synthesis and Endoplasmic Reticulum Stress Can Be Modulated by the Hepatitis C Virus Envelope Protein E2 through the Eukaryotic Initiation Factor 2α Kinase PERK. J. Virol. 2003, 77, 3578–3585. [Google Scholar] [CrossRef]
- Child, S.J.; Hakki, M.; De Niro, K.L.; Geballe, A.P. Evasion of Cellular Antiviral Responses by Human Cytomegalovirus TRS1 and IRS1. J. Virol. 2004, 78, 197–205. [Google Scholar] [CrossRef]
- Jordan, R.; Wang, L.; Graczyk, T.M.; Block, T.M.; Romano, P.R. Replication of a Cytopathic Strain of Bovine Viral Diarrhea Virus Activates PERK and Induces Endoplasmic Reticulum Stress-Mediated Apoptosis of MDBK Cells. J. Virol. 2002, 76, 9588–9599. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xin, X.; Wang, T.; Wan, J.; Ou, Y.; Yang, Z.; Yu, Q.; Zhu, L.; Guo, Y.; Wu, Y.; et al. Japanese Encephalitis Virus Induces Apoptosis and Encephalitis by Activating the PERK Pathway. J. Virol. 2019, 93, e00887-19. [Google Scholar] [CrossRef] [PubMed]
- Vilas-Boas, F.d.A.S.; da Silva, A.M.; de Sousa, L.P.; Lima, K.M.; Vago, J.P.; Bittencourt, L.F.F.; Dantas, A.E.; Gomes, D.A.; Vilela, M.C.; Teixeira, M.M.; et al. Impairment of Stress Granule Assembly via Inhibition of the EIF2alpha Phosphorylation Sensitizes Glioma Cells to Chemotherapeutic Agents. J. Neurooncol. 2016, 127, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Yu, S.; Xu, T.; Zhang, J.; Wu, S. Emerging Role of RNA Sensors in Tumor Microenvironment and Immunotherapy. J. Hematol. Oncol. J Hematol. Oncol. 2022, 15, 43. [Google Scholar] [CrossRef]
- Chen, Y.G.; Hur, S. Cellular Origins of DsRNA, Their Recognition and Consequences. Nat. Rev. Mol. Cell Biol. 2022, 23, 286–301. [Google Scholar] [CrossRef]
- Chong, K.L.; Feng, L.; Schappert, K.; Meurs, E.; Donahue, T.F.; Friesen, J.D.; Hovanessian, A.G.; Williams, B.R. Human P68 Kinase Exhibits Growth Suppression in Yeast and Homology to the Translational Regulator GCN2. EMBO J. 1992, 11, 1553–1562. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour Heterogeneity and Resistance to Cancer Therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Haines III, G.K.; Panos, R.J.; Bak, P.M.; Brown, T.; Zielinski, M.; Leyland, J.; Radosevich, J.A. Interferon-Responsive Protein Kinase (P68) and Proliferating Cell Nuclear Antigen Are Inversely Distributed in Head and Neck Squamous Cell Carcinoma. Tumor Biol. 1997, 19, 52–59. [Google Scholar] [CrossRef]
- Pataer, A.; Swisher, S.G.; Roth, J.A.; Logothetis, C.J.; Corn, P. Inhibition of RNA-Dependent Protein Kinase (PKR) Leads to Cancer Cell Death and Increases Chemosensitivity. Cancer Biol. Ther. 2009, 8, 245–252. [Google Scholar] [CrossRef]
- Donzé, O.; Deng, J.; Curran, J.; Sladek, R.; Picard, D.; Sonenberg, N. The Protein Kinase PKR: A Molecular Clock That Sequentially Activates Survival and Death Programs. EMBO J. 2004, 23, 564–571. [Google Scholar] [CrossRef]
- Xia, Y.; Shen, S.; Verma, I.M. NF-ΚB, an Active Player in Human Cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Min, J.-Y.; Li, S.; Sen, G.C.; Krug, R.M. A Site on the Influenza A Virus NS1 Protein Mediates Both Inhibition of PKR Activation and Temporal Regulation of Viral RNA Synthesis. Virology 2007, 363, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Schmechel, S.C.; Raghavan, A.; Abelson, M.; Reilly, C.; Katze, M.G.; Kaufman, R.J.; Bohjanen, P.R.; Schiff, L.A. Reovirus Induces and Benefits from an Integrated Cellular Stress Response. J. Virol. 2006, 80, 2019–2033. [Google Scholar] [CrossRef]
- Venticinque, L.; Meruelo, D. Sindbis Viral Vector Induced Apoptosis Requires Translational Inhibition and Signaling through Mcl-1 and Bak. Mol. Cancer 2010, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- McInerney, G.M.; Kedersha, N.L.; Kaufman, R.J.; Anderson, P.; Liljeström, P. Importance of EIF2α Phosphorylation and Stress Granule Assembly in Alphavirus Translation Regulation. Mol. Biol. Cell 2005, 16, 3753–3763. [Google Scholar] [CrossRef] [PubMed]
- Beauclair, G.; Streicher, F.; Chazal, M.; Bruni, D.; Lesage, S.; Gracias, S.; Bourgeau, S.; Sinigaglia, L.; Fujita, T.; Meurs, E.F.; et al. Retinoic Acid Inducible Gene I and Protein Kinase R, but Not Stress Granules, Mediate the Proinflammatory Response to Yellow Fever Virus. J. Virol. 2020, 94, e00403-20. [Google Scholar] [CrossRef]
- Gao, P.; Liu, Y.; Wang, H.; Chai, Y.; Weng, W.; Zhang, Y.; Zhou, L.; Ge, X.; Guo, X.; Han, J.; et al. Viral Evasion of PKR Restriction by Reprogramming Cellular Stress Granules. Proc. Natl. Acad. Sci. USA 2022, 119, e2201169119. [Google Scholar] [CrossRef]
- Back, S.H.; Scheuner, D.; Han, J.; Song, B.; Ribick, M.; Wang, J.; Gildersleeve, R.D.; Pennathur, S.; Kaufman, R.J. Translation Attenuation through EIF2α Phosphorylation Prevents Oxidative Stress and Maintains the Differentiated State in β Cells. Cell Metab. 2009, 10, 13–26. [Google Scholar] [CrossRef]
- Moenner, M.; Pluquet, O.; Bouchecareilh, M.; Chevet, E. Integrated Endoplasmic Reticulum Stress Responses in Cancer. Cancer Res. 2007, 67, 10631–10634. [Google Scholar] [CrossRef]
- He, B. Viruses, Endoplasmic Reticulum Stress, and Interferon Responses. Cell Death Differ. 2006, 13, 393–403. [Google Scholar] [CrossRef]
- Shacham, T.; Patel, C.; Lederkremer, G.Z. PERK Pathway and Neurodegenerative Disease: To Inhibit or to Activate? Biomolecules 2021, 11, 354. [Google Scholar] [CrossRef]
- Wang, P.; Han, L.; Yu, M.; Cao, Z.; Li, X.; Shao, Y.; Zhu, G. The Prognostic Value of PERK in Cancer and Its Relationship With Immune Cell Infiltration. Front. Mol. Biosci. 2021, 8, 648752. [Google Scholar] [CrossRef]
- Grabocka, E.; Bar-Sagi, D. Mutant KRAS Enhances Tumor Cell Fitness by Upregulating Stress Granules. Cell 2016, 167, 1803–1813.e12. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Bagchi, P.; Tsai, B. ER Functions Are Exploited by Viruses to Support Distinct Stages of Their Life Cycle. Biochem. Soc. Trans. 2020, 48, 2173–2184. [Google Scholar] [CrossRef] [PubMed]
- Su, H.-L.; Liao, C.-L.; Lin, Y.-L. Japanese Encephalitis Virus Infection Initiates Endoplasmic Reticulum Stress and an Unfolded Protein Response. J. Virol. 2002, 76, 4162–4171. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Fu, F.; Ma, Y.; Zhang, X.; Li, L.; Feng, L.; Liu, P. The PERK Arm of the Unfolded Protein Response Negatively Regulates Transmissible Gastroenteritis Virus Replication by Suppressing Protein Translation and Promoting Type I Interferon Production. J. Virol. 2018, 92, e00431-18. [Google Scholar] [CrossRef]
- Isler, J.A.; Skalet, A.H.; Alwine, J.C. Human Cytomegalovirus Infection Activates and Regulates the Unfolded Protein Response. J. Virol. 2005, 79, 6890–6899. [Google Scholar] [CrossRef]
- Chen, J.-J. Regulation of Protein Synthesis by the Heme-Regulated EIF2α Kinase: Relevance to Anemias. Blood 2006, 109, 2693–2699. [Google Scholar] [CrossRef]
- Ricketts, M.D.; Emptage, R.P.; Blobel, G.A.; Marmorstein, R. The Heme-Regulated Inhibitor Kinase Requires Dimerization for Heme-Sensing Activity. J. Biol. Chem. 2022, 298, 102451. [Google Scholar] [CrossRef]
- Liu, S.; Suragani, R.N.V.S.; Wang, F.; Han, A.; Zhao, W.; Andrews, N.C.; Chen, J.-J. The Function of Heme-Regulated EIF2α Kinase in Murine Iron Homeostasis and Macrophage Maturation. J. Clin. Investig. 2007, 117, 3296–3305. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Malireddi, R.K.S.; Gurung, P.; Kesavardhana, S.; Samir, P.; Burton, A.; Mummareddy, H.; Vogel, P.; Pelletier, S.; Burgula, S.; Kanneganti, T.-D. Innate Immune Priming in the Absence of TAK1 Drives RIPK1 Kinase Activity-Independent Pyroptosis, Apoptosis, Necroptosis, and Inflammatory Disease. J. Exp. Med. 2020, 217, jem.20191644. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper Induces Cell Death by Targeting Lipoylated TCA Cycle Proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Holze, C.; Michaudel, C.; Mackowiak, C.; Haas, D.A.; Benda, C.; Hubel, P.; Pennemann, F.L.; Schnepf, D.; Wettmarshausen, J.; Braun, M.; et al. Oxeiptosis, a ROS-Induced Caspase-Independent Apoptosis-like Cell-Death Pathway. Nat. Immunol. 2018, 19, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Fu, Y.; Shen, J.; Li, Y.; Liu, F.; Ning, B.; Zheng, Y.; Jiang, X. Inhibition of the PERK/TXNIP/NLRP3 Axis by Baicalin Reduces NLRP3 Inflammasome-Mediated Pyroptosis in Macrophages Infected with Mycobacterium Tuberculosis. Mediators Inflamm. 2021, 2021, 1805147. [Google Scholar] [CrossRef]
- Briard, B.; Fontaine, T.; Samir, P.; Place, D.E.; Muszkieta, L.; Malireddi, R.K.S.; Karki, R.; Christgen, S.; Bomme, P.; Vogel, P.; et al. Galactosaminogalactan Activates the Inflammasome to Provide Host Protection. Nature 2020, 588, 688–692. [Google Scholar] [CrossRef]
- Lu, B.; Nakamura, T.; Inouye, K.; Li, J.; Tang, Y.; Lundbäck, P.; Valdes-Ferrer, S.I.; Olofsson, P.S.; Kalb, T.; Roth, J.; et al. Novel Role of PKR in Inflammasome Activation and HMGB1 Release. Nature 2012, 488, 670–674. [Google Scholar] [CrossRef]
- Ravindran, R.; Loebbermann, J.; Nakaya, H.I.; Khan, N.; Ma, H.; Gama, L.; Machiah, D.K.; Lawson, B.; Hakimpour, P.; Wang, Y.; et al. The Amino Acid Sensor GCN2 Controls Gut Inflammation by Inhibiting Inflammasome Activation. Nature 2016, 531, 523–527. [Google Scholar] [CrossRef]
- Alim, I.; Caulfield, J.T.; Chen, Y.; Swarup, V.; Geschwind, D.H.; Ivanova, E.; Seravalli, J.; Ai, Y.; Sansing, L.H.; Ste Marie, E.J.; et al. Selenium Drives a Transcriptional Adaptive Program to Block Ferroptosis and Treat Stroke. Cell 2019, 177, 1262–1279.e25. [Google Scholar] [CrossRef]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.e21. [Google Scholar] [CrossRef]
- Ding, C.-K.C.; Rose, J.; Sun, T.; Wu, J.; Chen, P.-H.; Lin, C.-C.; Yang, W.-H.; Chen, K.-Y.; Lee, H.; Xu, E.; et al. MESH1 Is a Cytosolic NADPH Phosphatase That Regulates Ferroptosis. Nat. Metab. 2020, 2, 270–277. [Google Scholar] [CrossRef]
- Lin, C.-C.; Ding, C.-K.C.; Sun, T.; Wu, J.; Chen, K.-Y.; Zhou, P.; Chi, J.-T. The Regulation of Ferroptosis by MESH1 through the Activation of the Integrative Stress Response. Cell Death Dis. 2021, 12, 727. [Google Scholar] [CrossRef]
- Yu, Y.; Jiang, L.; Wang, H.; Shen, Z.; Cheng, Q.; Zhang, P.; Wang, J.; Wu, Q.; Fang, X.; Duan, L.; et al. Hepatic Transferrin Plays a Role in Systemic Iron Homeostasis and Liver Ferroptosis. Blood 2020, 136, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.W.; Amante, J.J.; Chhoy, P.; Elaimy, A.L.; Liu, H.; Zhu, L.J.; Baer, C.E.; Dixon, S.J.; Mercurio, A.M. Prominin2 Drives Ferroptosis Resistance by Stimulating Iron Export. Dev. Cell 2019, 51, 575–586.e4. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Qin, Q.; Li, K.; Li, H.; Song, C.; Li, Y.; Dai, M.; Lin, F.; Mao, Z.; Li, Q.; et al. PKR Suppress NLRP3-Pyroptosis Pathway in Lipopolysaccharide-Induced Acute Lung Injury Model of Mice. Biochem. Biophys. Res. Commun. 2019, 519, 8–14. [Google Scholar] [CrossRef]
- Jiang, Y.; Steinle, J.J. Epac1 Inhibits PKR to Reduce NLRP3 Inflammasome Proteins in Retinal Endothelial Cells. J. Inflamm. Res. 2019, 12, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Boriushkin, E.; Wang, J.J.; Li, J.; Bhatta, M.; Zhang, S.X. P58IPK Suppresses NLRP3 Inflammasome Activation and IL-1β Production via Inhibition of PKR in Macrophages. Sci. Rep. 2016, 6, 25013. [Google Scholar] [CrossRef]
- Yoshida, K.; Okamura, H.; Hiroshima, Y.; Abe, K.; Kido, J.-I.; Shinohara, Y.; Ozaki, K. PKR Induces the Expression of NLRP3 by Regulating the NF-ΚB Pathway in Porphyromonas Gingivalis-Infected Osteoblasts. Exp. Cell Res. 2017, 354, 57–64. [Google Scholar] [CrossRef]
- He, Y.; Franchi, L.; Núñez, G. The Protein Kinase PKR Is Critical for LPS-Induced INOS Production but Dispensable for Inflammasome Activation in Macrophages. Eur. J. Immunol. 2013, 43, 1147–1152. [Google Scholar] [CrossRef]
- Samir, P.; Kanneganti, T.-D. DDX3X Sits at the Crossroads of Liquid–Liquid and Prionoid Phase Transitions Arbitrating Life and Death Cell Fate Decisions in Stressed Cells. DNA Cell Biol. 2020, 39, 1091–1095. [Google Scholar] [CrossRef] [PubMed]
- de Reuver, R.; Verdonck, S.; Dierick, E.; Nemegeer, J.; Hessmann, E.; Ahmad, S.; Jans, M.; Blancke, G.; Van Nieuwerburgh, F.; Botzki, A.; et al. ADAR1 Prevents Autoinflammation by Suppressing Spontaneous ZBP1 Activation. Nature 2022, 607, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Zhang, T.; Balachandran, S. Detecting Z-RNA and Z-DNA in Mammalian Cells. In Methods in Molecular Biology; Humana: New York, NY, USA, 2023; Volume 2651, pp. 277–284. [Google Scholar] [CrossRef]
- Tang, Q.; Rigby, R.E.; Young, G.R.; Hvidt, A.K.; Davis, T.; Tan, T.K.; Bridgeman, A.; Townsend, A.R.; Kassiotis, G.; Rehwinkel, J. Adenosine-to-Inosine Editing of Endogenous Z-Form RNA by the Deaminase ADAR1 Prevents Spontaneous MAVS-Dependent Type I Interferon Responses. Immunity 2021, 54, 1961–1975.e5. [Google Scholar] [CrossRef]
- Maurano, M.; Snyder, J.M.; Connelly, C.; Henao-Mejia, J.; Sidrauski, C.; Stetson, D.B. Protein Kinase R and the Integrated Stress Response Drive Immunopathology Caused by Mutations in the RNA Deaminase ADAR1. Immunity 2021, 54, 1948–1960.e5. [Google Scholar] [CrossRef] [PubMed]
- Liddicoat, B.J.; Piskol, R.; Chalk, A.M.; Ramaswami, G.; Higuchi, M.; Hartner, J.C.; Li, J.B.; Seeburg, P.H.; Walkley, C.R. RNA Editing by ADAR1 Prevents MDA5 Sensing of Endogenous DsRNA as Nonself. Science 2015, 349, 1115–1120. [Google Scholar] [CrossRef]
- Zhang, T.; Yin, C.; Boyd, D.F.; Quarato, G.; Ingram, J.P.; Shubina, M.; Ragan, K.B.; Ishizuka, T.; Crawford, J.C.; Tummers, B.; et al. Influenza Virus Z-RNAs Induce ZBP1-Mediated Necroptosis. Cell 2020, 180, 1115–1129.e13. [Google Scholar] [CrossRef]
- Li, S.; Zhang, Y.; Guan, Z.; Ye, M.; Li, H.; You, M.; Zhou, Z.; Zhang, C.; Zhang, F.; Lu, B.; et al. SARS-CoV-2 Z-RNA Activates the ZBP1-RIPK3 Pathway to Promote Virus-Induced Inflammatory Responses. Cell Res. 2023, 33, 201–214. [Google Scholar] [CrossRef]
- Koehler, H.; Cotsmire, S.; Zhang, T.; Balachandran, S.; Upton, J.W.; Langland, J.; Kalman, D.; Jacobs, B.L.; Mocarski, E.S. Vaccinia Virus E3 Prevents Sensing of Z-RNA to Block ZBP1-Dependent Necroptosis. Cell Host Microbe 2021, 29, 1266–1276.e5. [Google Scholar] [CrossRef]
- Kuriakose, T.; Man, S.M.; Malireddi, R.K.S.; Karki, R.; Kesavardhana, S.; Place, D.E.; Neale, G.; Vogel, P.; Kanneganti, T.-D. ZBP1/DAI Is an Innate Sensor of Influenza Virus Triggering the NLRP3 Inflammasome and Programmed Cell Death Pathways. Sci. Immunol. 2016, 1, aag2045. [Google Scholar] [CrossRef]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. DAI/ZBP1/DLM-1 Complexes with RIP3 to Mediate Virus-Induced Programmed Necrosis That Is Targeted by Murine Cytomegalovirus VIRA. Cell Host Microbe 2012, 11, 290–297. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, H.; Zhao, Y.; Shan, L.; Lan, S. Fisetin-Induced Cell Death in Human Ovarian Cancer Cell Lines via Zbp1-Mediated Necroptosis. J. Ovarian Res. 2022, 15, 57. [Google Scholar] [CrossRef] [PubMed]
- Baik, J.Y.; Liu, Z.; Jiao, D.; Kwon, H.-J.; Yan, J.; Kadigamuwa, C.; Choe, M.; Lake, R.; Kruhlak, M.; Tandon, M.; et al. ZBP1 Not RIPK1 Mediates Tumor Necroptosis in Breast Cancer. Nat. Commun. 2021, 12, 2666. [Google Scholar] [CrossRef] [PubMed]
- Deigendesch, N.; Koch-Nolte, F.; Rothenburg, S. ZBP1 Subcellular Localization and Association with Stress Granules Is Controlled by Its Z-DNA Binding Domains. Nucleic Acids Res. 2006, 34, 5007–5020. [Google Scholar] [CrossRef] [PubMed]
- Sáez-Freire, M.D.M.; Blanco-Gómez, A.; Castillo-Lluva, S.; Gómez-Vecino, A.; Galvis-Jiménez, J.M.; Martín-Seisdedos, C.; Isidoro-García, M.; Hontecillas-Prieto, L.; García-Cenador, M.B.; García-Criado, F.J.; et al. The Biological Age Linked to Oxidative Stress Modifies Breast Cancer Aggressiveness. Free Radic. Biol. Med. 2018, 120, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Pratt, D.A. The Chemical Basis of Ferroptosis. Nat. Chem. Biol. 2019, 15, 1137–1147. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the Ferroptosis Regulator Gpx4 Triggers Acute Renal Failure in Mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Qi, Y.; Zhang, X.; Wu, Z.; Tian, M.; Chen, F.; Guan, W.; Zhang, S. Ferroptosis Regulation by Nutrient Signalling. Nutr. Res. Rev. 2022, 35, 282–294. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of Polyunsaturated Fatty Acids by Lipoxygenases Drives Ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef]
- Basu, M.; Courtney, S.C.; Brinton, M.A. Arsenite-Induced Stress Granule Formation Is Inhibited by Elevated Levels of Reduced Glutathione in West Nile Virus-Infected Cells. PLoS Pathog. 2017, 13, e1006240. [Google Scholar] [CrossRef]
- Fujikawa, D.; Nakamura, T.; Yoshioka, D.; Li, Z.; Moriizumi, H.; Taguchi, M.; Tokai-Nishizumi, N.; Kozuka-Hata, H.; Oyama, M.; Takekawa, M. Stress Granule Formation Inhibits Stress-Induced Apoptosis by Selectively Sequestering Executioner Caspases. Curr. Biol. 2023, 33, 1967–1981.e8. [Google Scholar] [CrossRef] [PubMed]
- Weissbach, R.; Scadden, A.D.J. Tudor-SN and ADAR1 Are Components of Cytoplasmic Stress Granules. RNA 2012, 18, 462–471. [Google Scholar] [CrossRef] [PubMed]
Figure 2.
Regulated cell death pathways: (A) Apoptosis. Ligand binding induces oligomerization of death receptors activating the extrinsic apoptosis cascade. Tumor necrosis factor receptor 1 (TNFR1), TNFR1/2-associated death domain protein (TRADD), Fas-associated death domain protein (FADD) and pro-caspase-8 are recruited to the oligomerized death receptors forming death-inducing signaling cascade (DISC). Autoproteolytic processing of pro-caspase 8 generates catalytically active initiator caspase 8, which cleaves executioner caspases, caspase-3 and caspase-7, completing extrinsic apoptosis [41,42,43]. Intrinsic apoptosis is triggered by various intracellular danger signals that induce an increase in mitochondrial outer membrane permeabilization (MOMPs). An increase in MOMPs releases cytochrome c in the cytosol. Cytochrome c binds with APAF1 and pro-caspase-9 forming an apoptosome signaling complex. Autoproteolytic processing of pro-caspase-9 in the apoptosome generates catalytically active initiator caspase-9, which cleaves executioner caspases, caspase-3 and caspase-7, leading to intrinsic apoptosis [41,42,43]. (B) Pyroptosis. Diverse PAMPs and DAMPs are sensed by different inflammasome sensors, NLRP1 [44], NLRP3 [45,46,47,48,49,50,51], NLRC4 [52,53,54], PYRIN [55,56], and AIM2 [57,58,59,60]. Activated inflammasome sensors mediate ASC (apoptosis-associated speck-like protein containing CARD) dependent recruitment of pro-Caspase 1 and subsequent autoprocessing generating active caspase-1 [61,62,63,64]. Active caspase-1 cleaves pro-interleukin-1β (proIL-1β) [65,66,67], pro-IL-18 [68,69] and Gasdermin D (GSDMD) [70,71]. The N-terminal fragment of GSDMD forms pyroptotic pores through which IL-1β and IL-18, along with intracellular content, are released [70,72]. (C) Necroptosis. TNFR1 signaling in the absence of caspase-8 recruits RIPK1 (receptor-interacting protein kinase 1) and RIPK3 forming a necroptosis-inducing complex called the necrosome [42,73]. MLKL (mixed lineage kinase domain-like pseudokinase) is phosphorylated at the necrosome and translocates to the cell membrane to form pores that cause osmotic lysis of cells [74,75,76].
Figure 2.
Regulated cell death pathways: (A) Apoptosis. Ligand binding induces oligomerization of death receptors activating the extrinsic apoptosis cascade. Tumor necrosis factor receptor 1 (TNFR1), TNFR1/2-associated death domain protein (TRADD), Fas-associated death domain protein (FADD) and pro-caspase-8 are recruited to the oligomerized death receptors forming death-inducing signaling cascade (DISC). Autoproteolytic processing of pro-caspase 8 generates catalytically active initiator caspase 8, which cleaves executioner caspases, caspase-3 and caspase-7, completing extrinsic apoptosis [41,42,43]. Intrinsic apoptosis is triggered by various intracellular danger signals that induce an increase in mitochondrial outer membrane permeabilization (MOMPs). An increase in MOMPs releases cytochrome c in the cytosol. Cytochrome c binds with APAF1 and pro-caspase-9 forming an apoptosome signaling complex. Autoproteolytic processing of pro-caspase-9 in the apoptosome generates catalytically active initiator caspase-9, which cleaves executioner caspases, caspase-3 and caspase-7, leading to intrinsic apoptosis [41,42,43]. (B) Pyroptosis. Diverse PAMPs and DAMPs are sensed by different inflammasome sensors, NLRP1 [44], NLRP3 [45,46,47,48,49,50,51], NLRC4 [52,53,54], PYRIN [55,56], and AIM2 [57,58,59,60]. Activated inflammasome sensors mediate ASC (apoptosis-associated speck-like protein containing CARD) dependent recruitment of pro-Caspase 1 and subsequent autoprocessing generating active caspase-1 [61,62,63,64]. Active caspase-1 cleaves pro-interleukin-1β (proIL-1β) [65,66,67], pro-IL-18 [68,69] and Gasdermin D (GSDMD) [70,71]. The N-terminal fragment of GSDMD forms pyroptotic pores through which IL-1β and IL-18, along with intracellular content, are released [70,72]. (C) Necroptosis. TNFR1 signaling in the absence of caspase-8 recruits RIPK1 (receptor-interacting protein kinase 1) and RIPK3 forming a necroptosis-inducing complex called the necrosome [42,73]. MLKL (mixed lineage kinase domain-like pseudokinase) is phosphorylated at the necrosome and translocates to the cell membrane to form pores that cause osmotic lysis of cells [74,75,76].

Figure 3.
Domain organization of stress kinases. The domain architecture of the four mammalian eIF2α kinases is represented as bars. GCN2 is 1649 amino acids long and has an N-terminal RWD domain (RING finger-containing protein, WD repeat-containing protein and yeast DEAD-like helicase), pseudokinase domain, catalytically active kinase domain (KD), histidyl-tRNA synthetase like (HisRS) domain and C-terminal domain (CTD) [80,81]. PKR is 551 amino acids long and has an N-terminal regulatory domain joined by a linker domain to the catalytic C-terminal kinase domain. The regulatory domain has two double-stranded RNA (dsRNA) binding motifs; dRBM1 and dRBM2 [28,85]. PERK is 1116 amino acids long and consists of an N-terminal regulatory lumenal domain and a C-terminal cytosolic kinase domain [86]. HRI is 630 amino acids in length and has an N-terminal domain, a central regulatory kinase insertion (KI) domain flanked by two kinase domains (Kinase I and Kinase II) and the C-terminal domain [86,87].
Figure 3.
Domain organization of stress kinases. The domain architecture of the four mammalian eIF2α kinases is represented as bars. GCN2 is 1649 amino acids long and has an N-terminal RWD domain (RING finger-containing protein, WD repeat-containing protein and yeast DEAD-like helicase), pseudokinase domain, catalytically active kinase domain (KD), histidyl-tRNA synthetase like (HisRS) domain and C-terminal domain (CTD) [80,81]. PKR is 551 amino acids long and has an N-terminal regulatory domain joined by a linker domain to the catalytic C-terminal kinase domain. The regulatory domain has two double-stranded RNA (dsRNA) binding motifs; dRBM1 and dRBM2 [28,85]. PERK is 1116 amino acids long and consists of an N-terminal regulatory lumenal domain and a C-terminal cytosolic kinase domain [86]. HRI is 630 amino acids in length and has an N-terminal domain, a central regulatory kinase insertion (KI) domain flanked by two kinase domains (Kinase I and Kinase II) and the C-terminal domain [86,87].

Figure 4.
Stress kinases in modulation of apoptosis in tumors. (A) Tumor cells have increased expression of GCN2 [90,91,92,93,94,95]. Nutrient deprivation in tumor microenvironment (TME) increases the pool of uncharged tRNAs, which binds to HisRS domain of GCN2. Activated GCN2 phosphorylates eIF2α leading to translational inhibition and promoting survival of tumor cells. In contrast, inhibition of GCN2 by anti-tumor drugs promoted tumor cell death [95,96]. (B) dsRNA generated from diverse cellular processes within the TME activates PKR. PKR activation can have tumorigenic [28,97,98,99,100,101,102] as well as tumor-suppressive roles [103,104,105,106]. How PKR promotes tumorigenesis is unknown. PKR-dependent eIF2α phosphorylation mediates apoptosis of the tumor cells [105,106,107] and promotes tumor suppression. (C) Stress in TME leads to accumulation of unfolded or misfolded proteins [108], which translocate to the endoplasmic reticulum (ER). An increase in misfolded proteins in the ER leads to dissociation of immunoglobulin binding protein (BiP) from the PERK and subsequently activates PERK [109]. Activated PERK phosphorylates eIF2α leading to formation of SGs. In addition, chemotherapeutic agents also favor eIF2α phosphorylation mediated formation of SGs [110,111,112,113]. SGs formation stalls translation at the initiation step, inhibits apoptosis and favors tumor cell survival. (D) Compounds such as N,N′-diarylureas activate HRI leading to eIF2α phosphorylation-dependent inhibition of oncogenesis [114,115,116]. Chemotherapeutic agents induce incomplete mitochondrial outer membrane permeabilization (iMOMPs), liberating suboptimal cytochrome c, which activates HRI kinase [117]. HRI kinase-mediated translational reprogramming generates drug-resistant cancer cells that promote tumorigenesis.
Figure 4.
Stress kinases in modulation of apoptosis in tumors. (A) Tumor cells have increased expression of GCN2 [90,91,92,93,94,95]. Nutrient deprivation in tumor microenvironment (TME) increases the pool of uncharged tRNAs, which binds to HisRS domain of GCN2. Activated GCN2 phosphorylates eIF2α leading to translational inhibition and promoting survival of tumor cells. In contrast, inhibition of GCN2 by anti-tumor drugs promoted tumor cell death [95,96]. (B) dsRNA generated from diverse cellular processes within the TME activates PKR. PKR activation can have tumorigenic [28,97,98,99,100,101,102] as well as tumor-suppressive roles [103,104,105,106]. How PKR promotes tumorigenesis is unknown. PKR-dependent eIF2α phosphorylation mediates apoptosis of the tumor cells [105,106,107] and promotes tumor suppression. (C) Stress in TME leads to accumulation of unfolded or misfolded proteins [108], which translocate to the endoplasmic reticulum (ER). An increase in misfolded proteins in the ER leads to dissociation of immunoglobulin binding protein (BiP) from the PERK and subsequently activates PERK [109]. Activated PERK phosphorylates eIF2α leading to formation of SGs. In addition, chemotherapeutic agents also favor eIF2α phosphorylation mediated formation of SGs [110,111,112,113]. SGs formation stalls translation at the initiation step, inhibits apoptosis and favors tumor cell survival. (D) Compounds such as N,N′-diarylureas activate HRI leading to eIF2α phosphorylation-dependent inhibition of oncogenesis [114,115,116]. Chemotherapeutic agents induce incomplete mitochondrial outer membrane permeabilization (iMOMPs), liberating suboptimal cytochrome c, which activates HRI kinase [117]. HRI kinase-mediated translational reprogramming generates drug-resistant cancer cells that promote tumorigenesis.

Figure 5.
Stress kinases in modulation of viral infection outcome. (A) Virus infection activates GCN2 leading to eIF2α phosphorylation mediated translation shutdown, which inhibits virus replication [121,122]. In contrast, HIV-1 proteases cleave GCN2, abrogating eIF2α phosphorylation mediated translation inhibition and promoting virus growth [122]. Moreover, GCN2 phosphorylation during SARS coronavirus 2 infection increases expression of the ACE2 (Angiotensin-converting enzyme 2) receptor [120] that increases SARS-CoV2 infectivity. (B) dsRNA generated during virus replication activates PKR. PKR-mediated eIF2α phosphorylation inhibits translation leading to the formation of stress granules favoring virus replication through poorly explored mechanisms. Rift valley fever virus [123,124] and mouse adenovirus type I [125] routes activated PKR for proteasomal degradation to favor its replication. Viral proteins such as influenza virus nonstructural protein 1 (NS1) [126], MERS-CoV p4a protein [127], mammalian orthoreovirus σ3 protein [128], and vaccinia virus E3L protein [129] sequestered viral dsRNA away from PKR abrogating PKR activation to favor virus replication. (C) Profound virus replication generates unfolded or misfolded proteins which translocate to the endoplasmic reticulum (ER). An increase in misfolded proteins in the ER leads to dissociation of immunoglobulin binding protein (BiP) from the PERK [109]. Hepatitis C virus E2 protein [130] and human cytomegalovirus TRSI protein [131] inhibit the kinase activity of PERK, abrogating eIF2α phosphorylation and favoring virus replication. The bovine viral diarrhea virus mediates PERK-dependent inhibition of anti-apoptotic protein Bcl-2, promoting apoptosis of infected cells [132]. In contrast, Japanese encephalitis virus induces stress granules in a PERK-dependent manner to inhibit apoptosis and promote virus replication [133]. In (B,C), each number represents the unique pathway being followed (example: number 1 is one pathway while number 2 represents the other pathway).
Figure 5.
Stress kinases in modulation of viral infection outcome. (A) Virus infection activates GCN2 leading to eIF2α phosphorylation mediated translation shutdown, which inhibits virus replication [121,122]. In contrast, HIV-1 proteases cleave GCN2, abrogating eIF2α phosphorylation mediated translation inhibition and promoting virus growth [122]. Moreover, GCN2 phosphorylation during SARS coronavirus 2 infection increases expression of the ACE2 (Angiotensin-converting enzyme 2) receptor [120] that increases SARS-CoV2 infectivity. (B) dsRNA generated during virus replication activates PKR. PKR-mediated eIF2α phosphorylation inhibits translation leading to the formation of stress granules favoring virus replication through poorly explored mechanisms. Rift valley fever virus [123,124] and mouse adenovirus type I [125] routes activated PKR for proteasomal degradation to favor its replication. Viral proteins such as influenza virus nonstructural protein 1 (NS1) [126], MERS-CoV p4a protein [127], mammalian orthoreovirus σ3 protein [128], and vaccinia virus E3L protein [129] sequestered viral dsRNA away from PKR abrogating PKR activation to favor virus replication. (C) Profound virus replication generates unfolded or misfolded proteins which translocate to the endoplasmic reticulum (ER). An increase in misfolded proteins in the ER leads to dissociation of immunoglobulin binding protein (BiP) from the PERK [109]. Hepatitis C virus E2 protein [130] and human cytomegalovirus TRSI protein [131] inhibit the kinase activity of PERK, abrogating eIF2α phosphorylation and favoring virus replication. The bovine viral diarrhea virus mediates PERK-dependent inhibition of anti-apoptotic protein Bcl-2, promoting apoptosis of infected cells [132]. In contrast, Japanese encephalitis virus induces stress granules in a PERK-dependent manner to inhibit apoptosis and promote virus replication [133]. In (B,C), each number represents the unique pathway being followed (example: number 1 is one pathway while number 2 represents the other pathway).

Figure 6.
RCD pathways and Stress kinases. (A) M. tuberculosis [167] and A. fumigatus [168] infection induces ER stress-mediated PERK phosphorylation. Double-stranded RNA (dsRNA) phosphorylated PKR [169]. Phosphorylated PERK and PKR can activate inflammasomes leading to pyroptosis [167,168,169]. In contrast, amino acid depletion during inflammation phosphorylates GCN2, which suppresses inflammasome activation, reducing pyroptosis [170]. Additionally, SGs formed in response to several stressors, including arsenite-induced oxidative stress sequester DDX3X and inhibit NLRP3 inflammasome activation [9]; however, whether HRI alone can suppress activation of the NLRP3 inflammasome or other stress kinases are also capable is unclear. (B) Oxidative stress phosphorylates HRI leading to translation arrest and formation of SGs. SGs act as a site for necrosome assembly that activates necroptosis [40]. (C) Selenium deficiency sensitizes cells to ferroptosis [171,172]; uncharged tRNA during selenium deficiency might activate the amino acid sensor GCN2 that possibly modulates the outcome of ferroptosis. Oxidative stress induces MESH1 (Metazoan SpoT Homologue 1), leading to cell death by ferroptosis [173,174]. In addition, MESH1 can inhibit PERK activation [174]; the crosstalk between PERK activation and ferroptosis is yet to be discovered. Cellular iron accumulation activates ferroptosis [175,176]. How stress kinases that sense oxidative stress or heme interconnect cellular stress with ferroptosis is unknown.
Figure 6.
RCD pathways and Stress kinases. (A) M. tuberculosis [167] and A. fumigatus [168] infection induces ER stress-mediated PERK phosphorylation. Double-stranded RNA (dsRNA) phosphorylated PKR [169]. Phosphorylated PERK and PKR can activate inflammasomes leading to pyroptosis [167,168,169]. In contrast, amino acid depletion during inflammation phosphorylates GCN2, which suppresses inflammasome activation, reducing pyroptosis [170]. Additionally, SGs formed in response to several stressors, including arsenite-induced oxidative stress sequester DDX3X and inhibit NLRP3 inflammasome activation [9]; however, whether HRI alone can suppress activation of the NLRP3 inflammasome or other stress kinases are also capable is unclear. (B) Oxidative stress phosphorylates HRI leading to translation arrest and formation of SGs. SGs act as a site for necrosome assembly that activates necroptosis [40]. (C) Selenium deficiency sensitizes cells to ferroptosis [171,172]; uncharged tRNA during selenium deficiency might activate the amino acid sensor GCN2 that possibly modulates the outcome of ferroptosis. Oxidative stress induces MESH1 (Metazoan SpoT Homologue 1), leading to cell death by ferroptosis [173,174]. In addition, MESH1 can inhibit PERK activation [174]; the crosstalk between PERK activation and ferroptosis is yet to be discovered. Cellular iron accumulation activates ferroptosis [175,176]. How stress kinases that sense oxidative stress or heme interconnect cellular stress with ferroptosis is unknown.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lamichhane, P.P.; Samir, P. Cellular Stress: Modulator of Regulated Cell Death. Biology 2023, 12, 1172. https://doi.org/10.3390/biology12091172
AMA Style
Lamichhane PP, Samir P. Cellular Stress: Modulator of Regulated Cell Death. Biology. 2023; 12(9):1172. https://doi.org/10.3390/biology12091172
Chicago/Turabian StyleLamichhane, Prem Prasad, and Parimal Samir. 2023. "Cellular Stress: Modulator of Regulated Cell Death" Biology 12, no. 9: 1172. https://doi.org/10.3390/biology12091172
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.