
Utilizing 5′ UTR Engineering Enables Fine-Tuning of Multiple Genes within Operons to Balance Metabolic Flux in Bacillus subtilis
 ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Strains, Plasmids, and Culture Conditions
2.2. Molecular Biology
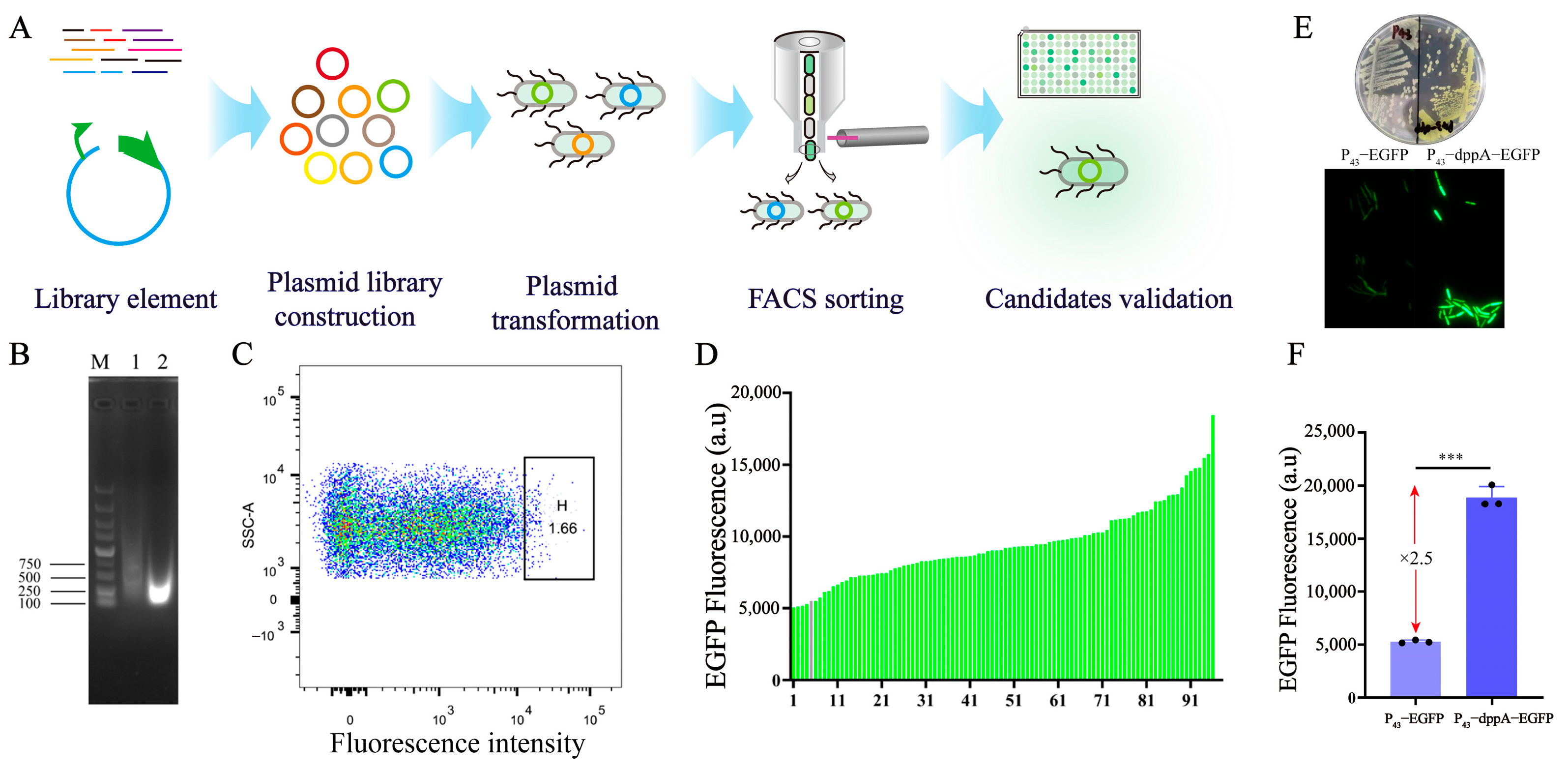
2.3. Screening and Characterization of 5′ UTR
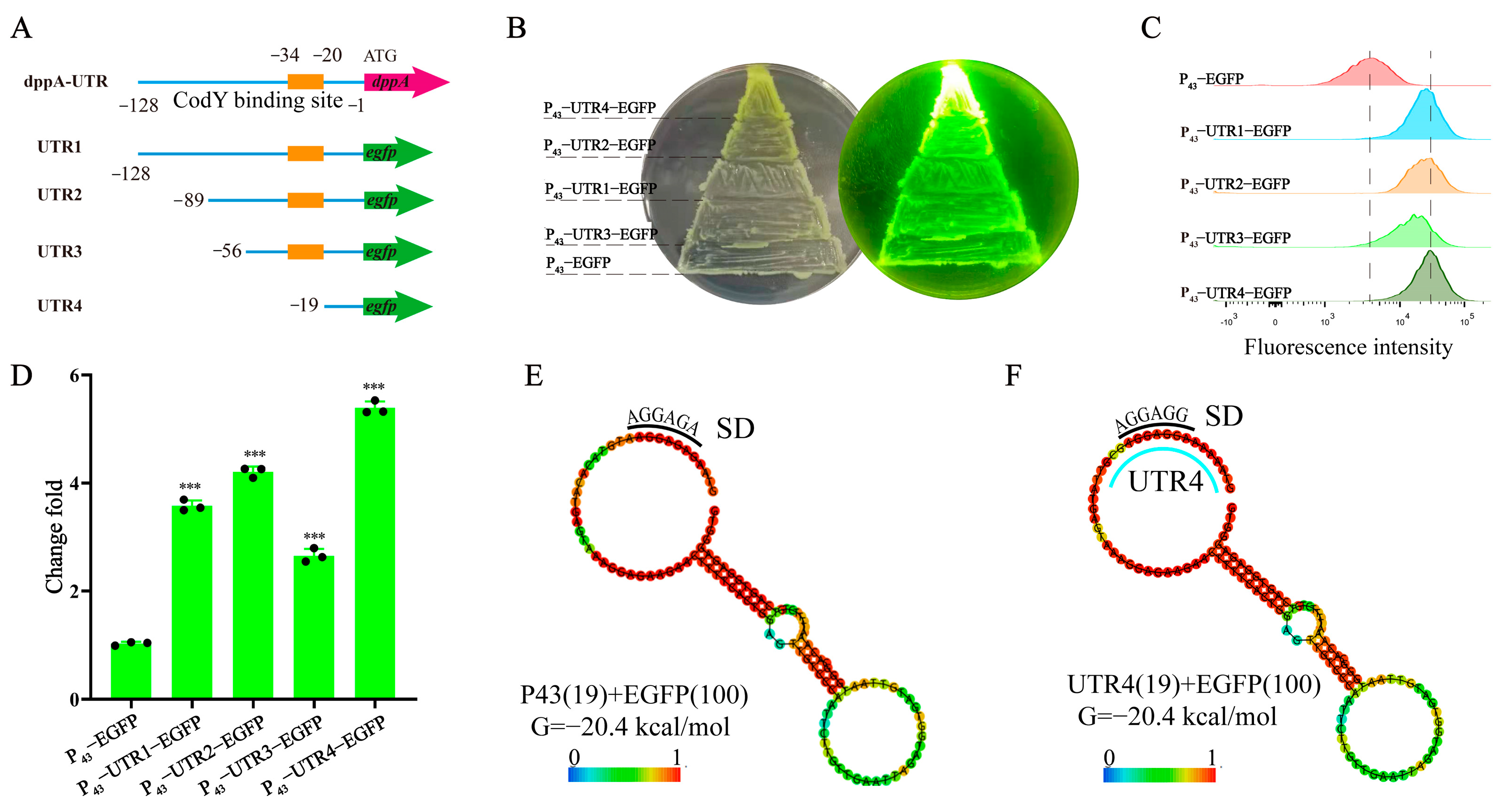
2.4. Identification of Effective 5′ UTR Sequences
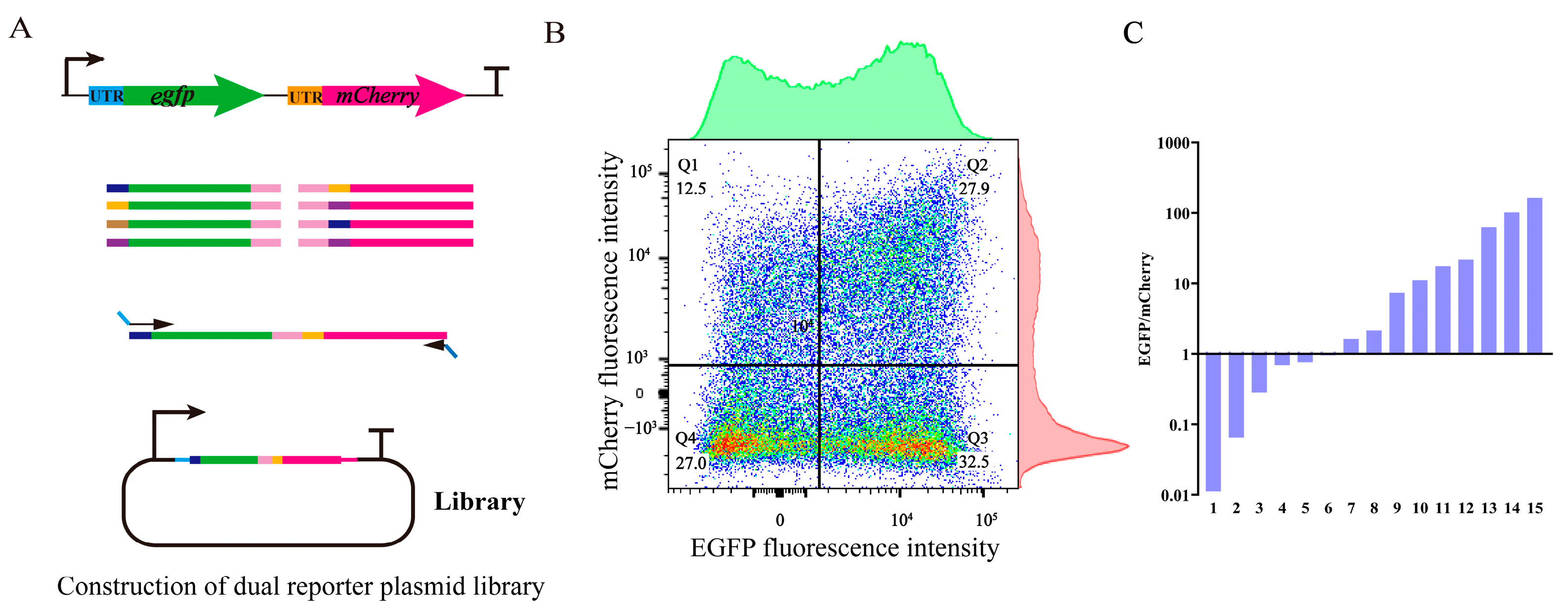
2.5. Construction of 5′ UTR Library
2.6. Flow Cytometer Sorting and Analysis
2.7. Rib Operon Library Construction
2.8. Fluorescence Assays
2.9. Cultivation in Shake Flasks
2.10. Analytical Methods
2.11. Statistical Analysis
3. Results
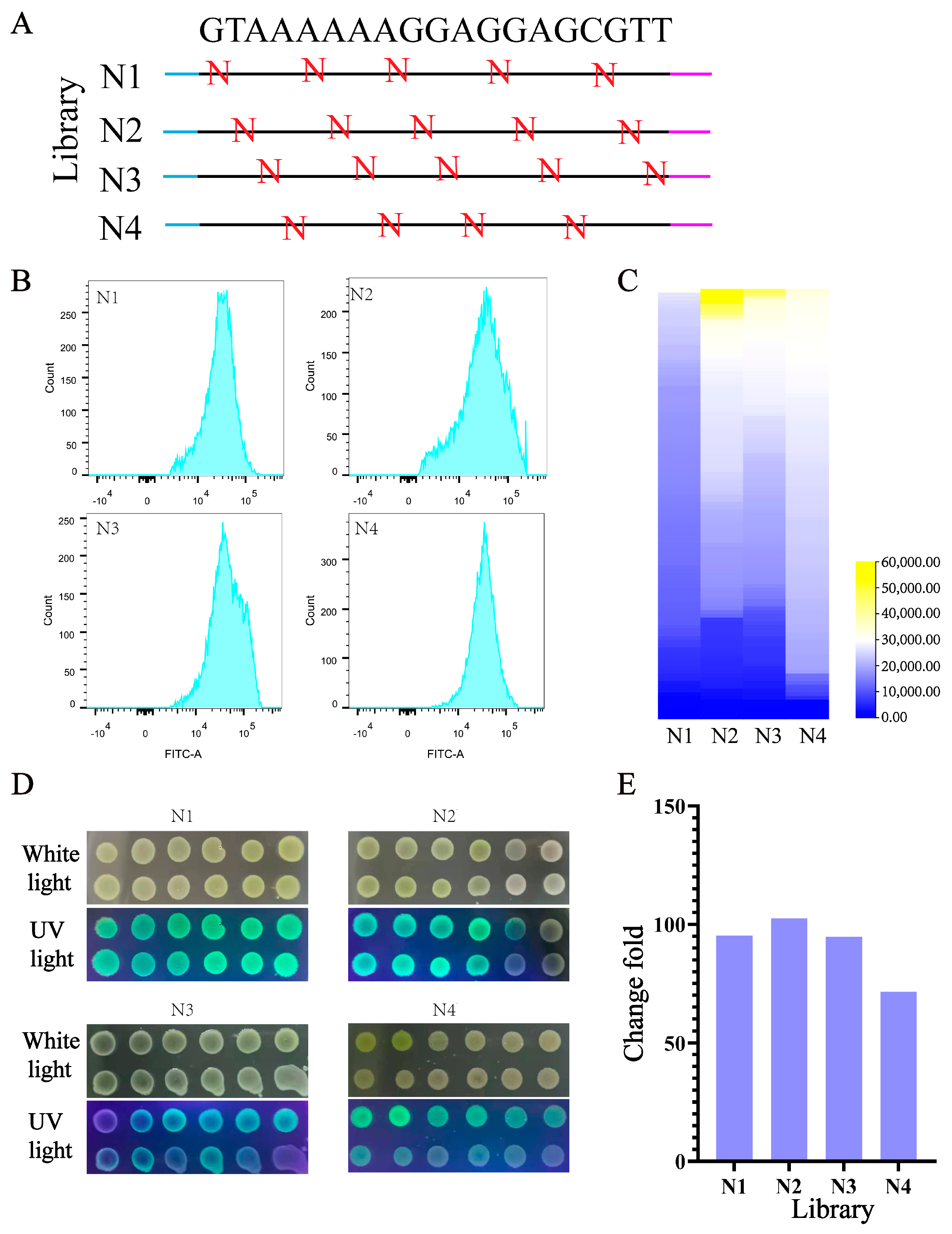
3.1. Screening and Identification of 5′ UTR
3.2. Identification of Effective 5′ UTR Sequences and mRNA Secondary Structure Analysis
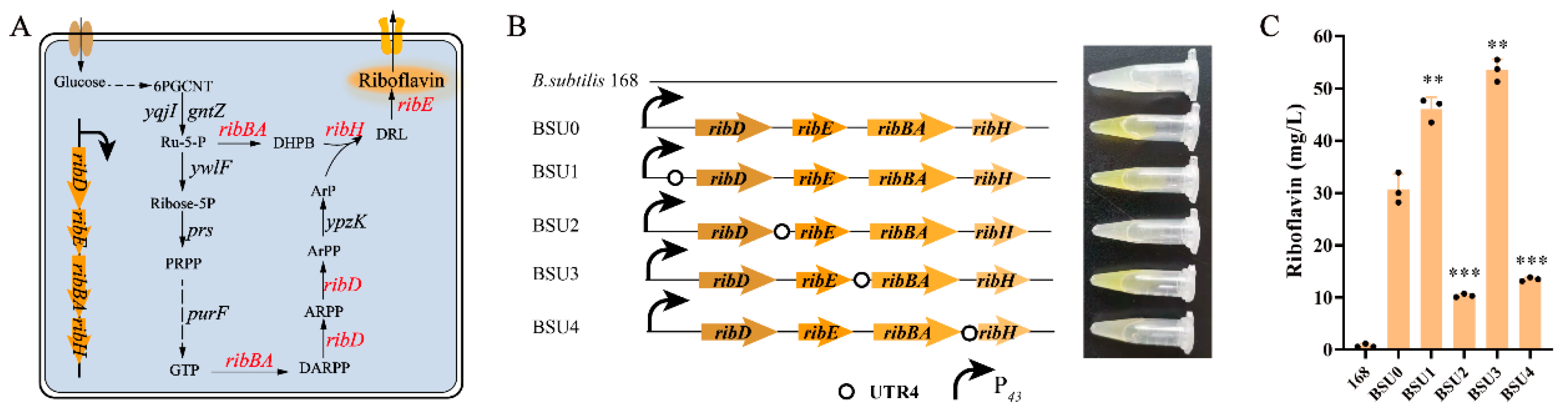
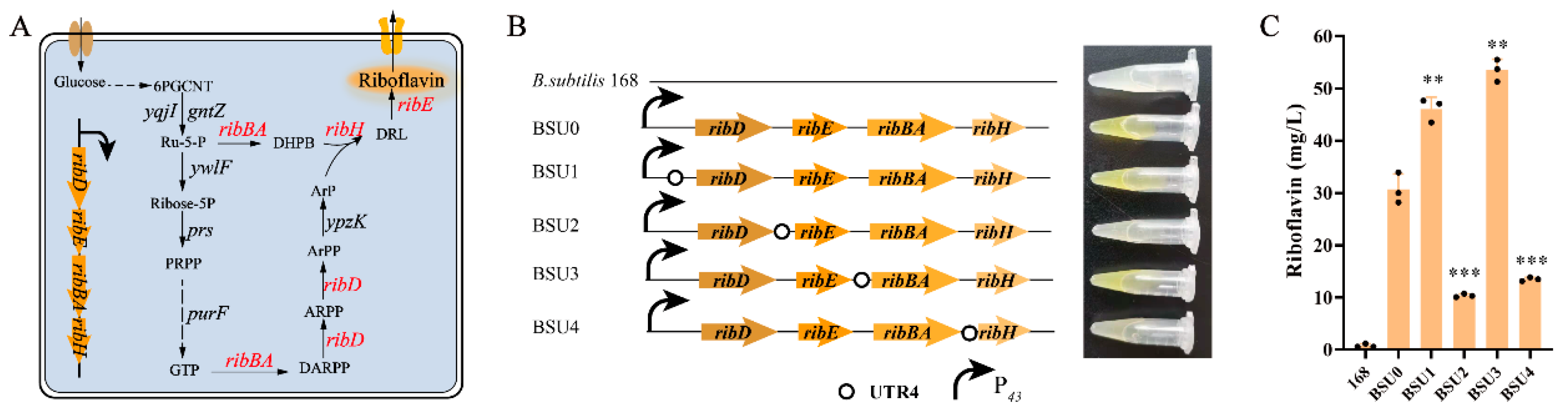
3.3. UTR4 Was Used to Increase rib Operon Gene Expression
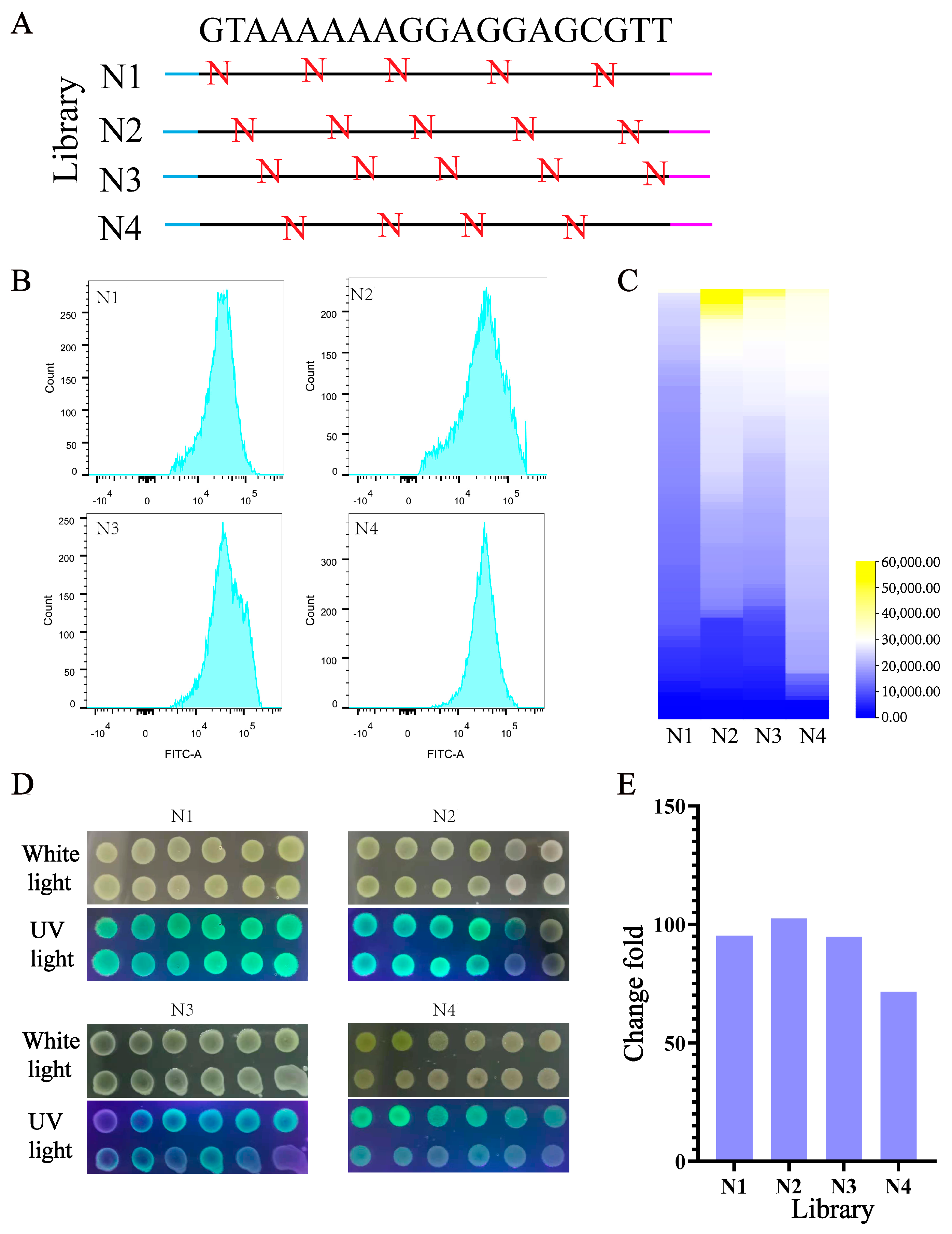
3.4. Design and Construction of UTR4 Library
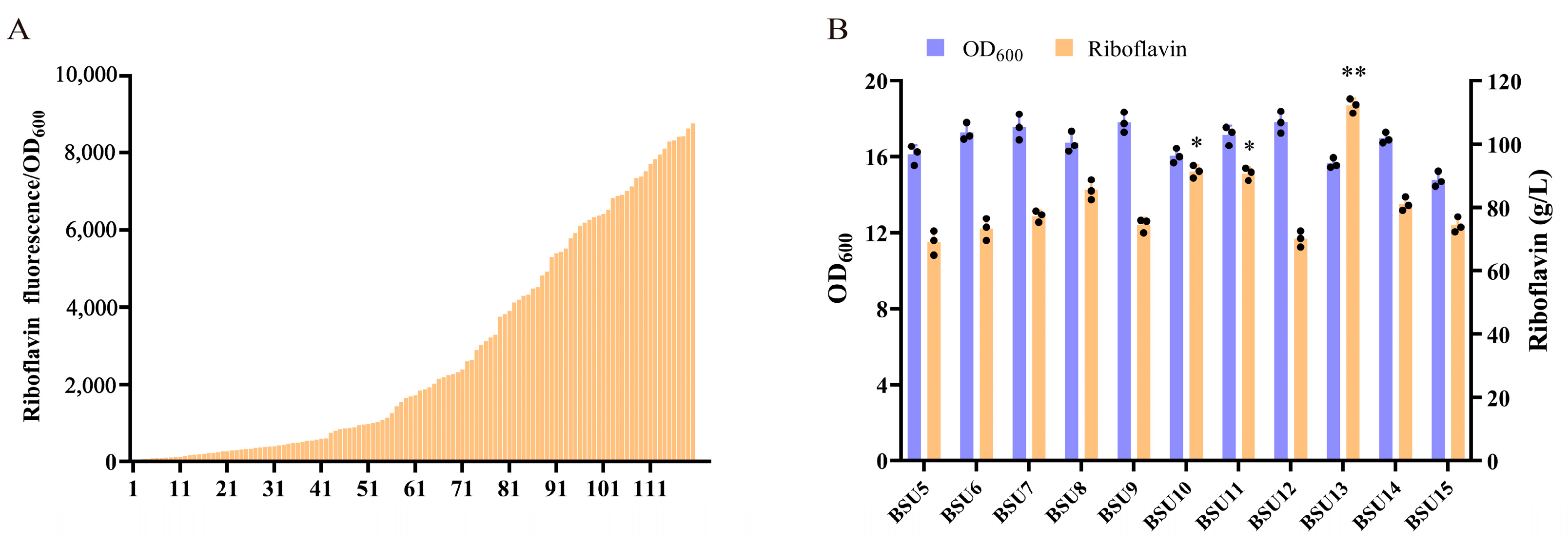
3.5. Tuning the Expression of Multiple Genes in Operons Using the UTR4 Library
3.6. The UTR4 Library Was Utilized to Regulate the Expression of Native rib Operon Genes and Construct Synthetic Operons
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McCarty, N.S.; Ledesma-Amaro, R. Synthetic Biology Tools to Engineer Microbial Communities for Biotechnology. Trends Biotechnol. 2019, 37, 181–197. [Google Scholar] [CrossRef]
- Quax, T.E.; Claassens, N.J.; Söll, D.; van der Oost, J. Codon Bias as a Means to Fine-Tune Gene Expression. Mol. Cell 2015, 59, 149–161. [Google Scholar] [CrossRef]
- Espah Borujeni, A.; Cetnar, D.; Farasat, I.; Smith, A.; Lundgren, N.; Salis, H.M. Precise quantification of translation inhibition by mRNA structures that overlap with the ribosomal footprint in N-terminal coding sequences. Nucleic Acids Res. 2017, 45, 5437–5448. [Google Scholar] [CrossRef] [PubMed]
- Egbert, R.G.; Klavins, E. Fine-tuning gene networks using simple sequence repeats. Proc. Natl. Acad. Sci. USA 2012, 109, 16817–16822. [Google Scholar] [CrossRef]
- Bentele, K.; Saffert, P.; Rauscher, R.; Ignatova, Z.; Blüthgen, N. Efficient translation initiation dictates codon usage at gene start. Mol. Syst. Biol. 2013, 9, 675. [Google Scholar] [CrossRef]
- Mao, Y.; Liu, H.; Liu, Y.; Tao, S. Deciphering the rules by which dynamics of mRNA secondary structure affect translation efficiency in Saccharomyces cerevisiae. Nucleic Acids Res. 2014, 42, 4813–4822. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Jiang, T.; Li, M.; Zou, Y.; Yan, Y. Fine-tuning gene expression for improved biosynthesis of natural products: From transcriptional to post-translational regulation. Biotechnol. Adv. 2022, 54, 107853. [Google Scholar] [CrossRef]
- Liu, X.; Gupta, S.T.P.; Bhimsaria, D.; Reed, J.L.; Rodríguez-Martínez, J.A.; Ansari, A.Z.; Raman, S. De novo design of programmable inducible promoters. Nucleic Acids Res. 2019, 47, 10452–10463. [Google Scholar] [CrossRef] [PubMed]
- Guiziou, S.; Sauveplane, V.; Chang, H.J.; Clerté, C.; Declerck, N.; Jules, M.; Bonnet, J. A part toolbox to tune genetic expression in Bacillus subtilis. Nucleic Acids Res. 2016, 44, 7495–7508. [Google Scholar] [CrossRef]
- Gingold, H.; Pilpel, Y. Determinants of translation efficiency and accuracy. Mol. Syst. Biol. 2011, 7, 481. [Google Scholar] [CrossRef]
- Kudla, G.; Murray, A.W.; Tollervey, D.; Plotkin, J.B. Coding-sequence determinants of gene expression in Escherichia coli. Science 2009, 324, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Peng, B.; Su, Z.; Liu, A.; Hu, Y.; Nomura, C.T.; Chen, S.; Wang, Q. Facilitating Protein Expression with Portable 5′-UTR Secondary Structures in Bacillus licheniformis. ACS Synth. Biol. 2020, 9, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Espah Borujeni, A.; Channarasappa, A.S.; Salis, H.M. Translation rate is controlled by coupled trade-offs between site accessibility, selective RNA unfolding and sliding at upstream standby sites. Nucleic Acids Res. 2014, 42, 2646–2659. [Google Scholar] [CrossRef]
- Braun, F.; Durand, S.; Condon, C. Initiating ribosomes and a 5′/3’-UTR interaction control ribonuclease action to tightly couple B. subtilis hbs mRNA stability with translation. Nucleic Acids Res. 2017, 45, 11386–11400. [Google Scholar] [CrossRef] [PubMed]
- Marzi, S.; Myasnikov, A.G.; Serganov, A.; Ehresmann, C.; Romby, P.; Yusupov, M.; Klaholz, B.P. Structured mRNAs regulate translation initiation by binding to the platform of the ribosome. Cell 2007, 130, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Viegas, S.C.; Apura, P.; Martínez-García, E.; de Lorenzo, V.; Arraiano, C.M. Modulating Heterologous Gene Expression with Portable mRNA-Stabilizing 5′-UTR Sequences. ACS Synth. Biol. 2018, 7, 2177–2188. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Harp, J.R.; Fozo, E.M. The 5′ UTR of the type I toxin ZorO can both inhibit and enhance translation. Nucleic Acids Res. 2017, 45, 4006–4020. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.D.; Kuo, S.T.; Chou, H.D. The diversity of Shine-Dalgarno sequences sheds light on the evolution of translation initiation. RNA Biol. 2021, 18, 1489–1500. [Google Scholar] [CrossRef] [PubMed]
- Simonetti, A.; Marzi, S.; Myasnikov, A.G.; Fabbretti, A.; Yusupov, M.; Gualerzi, C.O.; Klaholz, B.P. Structure of the 30S translation initiation complex. Nature 2008, 455, 416–420. [Google Scholar] [CrossRef]
- Jenner, L.; Romby, P.; Rees, B.; Schulze-Briese, C.; Springer, M.; Ehresmann, C.; Ehresmann, B.; Moras, D.; Yusupova, G.; Yusupov, M. Translational operator of mRNA on the ribosome: How repressor proteins exclude ribosome binding. Science 2005, 308, 120–123. [Google Scholar] [CrossRef]
- Tucker, B.J.; Breaker, R.R. Riboswitches as versatile gene control elements. Curr. Opin. Struct. Biol. 2005, 15, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Salis, H.M. The ribosome binding site calculator. Methods Enzymol. 2011, 498, 19–42. [Google Scholar] [CrossRef] [PubMed]
- Petersen, S.D.; Zhang, J.; Lee, J.S.; Jakociunas, T.; Grav, L.M.; Kildegaard, H.F.; Keasling, J.D.; Jensen, M.K. Modular 5′-UTR hexamers for context-independent tuning of protein expression in eukaryotes. Nucleic Acids Res. 2018, 46, e127. [Google Scholar] [CrossRef] [PubMed]
- Simonetti, A.; Marzi, S.; Jenner, L.; Myasnikov, A.; Romby, P.; Yusupova, G.; Klaholz, B.P.; Yusupov, M. A structural view of translation initiation in bacteria. Cell. Mol. Life Sci. 2009, 66, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Huang, C.; Mao, Y.; Zhou, S.; Deng, Y. Screening and Constructing a Library of Promoter-5′-UTR Complexes with Gradient Strength in Pediococcus acidilactici. ACS Synth. Biol. 2023, 12, 1794–1803. [Google Scholar] [CrossRef] [PubMed]
- Rondthaler, S.N.; Sarker, B.; Howitz, N.; Shah, I.; Andrews, L.B. Toolbox of Characterized Genetic Parts for Staphylococcus aureus. ACS Synth. Biol. 2024, 13, 103–118. [Google Scholar] [CrossRef]
- Abdulmalek, H.W.; Yazgan-Karataş, A. Improvement of Bacilysin Production in Bacillus subtilis by CRISPR/Cas9-Mediated Editing of the 5′-Untranslated Region of the bac Operon. J. Microbiol. Biotechnol. 2023, 33, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Mao, Z.; Guo, J.; Wei, L.; Ma, H.; Tang, Y.; Chen, T.; Wang, Z.; Zhao, X. Construction, Model-Based Analysis, and Characterization of a Promoter Library for Fine-Tuned Gene Expression in Bacillus subtilis. ACS Synth. Biol. 2018, 7, 1785–1797. [Google Scholar] [CrossRef]
- Zhang, X.; Lin, Y.; Wu, Q.; Wang, Y.; Chen, G.Q. Synthetic Biology and Genome-Editing Tools for Improving PHA Metabolic Engineering. Trends Biotechnol. 2020, 38, 689–700. [Google Scholar] [CrossRef]
- Zhang, M.; Song, J.; Xiao, J.; Jin, J.; Nomura, C.T.; Chen, S.; Wang, Q. Engineered multiple translation initiation sites: A novel tool to enhance protein production in Bacillus licheniformis and other industrially relevant bacteria. Nucleic Acids Res. 2022, 50, 11979–11990. [Google Scholar] [CrossRef]
- Kotopka, B.J.; Smolke, C.D. Model-driven generation of artificial yeast promoters. Nat. Commun. 2020, 11, 2113. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Liu, P.; Dai, Z.; Fan, F.; Zhang, X. Fine-tuning the expression of pathway gene in yeast using a regulatory library formed by fusing a synthetic minimal promoter with different Kozak variants. Microb. Cell Fact. 2021, 20, 148. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Zhai, W.; Liu, W.; Zhang, X.; Shi, J.S.; Zhang, X.; Xu, Z. Fine-Tuning Multi-Gene Clusters via Well-Characterized Gene Expression Regulatory Elements: Case Study of the Arginine Synthesis Pathway in C. glutamicum. ACS Synth. Biol. 2021, 10, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Tong, Y.; Li, Y.; Tao, J.; Rao, S.; Li, J.; Zhou, J.; Liu, S. Autoinduction Expression Modules for Regulating Gene Expression in Bacillus subtilis. ACS Synth. Biol. 2022, 11, 4220–4225. [Google Scholar] [CrossRef] [PubMed]
- Bareia, T.; Pollak, S.; Eldar, A. Self-sensing in Bacillus subtilis quorum-sensing systems. Nat. Microbiol. 2018, 3, 83–89. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Du, Y.; Pan, X.; Zhang, X.; Yang, T.; Rao, Z. Increased Production of Riboflavin by Coordinated Expression of Multiple Genes in Operons in Bacillus subtilis. ACS Synth. Biol. 2022, 11, 1801–1810. [Google Scholar] [CrossRef] [PubMed]
- Man, Z.W.; Rao, Z.M.; Cheng, Y.P.; Yang, T.W.; Zhang, X.; Xu, M.J.; Xu, Z.H. Enhanced riboflavin production by recombinant Bacillus subtilis RF1 through the optimization of agitation speed. World J. Microbiol. Biotechnol. 2014, 30, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Ingolia, N.T. Ribosome profiling: New views of translation, from single codons to genome scale. Nat. Rev. Genet. 2014, 15, 205–213. [Google Scholar] [CrossRef]
- Remaut, H.; Bompard-Gilles, C.; Goffin, C.; Frère, J.M.; Van Beeumen, J. Structure of the Bacillus subtilis D-aminopeptidase DppA reveals a novel self-compartmentalizing protease. Nat. Struct. Biol. 2001, 8, 674–678. [Google Scholar] [CrossRef]
- Belitsky, B.R.; Sonenshein, A.L. Genetic and biochemical analysis of CodY-binding sites in Bacillus subtilis. J. Bacteriol. 2008, 190, 1224–1236. [Google Scholar] [CrossRef]
- Rao, Y.; Li, P.; Xie, X.; Li, J.; Liao, Y.; Ma, X.; Cai, D.; Chen, S. Construction and Characterization of a Gradient Strength Promoter Library for Fine-Tuned Gene Expression in Bacillus licheniformis. ACS Synth. Biol. 2021, 10, 2331–2339. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lee, G.; Jeon, E.; Sohn, E.J.; Lee, Y.; Kang, H.; Lee, D.W.; Kim, D.H.; Hwang, I. The immediate upstream region of the 5′-UTR from the AUG start codon has a pronounced effect on the translational efficiency in Arabidopsis thaliana. Nucleic Acids Res. 2014, 42, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Hümbelin, M.; Griesser, V.; Keller, T.; Schurter, W.; Haiker, M.; Hohmann, H.P.; Ritz, H.; Richter, G.; Bacher, A.; van Loon, A.P.G.M. GTP cyclohydrolase II and 3,4-dihydroxy-2-butanone 4-phosphate synthase are rate-limiting enzymes in riboflavin synthesis of an industrial Bacillus subtilis strain used for riboflavin production. J. Ind. Microbiol. Biotechnol. 1999, 22, 1–7. [Google Scholar] [CrossRef]
- Jia, L.; Mao, Y.; Ji, Q.; Dersh, D.; Yewdell, J.W.; Qian, S.B. Decoding mRNA translatability and stability from the 5′ UTR. Nat. Struct. Mol. Biol. 2020, 27, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Pfleger, B.F.; Pitera, D.J.; Smolke, C.D.; Keasling, J.D. Combinatorial engineering of intergenic regions in operons tunes expression of multiple genes. Nat. Biotechnol. 2006, 24, 1027–1032. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, M.; Lu, Y.; Wu, N.; Chen, J.; Ji, Z.; Zhan, Y.; Ma, X.; Chen, J.; Cai, D.; et al. Metabolic engineering of Bacillus amyloliquefaciens for efficient production of α-glucosidase inhibitor1-deoxynojirimycin. Synth. Syst. Biotechnol. 2023, 8, 378–385. [Google Scholar] [CrossRef]
- Jin, L.Q.; Jin, W.R.; Ma, Z.C.; Shen, Q.; Cai, X.; Liu, Z.Q.; Zheng, Y.G. Promoter engineering strategies for the overproduction of valuable metabolites in microbes. Appl. Microbiol. Biotechnol. 2019, 103, 8725–8736. [Google Scholar] [CrossRef]
- Ryczek, N.; Łyś, A.; Makałowska, I. The Functional Meaning of 5′UTR in Protein-Coding Genes. Int. J. Mol. Sci. 2023, 24, 2976. [Google Scholar] [CrossRef]
- Zhou, S.; Ding, R.; Chen, J.; Du, G.; Li, H.; Zhou, J. Obtaining a Panel of Cascade Promoter-5′-UTR Complexes in Escherichia coli. ACS Synth. Biol. 2017, 6, 1065–1075. [Google Scholar] [CrossRef]
- Kucharova, V.; Skancke, J.; Brautaset, T.; Valla, S. Design and optimization of short DNA sequences that can be used as 5′ fusion partners for high-level expression of heterologous genes in Escherichia coli. Appl. Environ. Microbiol. 2013, 79, 6655–6664. [Google Scholar] [CrossRef]
- Li, J.; Liang, Q.; Song, W.; Marchisio, M.A. Nucleotides upstream of the Kozak sequence strongly influence gene expression in the yeast S. cerevisiae. J. Biol. Eng. 2017, 11, 25. [Google Scholar] [CrossRef]
- Kitamura, S.; Shimizu, H.; Toya, Y. Identification of a rate-limiting step in a metabolic pathway using the kinetic model and in vitro experiment. J. Biosci. Bioeng. 2021, 131, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Wang, Y.; Wang, Z.; Wang, G.; Liu, D.; Fu, J.; Chen, T.; Zhao, X. Deregulation of purine pathway in Bacillus subtilis and its use in riboflavin biosynthesis. Microb. Cell Fact. 2014, 13, 101. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains/Plasmids | Characteristics | Source |
|---|---|---|
| Strains | ||
| B. subtilis 168 | trpC2 | This lab |
| E. coli JM109 | The cloning host, recA1, endA1, thi, gyrA96, supE44, hsdR17∆ (lac-proAB)/F′ [traD36, proAB+, lacIq, lacZ∆ M15] | This lab |
| BSU0 | B. subtilis 168 derivate, P43-rib | This study |
| BSU1 | BSU0 derivate, ribD under the control of UTR4 via the plasmid pP43NMK | This study |
| BSU2 | BSU0 derivate, ribE under the control of UTR4 via the plasmid pP43NMK | This study |
| BSU3 | BSU0 derivate, ribBA under the control of UTR4 via the plasmid pP43NMK | This study |
| BSU5 | BSU0 derivate, each gene on the rib operon under the control of UTR4 via the plasmid pP43NMK | This study |
| BSU6 | B. subtilis 168 derivate, rib synthesis operator controlled by UTR4 library | This study |
| BSU7 | B. subtilis 168 derivate, rib synthesis operator controlled by UTR4 library | This study |
| BSU8 | B. subtilis 168 derivate, rib synthesis operator controlled by UTR4 library | This study |
| BSU9 | B. subtilis 168 derivate, rib synthesis operator controlled by UTR4 library | This study |
| BSU10 | B. subtilis 168 derivate, rib synthesis operator controlled by UTR4 library | This study |
| BSU11 | B. subtilis 168 derivate, rib synthesis operator controlled by UTR4 library | This study |
| BSU12 | B. subtilis 168 derivate, rib synthesis operator controlled by UTR4 library | This study |
| BSU13 | B. subtilis 168 derivate, rib synthesis operator controlled by UTR4 library | This study |
| BSU14 | B. subtilis 168 derivate, rib synthesis operator controlled by UTR4 library | This study |
| BSU15 | B. subtilis 168 derivate, rib synthesis operator controlled by UTR4 library | This study |
| Plasmids | ||
| pP43NMK | Ampr, Kanr, E. coli-B. subtilis shuttle vector | This lab |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
You, J.; Wang, Y.; Wang, K.; Du, Y.; Zhang, X.; Zhang, X.; Yang, T.; Pan, X.; Rao, Z. Utilizing 5′ UTR Engineering Enables Fine-Tuning of Multiple Genes within Operons to Balance Metabolic Flux in Bacillus subtilis. Biology 2024, 13, 277. https://doi.org/10.3390/biology13040277
You J, Wang Y, Wang K, Du Y, Zhang X, Zhang X, Yang T, Pan X, Rao Z. Utilizing 5′ UTR Engineering Enables Fine-Tuning of Multiple Genes within Operons to Balance Metabolic Flux in Bacillus subtilis. Biology. 2024; 13(4):277. https://doi.org/10.3390/biology13040277
Chicago/Turabian StyleYou, Jiajia, Yifan Wang, Kang Wang, Yuxuan Du, Xiaoling Zhang, Xian Zhang, Taowei Yang, Xuewei Pan, and Zhiming Rao. 2024. "Utilizing 5′ UTR Engineering Enables Fine-Tuning of Multiple Genes within Operons to Balance Metabolic Flux in Bacillus subtilis" Biology 13, no. 4: 277. https://doi.org/10.3390/biology13040277