
Modulation of Group I Ribozyme Activity by Cationic Porphyrins

 and
and 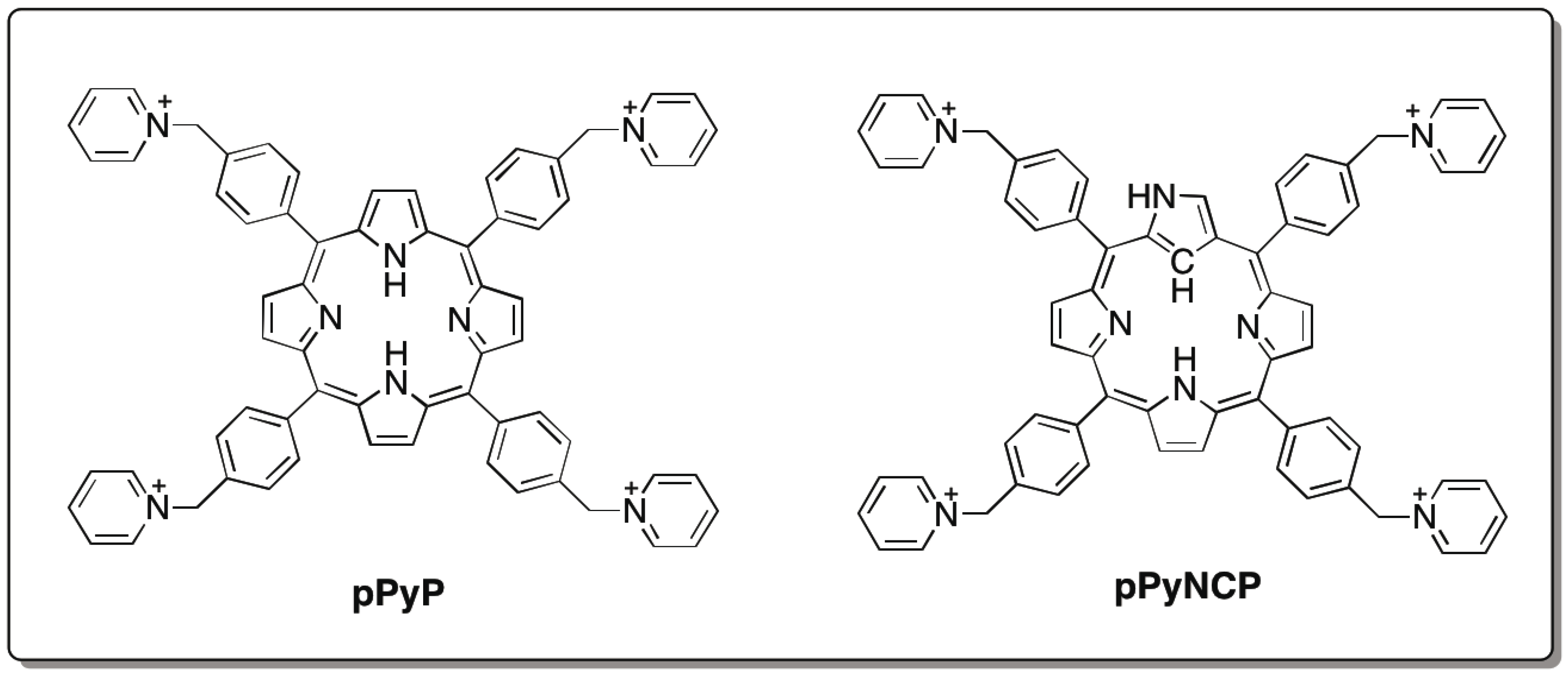{kind=link}
{kind=link}
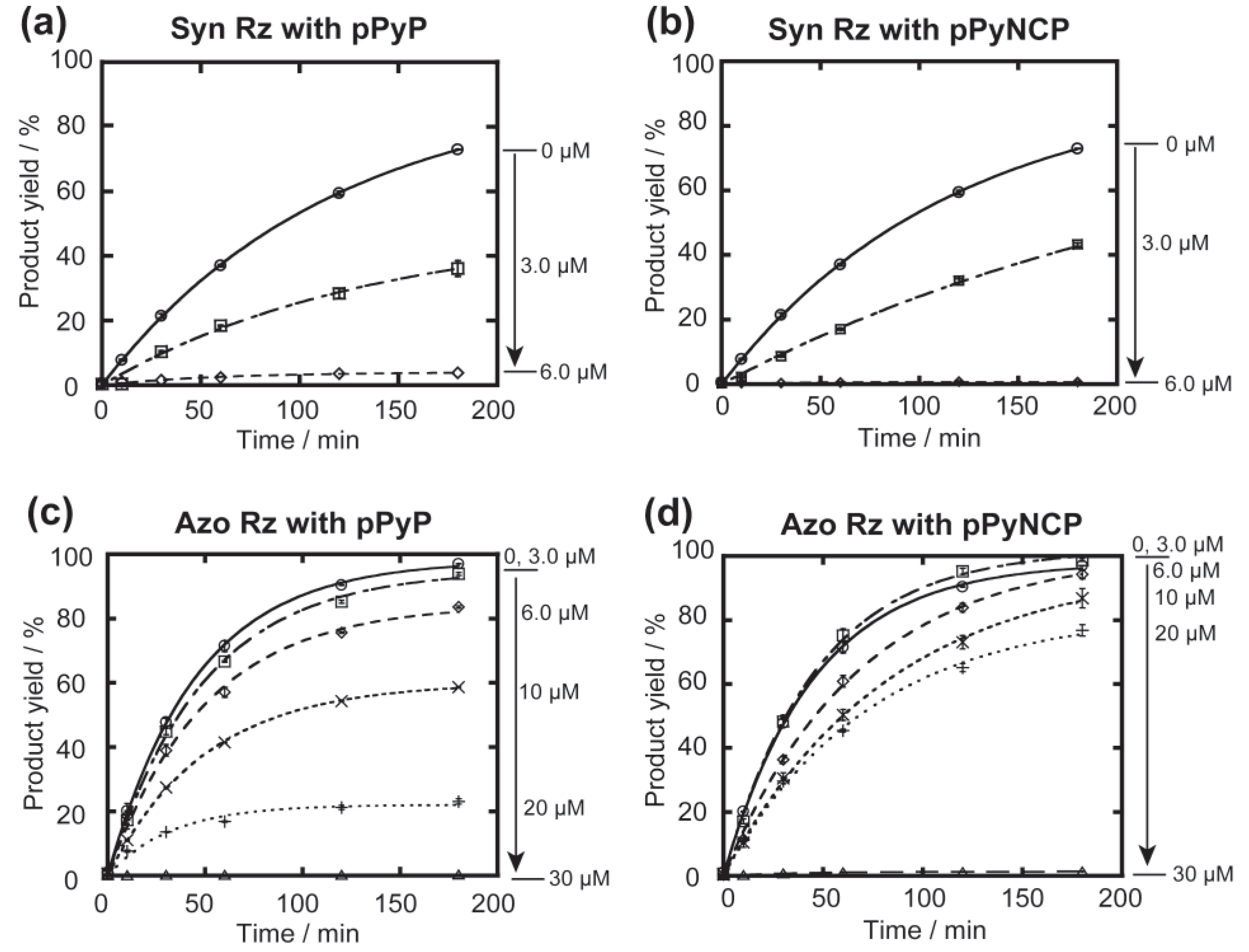{kind=link}
{kind=link}
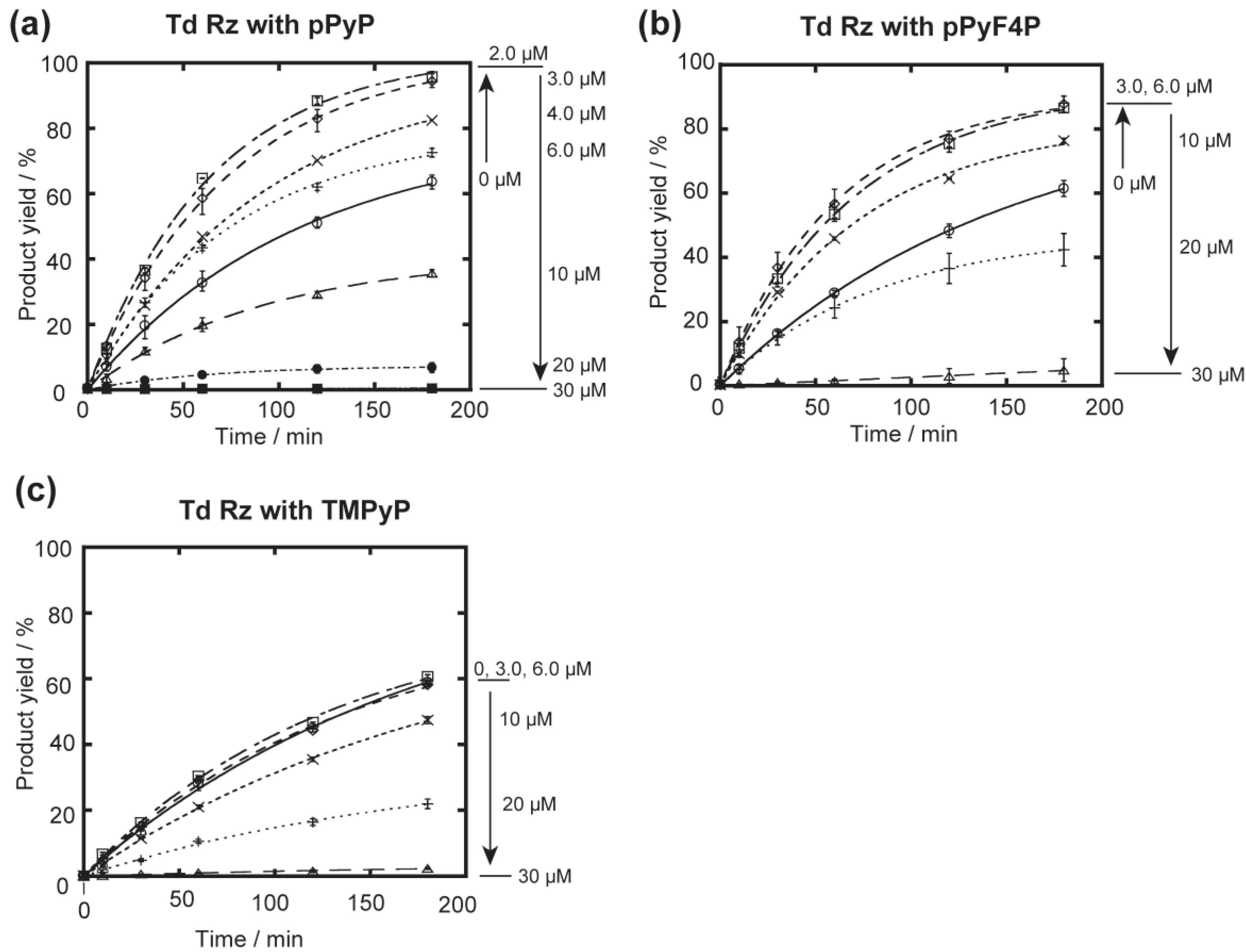{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Oligonucleotides
2.2. Porphyrin Compounds
2.3. Preparation of Ribozymes
2.4. GTP-Dependent Cleavage Reactions Catalyzed by the Group I Ribozymes
3. Results

3.1. Effects of Cationic Porphyrins on Group IC3 Ribozymes


3.2. Effects of the Cationic Porphyrin on the Group IC1 Ribozyme



4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Breaker, R.R. Prospects for riboswitch discovery and analysis. Mol. Cell 2011, 43, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Bastet, L.; Dubé, A.; Massé, E.; Lafontaine, D.A. New insights into riboswitch regulation mechanisms. Mol. Microbiol. 2011, 80, 1148–1154. [Google Scholar] [CrossRef] [PubMed]
- Tenson, T.; Mankin, A. Antibiotics and the ribosome. Mol. Microbiol. 2006, 59, 1664–1677. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.N. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat. Rev. Microbiol. 2014, 12, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Boer, D.R.; Canals, A.; Coll, M. DNA-binding drugs caught in action: The latest 3D pictures of drug-DNA complexes. Dalton Trans. 2009, 399–414. [Google Scholar] [CrossRef]
- Ali, A.; Bhattacharya, S. DNA binders in clinical trials and chemotherapy. Bioorg. Med. Chem. 2014, 22, 4506–4521. [Google Scholar] [CrossRef] [PubMed]
- Waring, M.J. Complex formation between ethidium bromide and nucleic acids. J. Mol. Biol. 1965, 13, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.C.; Jain, S.C.; Sobell, H.M. X-ray crystallographic visualization of drug-nucleic acid intercalative binding: Structure of an ethidium-dinucleoside monophosphate crystalline complex, Ethidium: 5-iodouridylyl (3'–5') adenosine. Proc. Natl. Acad. Sci. USA 1975, 72, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Fiel, R.J.; Howard, J.C.; Mark, E.H.; Datta Gupta, N. Interaction of DNA with a porphyrin ligand: Evidence for intercalation. Nucleic Acids Res. 1979, 6, 3093–3118. [Google Scholar] [CrossRef] [PubMed]
- Fiel, R.J. Porphyrin-nucleic acid interactions: A review. J. Biomol. Struct. Dyn. 1989, 6, 1259–1274. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.C.; Ulven, T. Macrocyclic G-quadruplex ligands. Curr. Med. Chem. 2010, 17, 3438–3448. [Google Scholar] [CrossRef] [PubMed]
- Romera, C.; Bombarde, O.; Bonnet, R.; Gomez, D.; Dumy, P.; Calsou, P.; Gwan, J.F.; Lin, J.H.; Defrancq, E.; Pratviel, G. Improvement of porphyrins for G-quadruplex DNA targeting. Biochimie 2011, 93, 1310–1317. [Google Scholar] [CrossRef] [PubMed]
- Rowland, G.B.; Barnett, K.; Dupont, J.I.; Akurathi, G.; Le, V.H.; Lewis, E.A. The effect of pyridyl substituents on the thermodynamics of porphyrin binding to G-quadruplex DNA. Bioorg. Med. Chem. 2013, 21, 7515–7522. [Google Scholar] [CrossRef] [PubMed]
- Ofer, N.; Weisman-Shomer, P.; Shklover, J.; Fry, M. The quadruplex r(CGG)n destabilizing cationic porphyrin TMPyP4 cooperates with hnRNPs to increase the translation efficiency of fragile X premutation mRNA. Nucleic Acids Res. 2009, 37, 2712–2722. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.J.; Wingate, K.L.; Silwal, J.; Leeper, T.C.; Basu, S. The porphyrin TmPyP4 unfolds the extremely stable G-quadruplex in MT3-MMP mRNA and alleviates its repressive effect to enhance translation in eukaryotic cells. Nucleic Acids Res. 2012, 40, 4137–4145. [Google Scholar] [CrossRef] [PubMed]
- Zamiri, B.; Reddy, K.; Macgregor, R.B., Jr.; Pearson, C.E. TMPyP4 porphyrin distorts RNA G-quadruplex structures of the disease-associated r(GGGGCC)n repeat of the C9orf72 gene and blocks interaction of RNA-binding proteins. J. Biol. Chem. 2014, 289, 4653–4659. [Google Scholar] [CrossRef] [PubMed]
- Ghazaryan, A.A.; Dalyan, Y.B.; Haroutiunian, S.G.; Vardanyan, V.I.; Ghazaryan, R.K.; Chalikian, T.V. Thermodynamics of interactions of TAlPyP4 and AgTAlPyP4 porphyrins with poly(rA)poly(rU) and poly(rI)poly(rC) duplexes. J. Biomol. Struct. Dyn. 2006, 24, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Ghazaryan, A.A.; Dalyan, Y.B.; Haroutiunian, S.G.; Tikhomirova, A.; Taulier, N.; Wells, J.W.; Chalikian, T.V. Thermodynamics of interactions of water-soluble porphyrins with RNA duplexes. J. Am. Chem. Soc. 2006, 128, 1914–1921. [Google Scholar] [CrossRef] [PubMed]
- Dalyan, Y.; Vardanyan, I.; Chavushyan, A.; Balayan, G. Peculiarities of interaction of porphyrins with tRNA at low ionic strength. J. Biomol. Struct. Dyn. 2010, 28, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Birdsall, W.J.; Anderson, W.R., Jr.; Foster, N. Studies of interactions of porphyrins with transfer RNA by high-resolution NMR. Biochim. Biophys. Acta 1989, 1007, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.; Rogert, M.C.; Tanaka, T.; Kikuchi, Y.; Bichenkova, E.V.; Wilton, A.N.; Gbaj, A.; Douglas, K.T. Porphyrins and porphines bind strongly and specifically to tRNA, precursor tRNA and to M1 RNA and inhibit the ribonuclease P ribozyme reaction. Biochim. Biophys. Acta 2005, 1730, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Celander, D.W.; Nussbaum, J.M. Efficient modification of RNA by porphyrin cation photochemistry: Monitoring the folding of coaxially stacked RNA helices in tRNA(Phe) and the human immunodeficiency virus type 1 rev response element RNA. Biochemistry 1996, 35, 12061–12069. [Google Scholar] [CrossRef] [PubMed]
- Ikawa, Y.; Moriyama, S.; Harada, H.; Furuta, H. Acid-base properties and DNA-binding of water soluble N-confused porphyrins with cationic side-arms. Org. Biomol. Chem. 2008, 6, 4157–4166. [Google Scholar] [CrossRef] [PubMed]
- Ikawa, Y.; Katsumata, S.; Sakashita, R.; Furuta, H. Spectrometric detection of DNA by Bis-Zn2+ complex of a water-soluble doubly N-confused hexaphyrin. Chem. Lett. 2014, 43, 1929–1931. [Google Scholar] [CrossRef]
- Williamson, C.L.; Desai, N.M.; Burke, J.M. Compensatory mutations demonstrate that P8 and P6 are RNA secondary structure elements important for processing of a group I intron. Nucleic Acids Res. 1989, 17, 675–689. [Google Scholar] [CrossRef] [PubMed]
- Heuer, T.S.; Chandry, P.S.; Belfort, M.; Celander, D.W.; Cech, T.R. Folding of group I introns from bacteriophage T4 involves internalization of the catalytic core. Proc. Natl. Acad. Sci. USA 1991, 88, 11105–11109. [Google Scholar] [CrossRef] [PubMed]
- Ikawa, Y.; Naito, D.; Shiraishi, H.; Inoue, T. Structure-function relationships of two closely related group IC3 intron ribozymes from Azoarcus and Synechococcus pre-tRNA. Nucleic Acids Res. 2000, 28, 3269–3277. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Uno, T.; Ishikawa, Y. Stabilization of guanine quadruplex DNA by the binding of porphyrins with cationic side arms. Bioorg. Med. Chem. 2005, 13, 2423–2430. [Google Scholar] [CrossRef] [PubMed]
- Woodson, S.A. Structure and assembly of group I introns. Curr. Opin. Struct. Biol. 2005, 15, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Vicens, Q.; Cech, T.R. Atomic level architecture of group I introns revealed. Trends Biochem. Sci. 2006, 31, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Cech, T.R. Self-splicing of group I introns. Annu. Rev. Biochem. 1990, 59, 543–568. [Google Scholar] [CrossRef] [PubMed]
- Zaug, A.J.; Been, M.D.; Cech, T.R. The Tetrahymena ribozyme acts like an RNA restriction endonuclease. Nature 1986, 324, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Zaug, A.J.; Dávila-Aponte, J.A.; Cech, T.R. Catalysis of RNA cleavage by a ribozyme derived from the group I intron of Anabaena pre-tRNA(Leu). Biochemistry 1994, 33, 14935–14947. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.Y.; Davidson, L.A.; Pico, S. Characterization of the Azoarcus ribozyme: Tight binding to guanosine and substrate by an unusually small group I ribozyme. Biochim. Biophys. Acta 1999, 1489, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Pichler, A.; Schroeder, R. Folding problems of the 5' splice site containing the P1 stem of the group I thymidylate synthase intron: Substrate binding inhibition in vitro and mis-splicing in vivo. J. Biol. Chem. 2002, 277, 17987–17993. [Google Scholar] [CrossRef] [PubMed]
- Ikawa, Y. Effects of a cationic N-confused porphyrin on the GTP-dependent cleavage reactions catalyzed by the Td ribozyme. University of Toyama: Toyama, Japan, Unpublished data; 2014. [Google Scholar]
- Rezler, E.M.; Seenisamy, J.; Bashyam, S.; Kim, M.Y.; White, E.; Wilson, W.D.; Hurley, L.H. Telomestatin and diseleno sapphyrin bind selectively to two different forms of the human telomeric G-quadruplex structure. J. Am. Chem. Soc. 2005, 127, 9439–9447. [Google Scholar] [CrossRef] [PubMed]
- Conn, M.M.; Prudent, J.R.; Schultz, P.G. Porphyrin metalation catalyzed by a small RNA molecule. J. Am. Chem. Soc. 1996, 118, 7012–7013. [Google Scholar] [CrossRef]
- Travascio, P.; Bennet, A.J.; Wang, D.Y.; Sen, D. A ribozyme and a catalytic DNA with peroxidase activity: Active sites versus cofactor-binding sites. Chem. Biol. 1999, 6, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Kagahara, T.; Abe, H.; Ito, Y. In vitro selection of hemin-binding catalytic RNA. Bioorg. Med. Chem. Lett. 2009, 19, 1484–1487. [Google Scholar] [CrossRef] [PubMed]
- Poon, L.C.; Methot, S.P.; Morabi-Pazooki, W.; Pio, F.; Bennet, A.J.; Sen, D. Guanine-rich RNAs and DNAs that bind heme robustly catalyze oxygen transfer reactions. J. Am. Chem. Soc. 2011, 133, 1877–1884. [Google Scholar] [CrossRef] [PubMed]
- Schön, A.; Krupp, G.; Gough, S.; Berry-Lowe, S.; Kannangara, C.G.; Söll, D. The RNA required in the first step of chlorophyll biosynthesis is a chloroplast glutamate tRNA. Nature 1986, 322, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Benner, S.A.; Ellington, A.D.; Tauer, A. Modern metabolism as a palimpsest of the RNA world. Proc. Natl. Acad. Sci. USA 1989, 86, 7054–7058. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsumura, S.; Ito, T.; Tanaka, T.; Furuta, H.; Ikawa, Y. Modulation of Group I Ribozyme Activity by Cationic Porphyrins. Biology 2015, 4, 251-263. https://doi.org/10.3390/biology4020251
Matsumura S, Ito T, Tanaka T, Furuta H, Ikawa Y. Modulation of Group I Ribozyme Activity by Cationic Porphyrins. Biology. 2015; 4(2):251-263. https://doi.org/10.3390/biology4020251
Chicago/Turabian StyleMatsumura, Shigeyoshi, Tatsunobu Ito, Takahiro Tanaka, Hiroyuki Furuta, and Yoshiya Ikawa. 2015. "Modulation of Group I Ribozyme Activity by Cationic Porphyrins" Biology 4, no. 2: 251-263. https://doi.org/10.3390/biology4020251