Tinkering and the Origins of Heritable Anatomical Variation in Vertebrates

Abstract
:1. Introduction
2. Anatomical Variation in Vertebrates
2.1. Variation within Crossbreeding Populations
2.2. Variation Across Non-Breeding but Related Vertebrate Groups
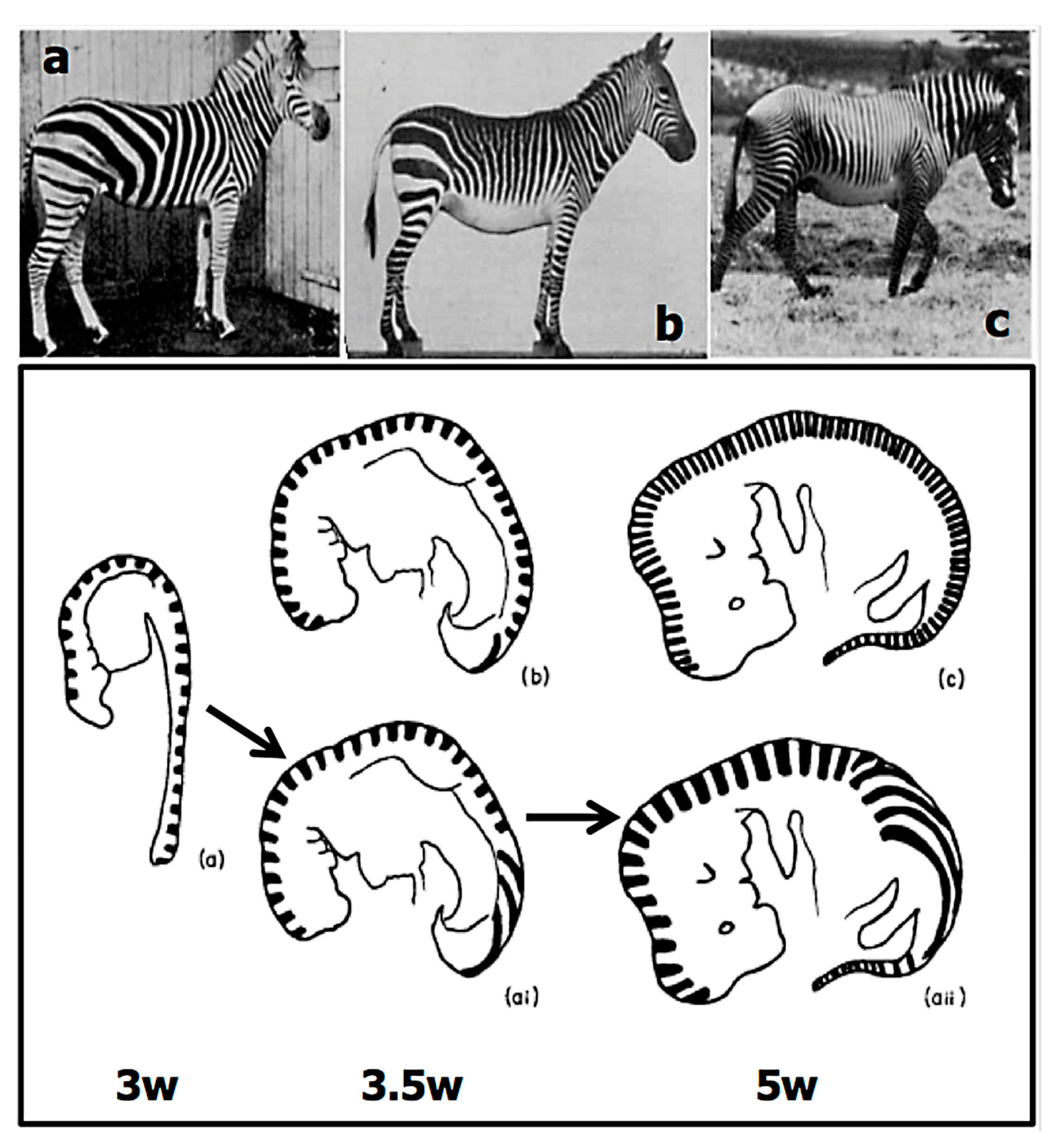
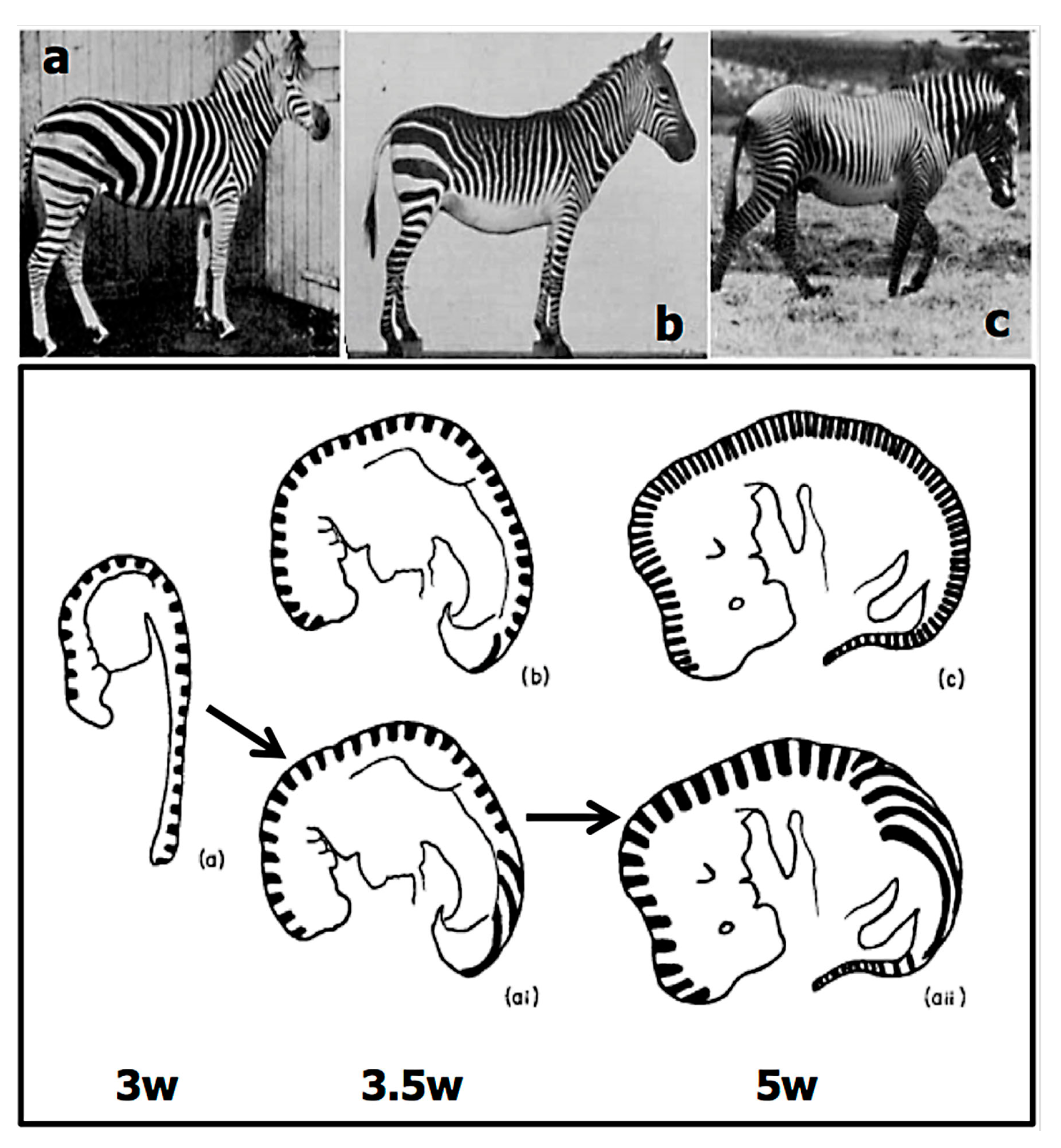
2.2.1. Size
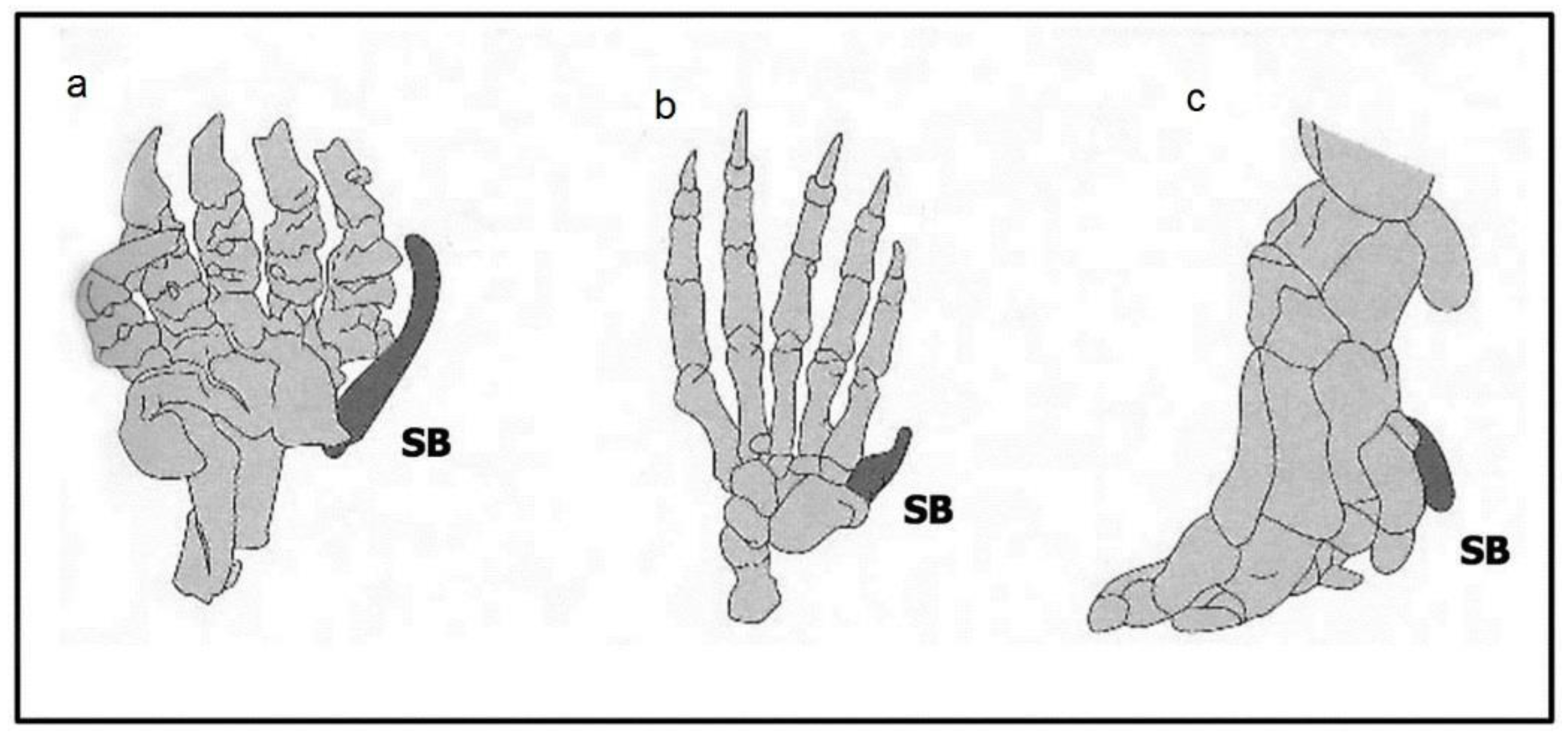
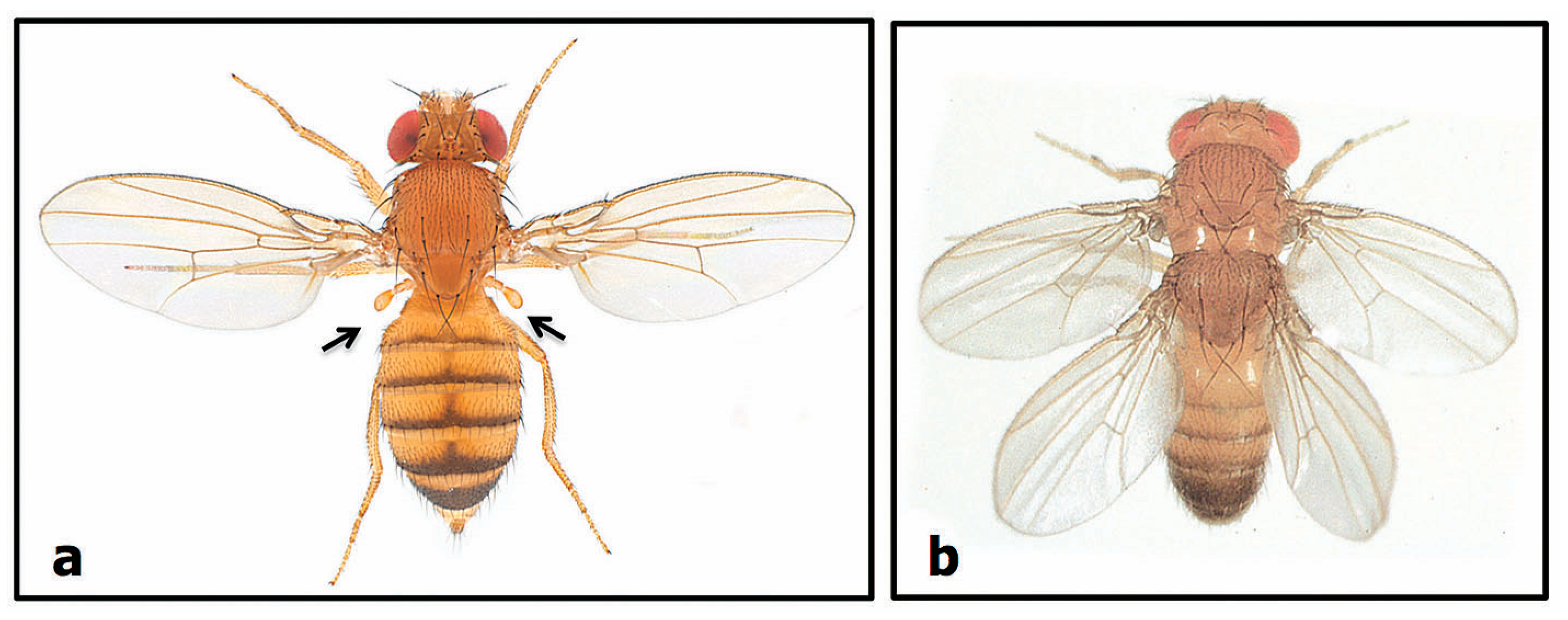
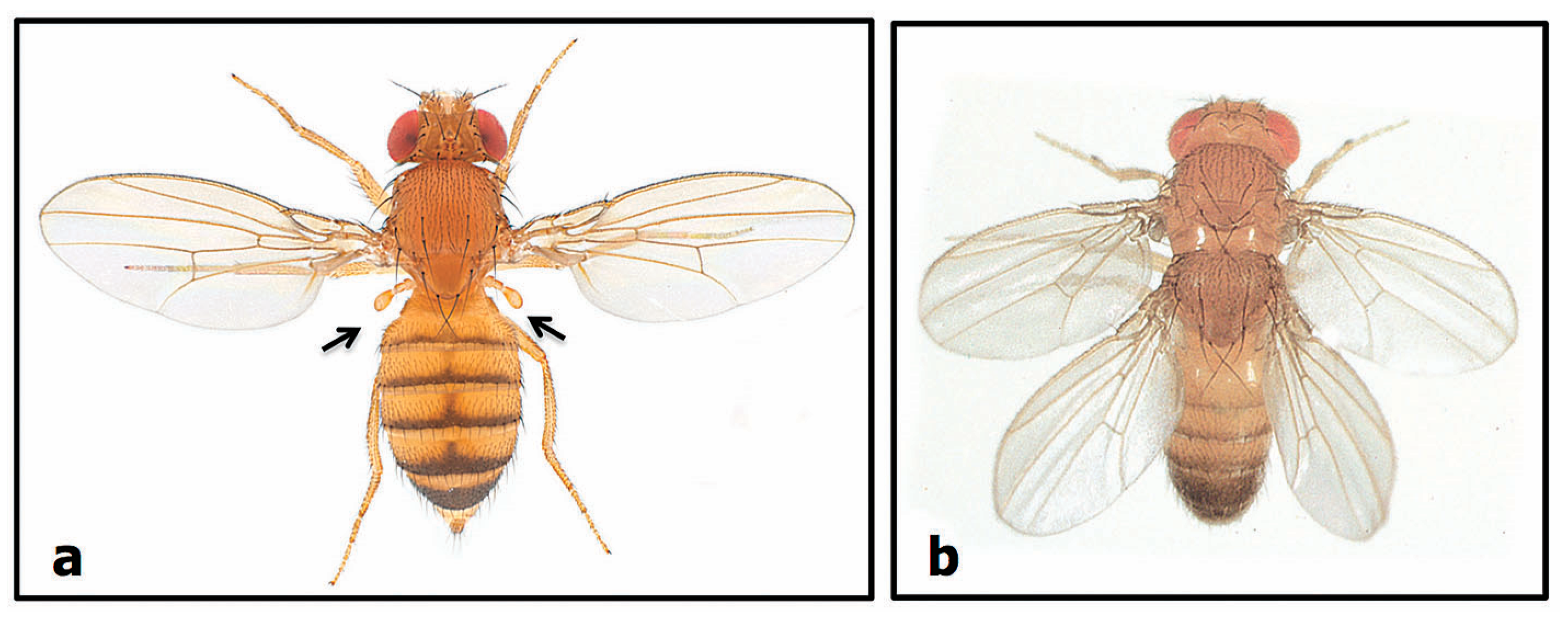
2.2.2. Number Variants
2.3. Variation in the Fossil Record
2.4. The Experimental Data on Variation
2.5. Implications of the Evidence on Variation
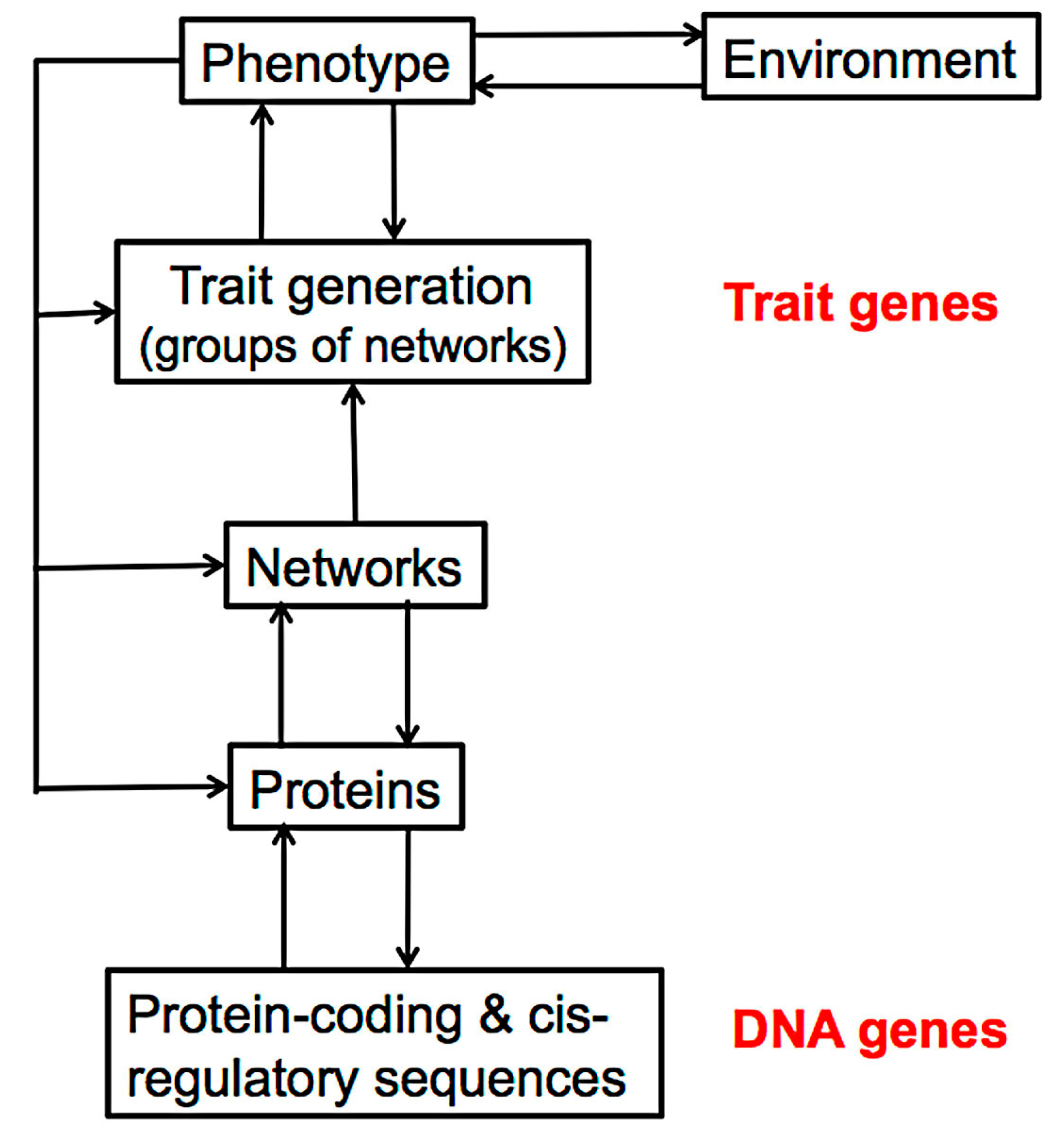
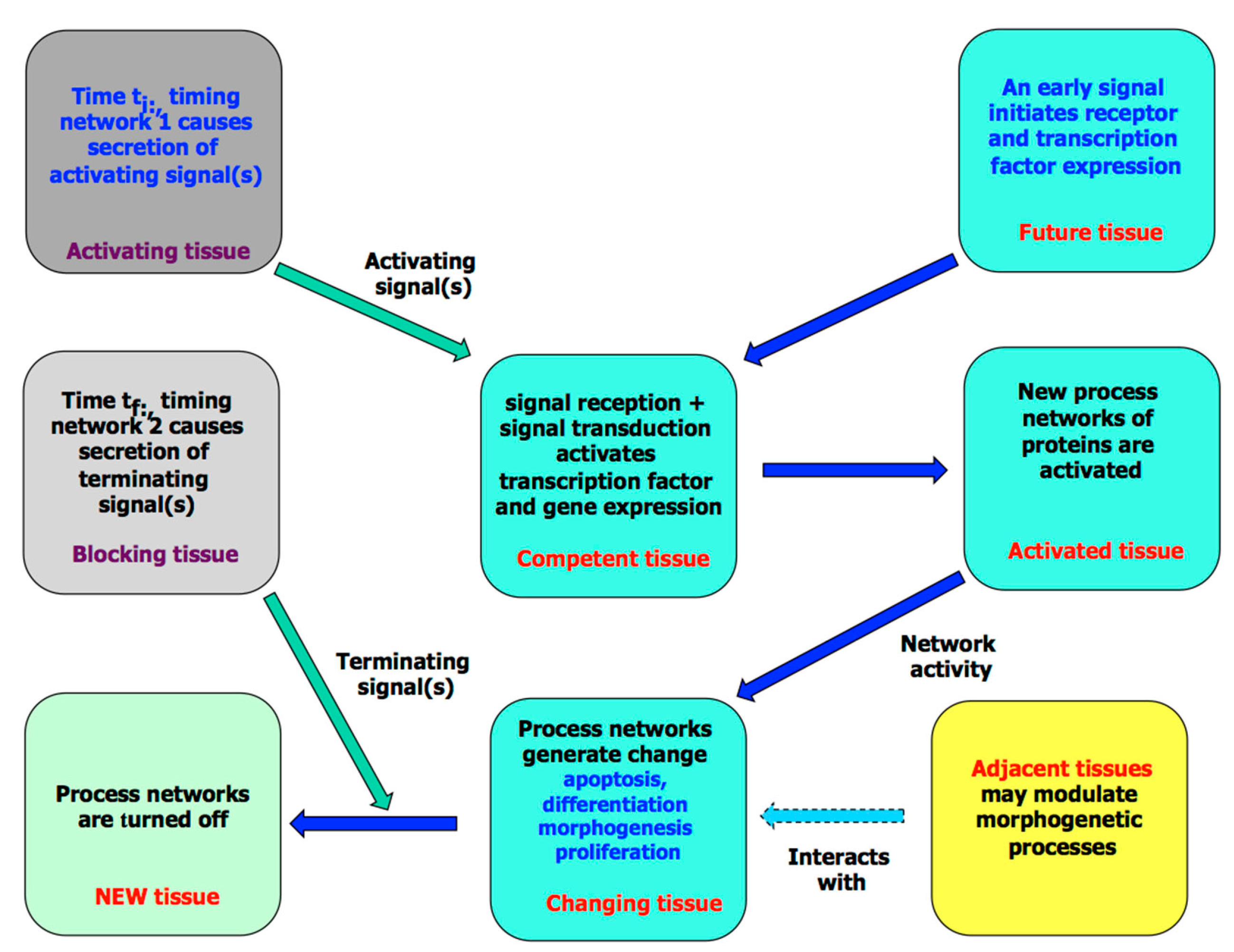
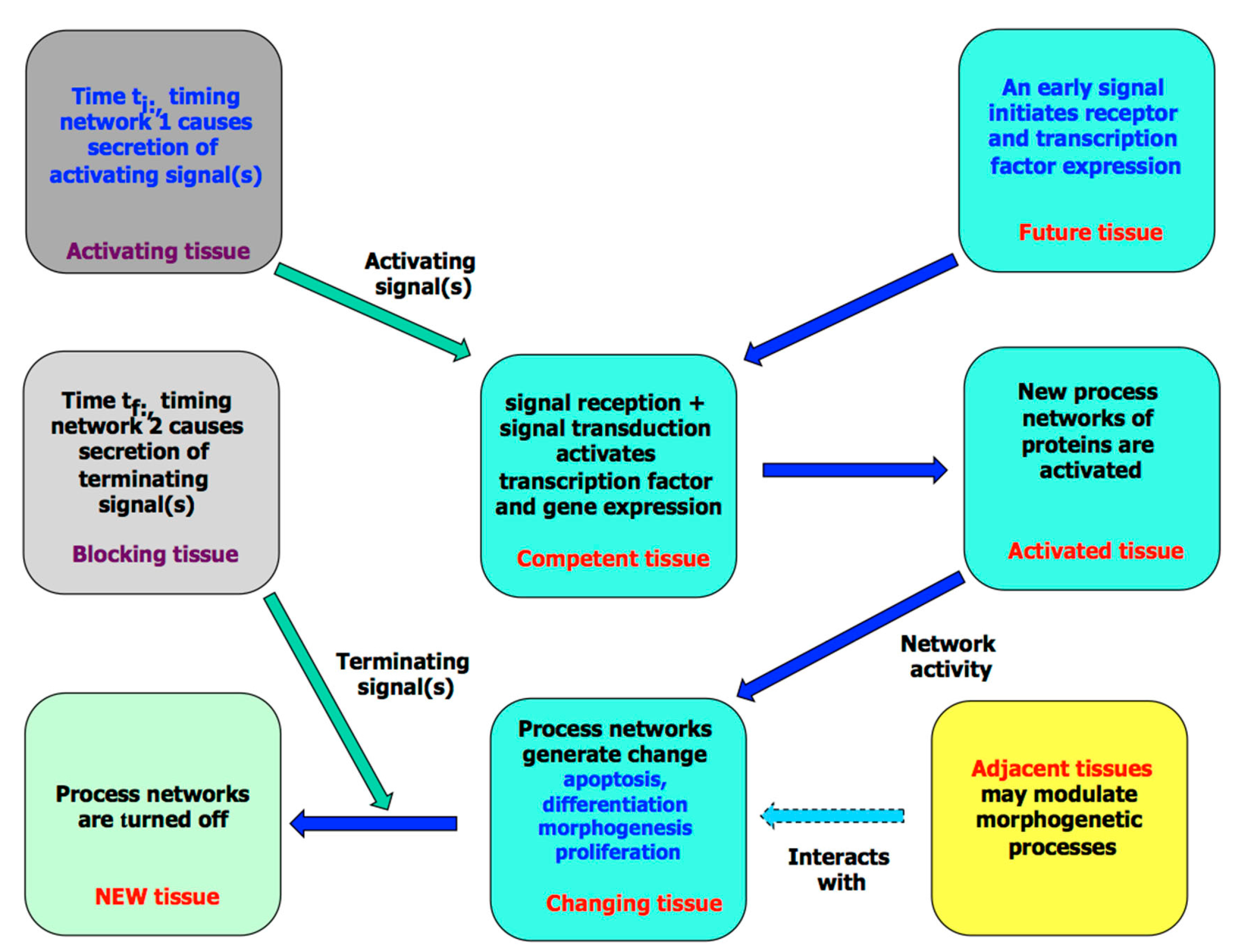
3. The Molecular Processes of Tissue Development
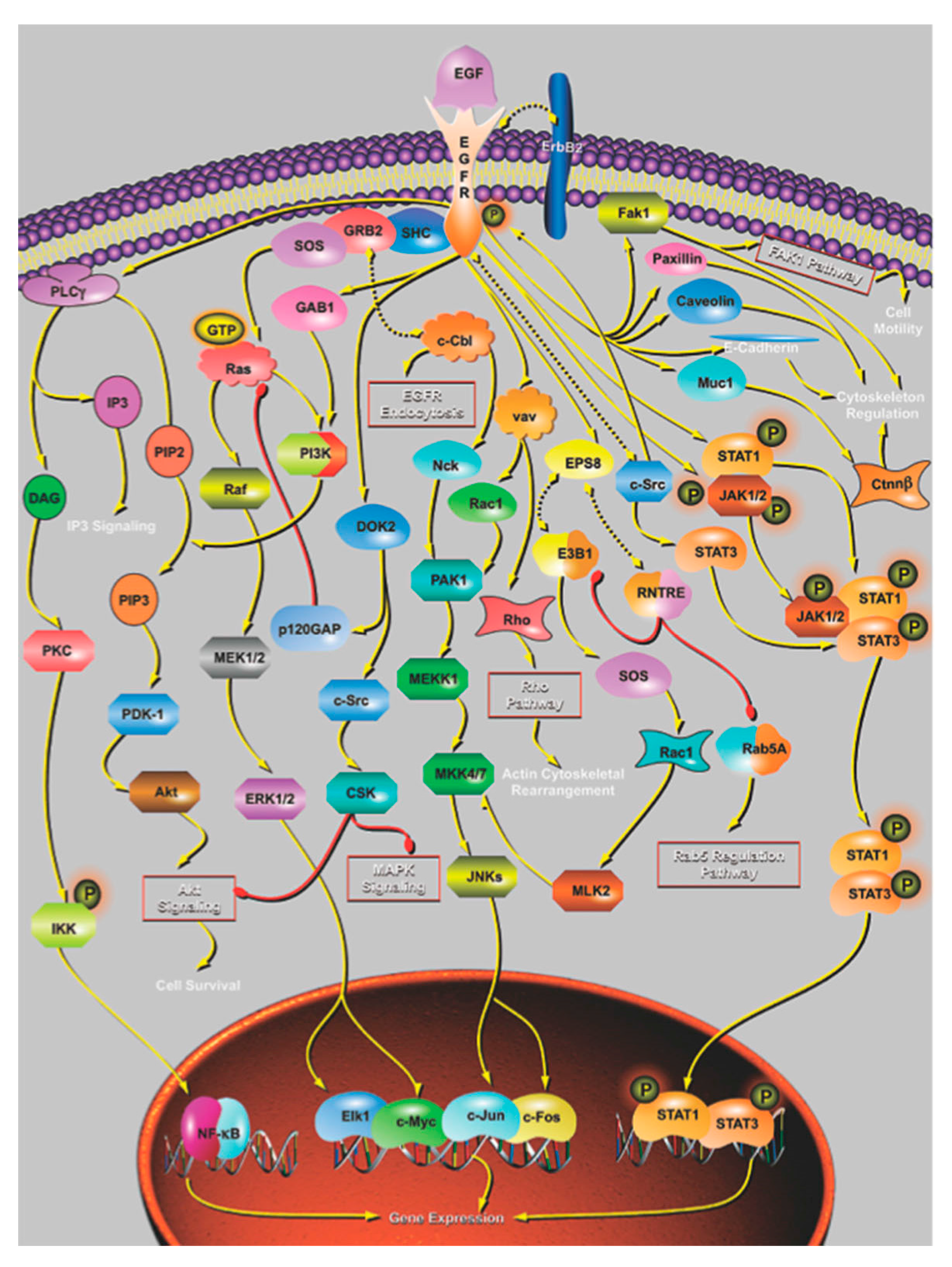
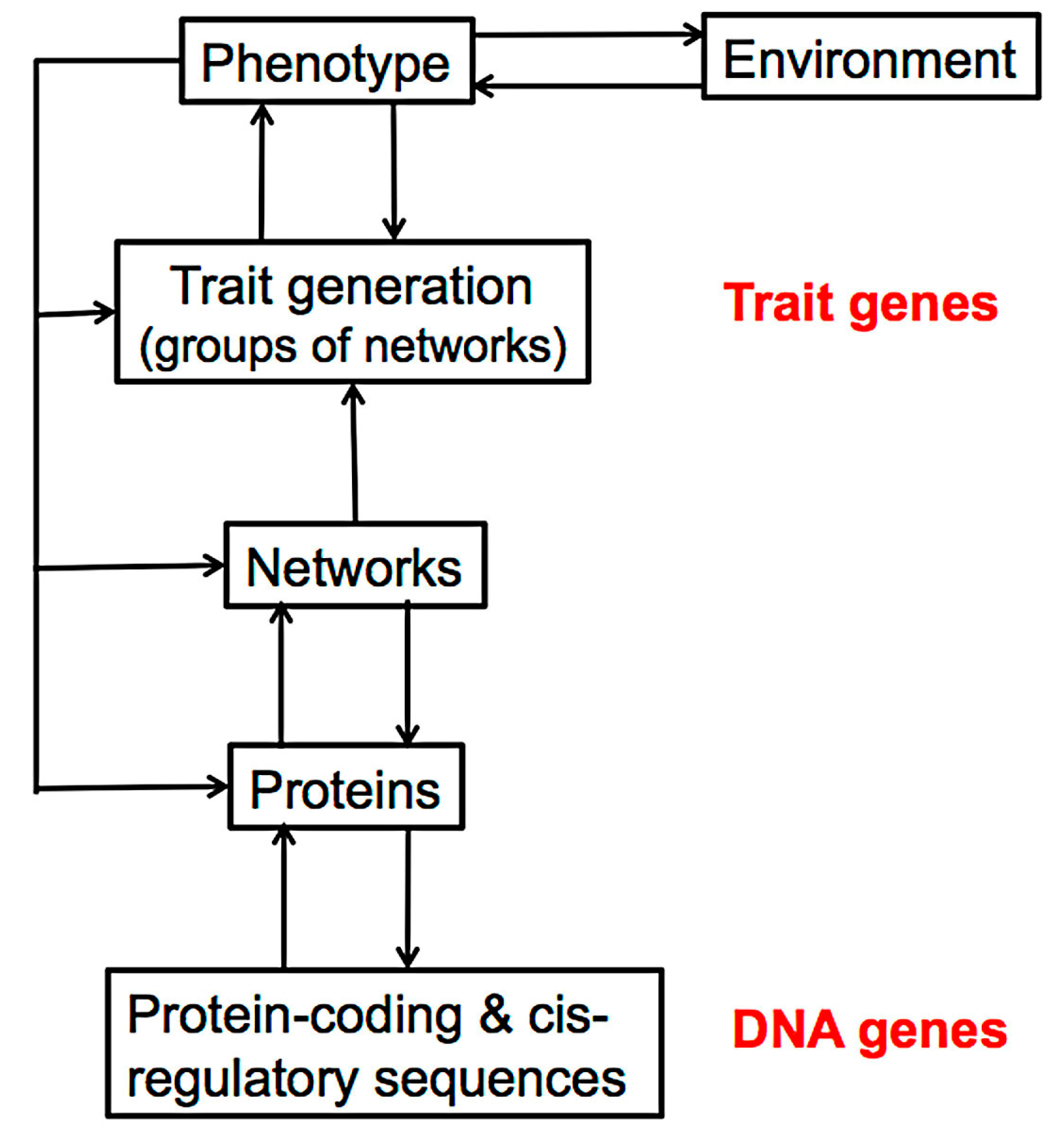
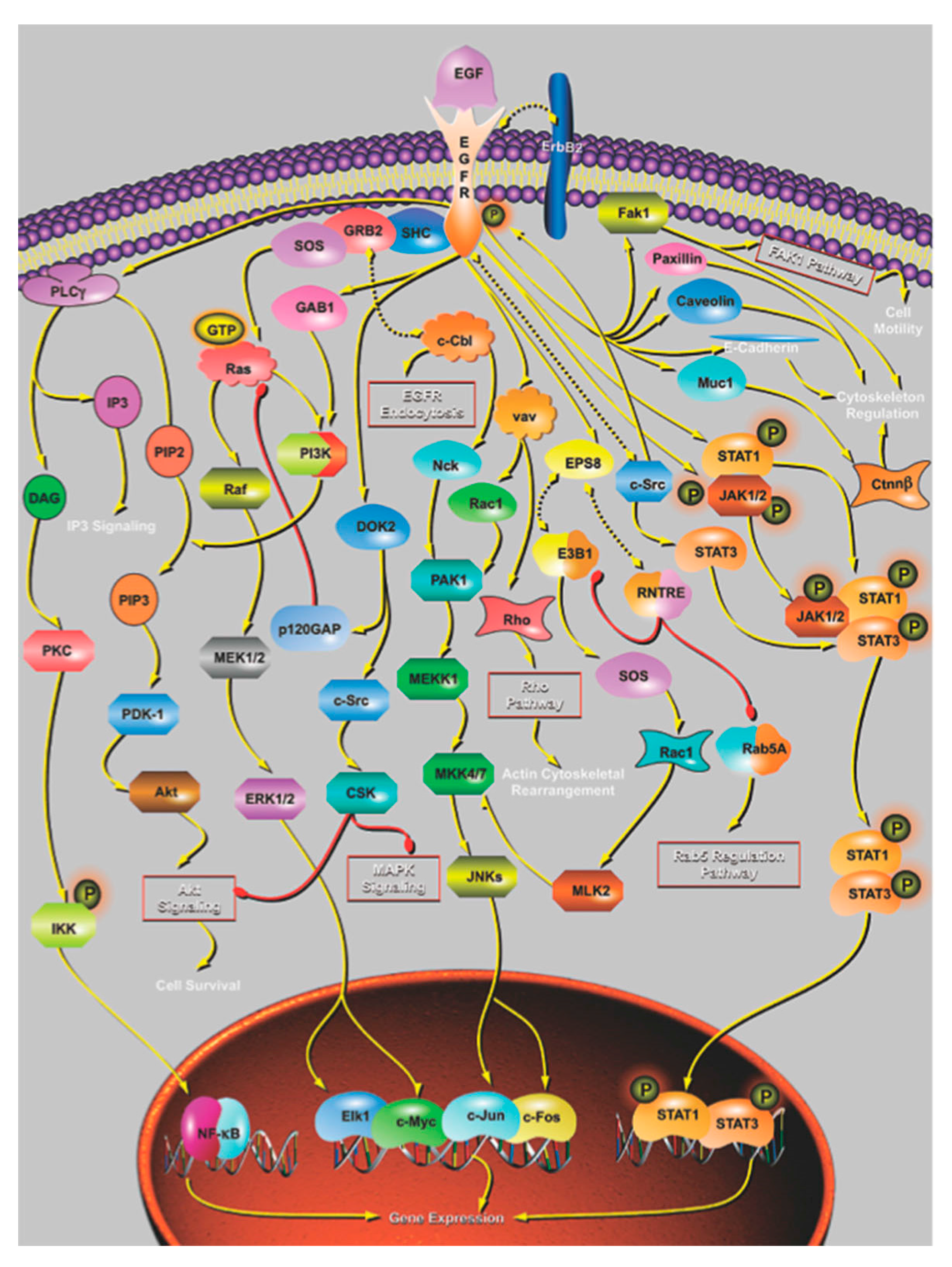
4. The Effect of Mutation on Proteins and Protein Networks
5. Discussion
The Speed of Evolutionary Change
Acknowledgment
Conflicts of Interest
References
- Darwin, C.R. On the Origin of Species by Means of Natural Selection or the Preservation of Favoured Races in the Struggle for Life; John Murray: London, UK, 1859; ISBN 9780486450063. [Google Scholar]
- Huxley, T.H. Letter of July 7, 1857, quoted in W. E. Friedman, W.E.; Dingle P.K. Charles Darwin and the Origin of Plant Evolutionary Developmental Biology. Plant Cell 2011, 23, 1194–1207. [Google Scholar]
- Merlin, F. Evolutionary chance mutation: A defense of the modern synthesis’ consensus view. Philos. Theor. Biol. 2010, 2, e103. [Google Scholar] [CrossRef] [Green Version]
- Bard, J.B.L. Principles of Evolution; Garland Press: New York, NY, USA, 2017; ISBN 9780815345398. [Google Scholar]
- Transformations of Lamarckism; Gissis, S.B.; Jablonka, E. (Eds.) MIT Press: Cambridge, MA, USA, 2011; ISBN 9780262015141. [Google Scholar]
- Bard, J. A systems biology representation of developmental anatomy. J. Anat. 2011, 218, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, S.F.; Epel, D. Ecological Developmental Biology, 2nd ed.; Sinauer Press: Sunderland, MA, USA, 2015; ISBN 978-0878932993. [Google Scholar]
- Salazar-Ciudad, I. Looking at the origin of phenotypic variation from pattern formation gene networks. J. Biosci. 2009, 34, 573–587. [Google Scholar] [CrossRef] [PubMed]
- Bard, J. A systems biology view of evolutionary genetics. Bioessays 2010, 32, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Jacob, F. Evolution and tinkering. Science 1977, 196, 1161–1166. [Google Scholar] [CrossRef] [PubMed]
- Theunissen, B. Darwin and his pigeons. The analogy between artificial and natural selection revisited. J. Hist. Biol. 2012, 45, 179–212. [Google Scholar] [PubMed]
- Wayne, R.K.; vonHoldt, B.M. Evolutionary genomics of dog domestication. Mamm. Genome 2012, 23, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Drake, A.G.; Klingenberg, C.P. Large-scale diversification of skull shape in domestic dogs: Disparity and modularity. Am. Nat. 2010, 175, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, S.F. Developmental Biology, 10th ed.; Sinauer Press: Sunderland, MA, USA, 2014; ISBN 0878939784. [Google Scholar]
- Haworth, K.E.; Healy, C.; McGonnell, I.M.; Binns, M.; Sharpe, P.T. Characterisation of the genomic canine Fgf8 locus and screen for genetic variants in 4 dogs with different face types. DNA Seq. 2007, 18, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Cresko, W.A.; Armores, A.; Wilson, C. Parallel genetic basis for repeated evolution of armor loss in Alaskan threespine stickleback populations. Proc. Natl. Acad. Sci. USA 2004, 101, 6050–6055. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.F.; Marks, M.E.; Jones, F.C. Adaptive evolution of pelvic reduction in sticklebacks by recurrent deletion of a Pitx1 enhancer. Science 2010, 327, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Schluter, D.; Conte, G.L. Genetics and ecological speciation. Proc. Nat. Acad. Sci. USA 2009, 106, 9955–9962. [Google Scholar] [CrossRef] [PubMed]
- Kardong, K. Vertebrates: Comparative Anatomy, Function, Evolution, 7th ed.; McGraw-Hill Education: New York, NY, USA, 2014; ISBN 9780078023026. [Google Scholar]
- Fleming, P.A.; Verburgt, L.; Scantlebury, M.; Medger, K.; Bateman, P.W. Jettisoning ballast or fuel? Caudal autotomy and locomotory energetics of the Cape dwarf gecko Lygodactylus capensis (Gekkonidae). Physiol. Biochem. Zool. 2009, 82, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Hagen, I.J.; Donnellan, S.C.; Bull, C.M. Phylogeography of the prehensile-tailed skink Corucia zebrata on the Solomon Archipelago. Ecol. Evol. 2012, 2, 1220–1234. [Google Scholar] [CrossRef] [PubMed]
- Field, D.J.; Campbell-Malone, R.; Goldbogen, J.A.; Shadwick, R.E. Quantitative computed tomography of humpback whale (Megaptera novaeangliae) mandibles: Mechanical implications for rorqual lunge-feeding. Anat. Rec. 2010, 293, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Hornbruch, A.; Wolpert, L. Cell division in the early growth and morphogenesis of the chick limb. Nature 1970, 226, 764–766. [Google Scholar] [CrossRef] [PubMed]
- Grisham, J.W. A morphologic study of deoxyribonucleic acid synthesis and cell proliferation in regenerating rat liver; autoradiography with thymidine-H3. Cancer Res. 1962, 22, 842–849. [Google Scholar] [PubMed]
- Head, J.J.; Polly, P.D. Dissociation of somatic growth from segmentation drives gigantism in snakes. Biol. Lett. 2007, 3, 296–298. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.C.; Nelson, C.E.; Morgan, B.A.; Tabin, C. Hox genes and the evolution of vertebrate axial morphology. Development 1995, 12, 333–346. [Google Scholar]
- Mallo, M.; Wellik, D.M.; Deschamps, J. Hox genes and regional patterning of the vertebrate body plan. Dev. Biol. 2010, 344, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Gomez, C.; Ozbudak, E.M.; Wunderlich, J.; Baumann, D.; Lewis, J.; Pourquié, O. Control of segment number in vertebrate embryos. Nature 2008, 454, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Leysen, H.; Roos, G.; Adriaens, D. Morphological variation in head shape of pipefishes and seahorses in relation to snout length and developmental growth. J. Morphol. 2011, 272, 1259–1270. [Google Scholar] [CrossRef] [PubMed]
- Richard, P.R.; Orr, J.R.; Black, S.R.; Nweeia, M.T.; Eichmiller, F.C.; Hauschka, P.V.; Tyler, E.; Mead, J.G.; Potter, C.W.; Angnatsiak, D.P. Vestigial tooth anatomy and tusk nomenclature for monodon monoceros. Anat. Rec. 2012, 295, 1006–1016. [Google Scholar]
- Balic, A.; Thesleff, I. Tissue Interactions Regulating Tooth Development and Renewal. Curr. Top. Dev. Biol. 2015, 115, 157–186. [Google Scholar] [PubMed]
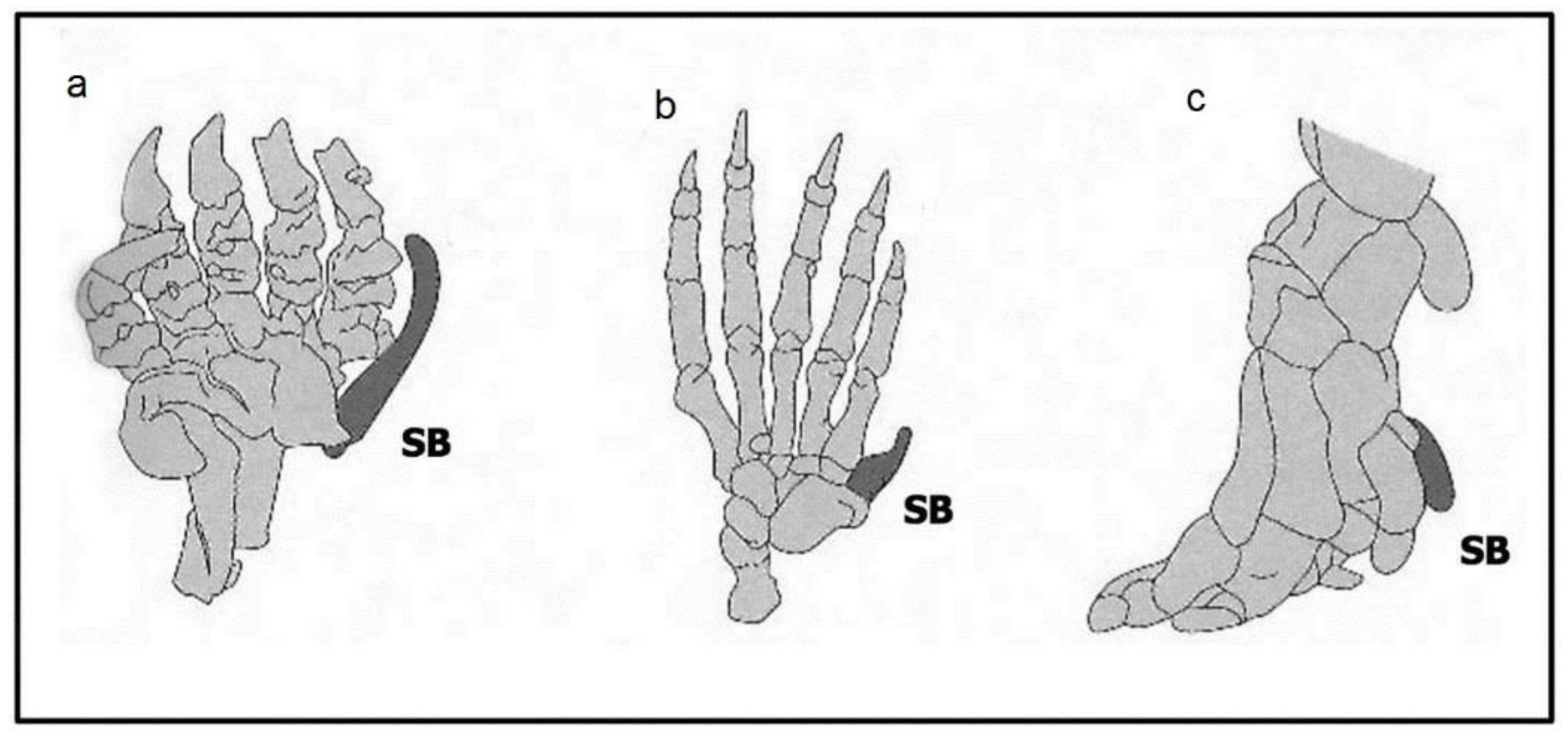
- Saxena, A.; Towers, M.; Cooper, K.L. The origins, scaling and loss of tetrapod digits. Philos. Trans. R. Soc. B 2017, 372, 20150482. [Google Scholar] [CrossRef] [PubMed]
- Mitgutsch, C.; Richardson, M.K.; Jiménez, R. Circumventing the polydactyly 'constraint' the mole's ‘thumb’. Biol. Lett. 2012, 9, 4–77. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.R.; Delmer, C.; Miller, C.E. From flat foot to fat foot structure ontogeny function and evolution of elephant. Science 2011, 33, 1699–1703. [Google Scholar] [CrossRef] [PubMed]
- Bard, J.B.L. A unity underlying the different zebra striping patterns. J. Zool. 2007, 187, 527–539. [Google Scholar]
- Barrio, R.A.; Varea, C.; Aragón, J.L.; Maini, P.K. A two-dimensional numerical study of spatial pattern formation in interacting Turing systems. Bull. Math. Biol. 1999, 61, 483–505. [Google Scholar] [CrossRef] [PubMed]
- Bard, J.B.L. A model generating aspects of zebra and other mammalian coat patterns. J. Theor. Biol. 1981, 93, 363–385. [Google Scholar] [CrossRef]
- Clack, J. Gaining ground: The Origin and Evolution of the Tetrapods, 2nd ed.; University Press: Bloomington, IN, USA, 2012; ISBN 9780253356758. [Google Scholar]
- Zhu, M.; Yu, X.; Ahlberg, P.E. A Silurian placoderm with osteichthyan-like marginal jaw bones. Nature 2013, 502, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Laurin, M.; Meunier, F.J.; Germain, D.; Lemoine, M. A microanatomical and histological study of the paired fin skeleton of the devonian sarcopterygian, eusthenopteron foordi. J. Palaeontol. 2007, 81, 143–153. [Google Scholar] [CrossRef]
- Johanson, Z.; Joss, J.; Boisvert, C.A.; Ericsson, R.; Sutija, M.; Ahlberg, P.E. Fish fingers: Digit homologues in sarcopterygian fish fins. J. Exp. Zool. B Mol. Dev. Evol. 2007, 308, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, C.A.; Mark-Kurik, E.; Ahlberg, P.E. The pectoral fin of Panderichthys and the origin of digits. Nature 2008, 456, 636–638. [Google Scholar] [CrossRef] [PubMed]
- Shubin, N.H.; Daeschler, E.B.; Jenkins, F.A., Jr. Pelvic girdle and fin of Tiktaalik roseae. Proc. Natl. Acad. Sci. USA 2014, 111, 893–899. [Google Scholar] [PubMed]
- Pierce, S.E.; Clack, J.A.; Hutchinson, J.R. Three-dimensional limb joint mobility in the early tetrapod Ichthyostega. Nature 2012, 486, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Coates, M.I. The origin of vertebrate limbs. Development 1994, 169–180. [Google Scholar] [PubMed]
- Lebedev, O.A.; Coates, M.I. The postcranial skeleton of the Devonian tetrapod Tulerpeton curtum Lebedev. Zool. J. 1995, 114, 307–348. [Google Scholar] [CrossRef]
- Clack, J.A. An early tetrapod from ‘Romer’s Gap’. Nature 2002, 418, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Cole, L.K.; Ross, L.S. Apoptosis in the developing zebrafish embryo. Dev. Biol. 2001, 240, 123–142. [Google Scholar] [CrossRef] [PubMed]
- Rice, W.R.; Salt, G.W. The evolution of reproductive isolation as a correlated character under sympatric conditions: Experimental evidence. Evolution 1990, 44, 1140–1152. [Google Scholar] [CrossRef] [PubMed]
- Waddington, C.H. Genetic assimilation of the bithorax phenotype. Evolution 1953, 10, 1–13. [Google Scholar] [CrossRef]
- Pavlopoulos, A.; Akam, M. Hox gene Ultrabithorax regulates distinct sets of target genes at successive stages of Drosophila haltere morphogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 2855–2860. [Google Scholar] [CrossRef] [PubMed]
- Alibardi, L. Review: Cornification, morphogenesis and evolution of feathers. Protoplasma 2017, 254, 1259–1281. [Google Scholar] [CrossRef] [PubMed]
- Takarada, T.; Hinoi, E.; Nakazato, R. An analysis of skeletal development in osteoblast-specific and chondrocyte-specific runt-related transcription factor-2 (Runx2) knockout mice. J. Bone Miner. Res. 2013, 28, 2064–2069. [Google Scholar] [CrossRef] [PubMed]
- Toro, R.; Burnod, Y. A morphogenetic model for the development of cortical convolutions. Cereb. Cortex 2005, 15, 1900–1913. [Google Scholar] [CrossRef] [PubMed]
- Denes, A.S.; Jékely, G.; Steinmetz, P.R. Molecular architecture of annelid nerve cord supports common origin of nervous system centralization in bilateria. Cell 2007, 129, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Giurumescu, C.A.; Sternberg, P.W.; Asthagiri, A.R. Intercellular coupling amplifies fate segregation during Caenorhabditis elegans vulval development. Proc. Natl. Acad. Sci. USA 2006, 103, 1331–1336. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.P.; Crampin, E.J. Development of models of active ion transport for whole-cell. Mol. Biol. 2004, 85, 387–405. [Google Scholar]
- Shoval, O.; Alon, U. SnapShot: Network motifs. Cell 2010, 143. [Google Scholar] [CrossRef] [PubMed]
- Ihekwaba, A.E.; Nguyen, P.T.; Priami, C. Elucidation of functional consequences of signaling pathway interactions. BMC Bioinf. 2009, 10, 370. [Google Scholar] [CrossRef] [PubMed]
- Aulehla, A.; Pourquié, O. Signaling gradients during paraxial mesoderm development. Cold Spring Harb. Perspect. Biol. 2010, 2, a000869. [Google Scholar] [CrossRef] [PubMed]
- Hubaud, A.; Pourquié, O. Signalling dynamics in vertebrate segmentation. Nat. Rev. Mol. Cell Biol. 2014, 15, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Harashima, H.; Dissmeyer, N.; Schnittger, A. Cell cycle control across the eukaryotic kingdom. Trends Cell Biol. 2013, 23, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Bollinger, T.; Schibler, U. Circadian rhythms—From genes to physiology and disease. Swiss Med. Wkly. 2014. [Google Scholar] [CrossRef] [PubMed]
- Turing, A.M. The chemical basis of morphogenesis. Phil. Trans. R. Soc. B 1952, 237, 37–72. [Google Scholar] [CrossRef]
- Glimm, T.; Headon, D.; Kiskowski, M.A. Computational and mathematical models of chondrogenesis in vertebrate limbs. Birth Defects Res. C. 2012, 96, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, J.A. Evolution: A View from the 21st Century; FT Press: Upper Saddle River, NJ, USA, 2011; ISBN 9780133435535. [Google Scholar]
- Carroll, S.B. Evolution at two levels: On genes and form. PLoS Biol. 2005, 3, e245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, V.J.; May, G.; Wagner, G.P. Regulatory evolution through divergence of a phosphoswitch in the transcription factor CEBPB. Nature 2011, 480, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Williamson, I.; Lettice, L.A.; Hill, R.E.; Bickmore, W.A. Shh and ZRS enhancer colocalisation is specific to the zone of polarising activity. Development 2016, 143, 2994–3001. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, A.S. Genetic networks as transmitting and amplifying devices for natural genetic tinkering. Novartis Found. Symp. 2007, 284, 71–86. [Google Scholar] [PubMed]
- Yates, L.; McMurray, F.; Zhang, Y.; Greenfield, A.; Moffatt, M.; Cookson, W.; Dean, C. ENU mutagenesis as a tool for understanding lung development and disease. Biochem. Soc. Trans. 2009, 37, 838–842. [Google Scholar] [CrossRef] [PubMed]
- Keyte, A.L.; Smith, K.K. Heterochrony and developmental timing mechanisms: Changing ontogenies in evolution. Semin. Cell Dev. Biol. 2014, 34, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Chew, K.Y.; Shaw, G.; Yu, H.; Pask, P.J.; Renfree, M.B. Heterochrony in the regulation of the developing marsupial limb. Dev. Dyn. 2014, 243, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Holleley, C.E.; O’Meally, D.; Sarre, S.D.; Marshall Graves, J.A.; Ezaz, T.; Matsubara, K.; Azad, B.; Zhang, X.; Georges, A. Sex reversal triggers the rapid transition from genetic to temperature-dependent sex. Nature 2015, 523, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, D. Evolution of the Human Head; Harvard University Press: Cambridge, MA, USA, 2011; ISBN 9780674046368. [Google Scholar]
- Goodwin, Z.A.; de Guzman Strong, C. Recent Positive Selection in Genes of the Mammalian Epidermal Differentiation Complex Locus. Front. Genet. 2017, 7, 227. [Google Scholar] [CrossRef] [PubMed]
- Jobling, M.; Hollox, E.; Hurles, M.; Kivisild, T.; Tyler-Smith, C. Human Evolutionary Genetics, 2nd ed.; Garland Press: New York, NY, USA, 2013; ISBN 9780815341482. [Google Scholar]
- Pagani, L.; Schiffels, S.; Gurdasani, D.; Danecek, P.; Scally, A.; Chen, Y.; Xue, Y.; Haber, M.; Ekong, R.; Oljira, T.; et al. Tracing the route of modern humans out of Africa by using 225 human genome sequences from Ethiopians and Egyptians. Am. J. Hum. Gene. 2015, 96, 986–991. [Google Scholar] [CrossRef] [PubMed]
- Oppenheimer, S. Out-of-Africa the peopling of continents and islands: Tracing uniparental gene trees across the map. Phil. Trans. R. Soc. B 2012, 367, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, A.A.; Mattern, B.C.; Claes, P.; McEcoy, B.; Hughes, C.; Shriver, M. Investigating the case of human nose shape and climate adaptation. PLoS Genet. 2017, 13, e1006616. [Google Scholar]
- Li, H.; Durbin, R. Inference of human population history from individual whole-genome sequences. Nature 2011, 475, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.M.; West, C.H.; Emery, M.S.; Bonsall, R.W.; Moore, J.P.; Boss-Williams, K.A. Rats selectively-bred for behavior related to affective disorders: Proclivity for intake of alcohol and drugs of abuse, and measures of brain monoamines. Biochem. Pharmacol. 2008, 75, 134–159. [Google Scholar] [CrossRef] [PubMed]
- Darwin, C.R. The Variation of Animals and Plants under Domestication; John Murray: London, UK, 1868; ISBN 9781108014229. [Google Scholar]
- Bulmer, M.G. Francis Galton: Pioneer of Heredity and Biometry; Johns Hopkins University Press: Baltimore, MA, USA, 2003; ISBN 9780801874031. [Google Scholar]
- Hajkova, P.; Erhardt, S.; Lane, N.; Haaf, T.; El-Maarri, O.; Reik, W.; Walter, J.; Surani, M.A. Epigenetic reprogramming in mouse primordial germ cells. Mech. Dev. 2002, 117, 15–23. [Google Scholar] [CrossRef]
- Monk, D. Germline-derived DNA methylation and early embryo epigenetic reprogramming: The selected survival of imprints. Int. J. Biochem. Cell Biol. 2015, 67, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Tran, D.A.; Li, A.X.; Warden, C.; Bai, A.Y.; Singh, P.; Wu, X.; Pfeifer, G.P.; Szabó, P.E. Deleterious effects of endocrine disruptors are corrected in the mammalian germline by epigenome reprogramming. Genome Biol. 2015, 16, 59. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A. Transgenerational epigenetics: Integrating soma to germline communication with gametic inheritance. Mech. Ageing Dev. 2017, 163, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Nelson, V.R.; Heaney, J.D.; Tesar, P.J.; Davidson, N.O.; Nadeau, JH. Transgenerational epigenetic effects of Apobec1 deficiency on testicular germ cell tumor susceptibility and embryonic viability. Proc. Natl. Acad. Sci. USA 2012, 109, E2766–E2773. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Schuler, M.A.; Berenbaum, M.R. Molecular mechanisms of metabolic resistance to synthetic and natural xenobiotics. Ann. Rev. Entomol. 2007, 52, 231–253. [Google Scholar] [CrossRef] [PubMed]
- Jablonka, E.; Lamb, M.J. The inheritance of acquired epigenetic variations. Int. J. Epidemiol. 2015, 44, 94–103. [Google Scholar] [CrossRef]
- Charlesworth, D.; Barton, N.H.; Charlesworth, B. The sources of adaptive variation. Proc. R. Soc. B 2017, 284, 20162864. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Mya | Family | Novel Features | Changed Processes? | Ref. |
|---|---|---|---|---|---|
| Kenichthys | c.395 | sarcopterygian | Earliest fish with nostrils linking to the oral cavity. Normal sarcopterygian fins with short limb bones. | Open nostrils due patterning changes inducing morphogenesis and apoptosis? | [39] |
| Eusthenopteron | c.385 | sarcopterygian | Fish with internal nasal adaptations and labyrinthodont teeth; the bones of pectoral and pelvic fins had growth plates for lengthening. | Repatterning of growth of teeth and of upper limb bones | [40] |
| Sauripterus | c.380 | sarcopterygian | A fish whose fins had both radials and primitive digits | Repatterning of distal bone organization | [41] |
| Panderychthys | c.380 | sarcopterygian | A fish with four unjointed digit-like bones and a tetrapod cranium | Repatterning of limb bones and cranium | [42] |
| Tiktaalik | c.375 | sarcopterygian | A fish with amphibian features. The pectoral fin had basic wrist bone but rays not digits. It also had a flexible neck, lungs, and basic pectoral and pelvic girdles | Novel bones and repatterning of existing bones. Major patterning & morphogenetic changes that turned gills into lungs | [43] |
| Ichthyostega | c.374 | labyrinthodont | An intermediate species with amphibian-type lungs, strong ribs and fore and hind limbs (7 jointed toes) + fish gills & tail − able to clamber on land | Pattern formation + numbering | [44] |
| Acanthostega | c.365 | labyrinthodont | A very early amphibian with 8 forelimb & 7 hind limb jointed digits, non-weight-bearing forelimbs, and a complete pelvic girdle | Pattern formation leading to numbering changes + apoptosis of tail | [45] |
| Tulerpeton | c.365 | labyrinthodont | This species had 6 jointed digits, powerful “wading” limbs, pectoral girdle, lungs and no gills. This was clearly an amphibian. | Pattern formation + numbering | [46] |
| Perdepes | c.348 | 1st land tetrapod | An amphibian with 5 (+1?) digits. Land-adapted feet | Pattern formation | [47] |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bard, J.B.L. Tinkering and the Origins of Heritable Anatomical Variation in Vertebrates. Biology 2018, 7, 20. https://doi.org/10.3390/biology7010020
Bard JBL. Tinkering and the Origins of Heritable Anatomical Variation in Vertebrates. Biology. 2018; 7(1):20. https://doi.org/10.3390/biology7010020
Chicago/Turabian StyleBard, Jonathan B. L. 2018. "Tinkering and the Origins of Heritable Anatomical Variation in Vertebrates" Biology 7, no. 1: 20. https://doi.org/10.3390/biology7010020
APA StyleBard, J. B. L. (2018). Tinkering and the Origins of Heritable Anatomical Variation in Vertebrates. Biology, 7(1), 20. https://doi.org/10.3390/biology7010020