1. Introduction
Melanin is synthesized in melanosomes, which are organelles found in the pigment cells (melanocytes) of the basal layer of the epidermis. Within melanosomes, the enzyme tyrosinase catalyzes the conversion of tyrosine to 3,4-dihydroxyphenylalanine (DOPA) and DOPA into dopaquinone. After dopaquinone formation, the order of appearance in the eumelanin formation pathway is, first, dopachrome, which evolves to 5,6-dihydroxy-1H-indole (DHI), through spontaneous decarboxylation, or to 5,6-dihydroxyindole-2-carboxylic acid (DHICA), catalyzed by dopachrome tautomerase (Dct) [
1]. Melanogenesis can culminate in eumelanin through oxidative polymerization reactions following the formation of DHI and DHICA [
1].
In Japan, active skin-lightening ingredients of formulated medicated whitening cosmetics (quasi-drugs) can suppress melanogenesis by inhibiting tyrosinase activity (as is the case with kojic acid, arbutin, and rucinol) or by accelerating the degradation of tyrosinase (as is the case with linoleic acid) [
2,
3,
4]. However, agents, such as rhododendrol and magnolignan, are no longer used; they release hydroxyl radicals from tyrosinase and are associated with strong cytotoxicity to melanocytes and the potential to induce leukoderma [
5,
6].
Generally, the three-dimensional structure of an enzyme is prone to change with alteration in conditions such as heat, pH, and salinity, or its solvent. When that alteration is too great, the enzyme degenerates and becomes unable to retain its correct structure and is rendered inactive. This alteration applies to the case of tyrosinase, as shown in Himalayan rabbits and mice. The heads, paws, and tails of these animals are exposed to lower temperatures than other parts of their bodies, and these parts display regions of black fur called Himalayan markings. These animals have regions of dark coloration when they are raised in low-temperature zones, and these regions become lighter when they are raised in high-temperature zones. The Himalayan markings represent a dominant allele of the tyrosine gene called the
ch gene. Bearers of this
ch gene have a temperature-sensitive tyrosinase mutation and they experience loss of tyrosinase activity, lack melanin formation, and become white at high temperatures. Specifically, the body parts that are kept warm are white and body parts that are exposed to a low temperature are darkly pigmented. Due to this mutation, the animals have a white body trunk, where tyrosinase enzyme activity is lost and no melanogenesis occurs, and this area is kept warm, but their head, paws, and tails, which are exposed to the cold, retain tyrosinase activity and melanin-formation capacity, and these regions have dark pigment as a result [
7]. The distinctive markings of Siamese cats are produced by a similar mechanism [
8]. The temperature-sensitive form of oculocutaneous albinism type 1 (OCA1-TS; one of the three categories of this condition together with OCA1A and OCAB1) is characterized by loss of tyrosinase activity at temperatures above 35 °C, and the resulting diffused areas of melanin accumulation are related to differential temperatures for parts of the body [
9,
10].
The actions of tyrosinase and environment-related loss or fluctuation in the activity described above give rise to another question: can changes in pH suppress melanogenesis? Research on this question has advanced with a focus on the pH of the intramelanosomal environment. The optimum pH for tyrosinase is between 7 and 8, and reportedly this pH range does not vary between light-skinned people (Caucasians) and dark-skinned people (people of African descent) [
11]. Racial differences in skin color are known to be caused by epidermal melanosome genetic characteristics, with reports that light-skinned people have less melanin and an acidic intramelanosomal pH, whereas dark-skinned people have more melanin and a neutral intramelanosomal pH [
12,
13].
Melanosome pH is regulated by vacuolar-type H
+-ATPase (V-ATPase), which mediates hydrogen ion (H
+) influx, and sodium-hydrogen (Na
+/H
+) exchangers, which mediate H
+ extrusion. Light-skinned people have plentiful melanosome-membrane V-ATPase, which enables large-scale H
+ influx, and few Na
+/H
+ exchangers, which restrict extrusion of H
+ from the melanosome and preserve large quantities of inwardly-transported H
+ and, thus, retains acidity. This is a stark contrast to the situation in dark-skinned people, who have little melanosome-membrane V-ATPase and limited H
+ influx, and numerous Na
+/H
+ exchangers, with consequently large-scale H
+ extrusion from the melanosome, which has a small quantity of H
+ and retains a neutral pH [
11,
13].
Based on this background, we investigated the potential for pH-dependent tyrosinase degradation with a focus on the low pH of the intramelanosomal environment-induced suppression of melanogenesis. We also investigated whether the mechanism of the anti-melanogenic agent propylparaben is implicated in melanosomal pH regulation at non-cytotoxic concentrations. In this report, we describe both of these investigations.
2. Materials and Methods
2.1. Materials
p-Hydroxybenzoic acid methyl ester (methylparaben), p-hydroxybenzoic acid ethyl ester (ethylparaben) and p-hydroxybenzoic acid butyl ester (butylparaben) were purchased from Wako Pure Chemical Industries (Osaka, Japan). p-Hydroxybenzoic acid propyl ester (propylparaben) was from Ueno Fine Chemicals Industry (Osaka, Japan). Bafilomycin A1, aprotinin from bovine lung, phenylmethane sulfonyl fluoride (PMSF), leupeptin hemisulfate salt, E-64, and pepstatin A were purchased from Sigma-Aldrich Corp. (St. Louis, MO, USA). DAMP {N-(3-[(2,4-dinitrophenyl)amino]propyl)-N-(3-aminopropyl)methylamine dihydrochloride} was from Oxford Biomedical Research, Inc. (Oxford, MI, USA). The anti-tyrosinase antibody, anti-tyrosinase-related proteins-1 (Trp-1) antibody, and anti-Dct antibody were purchased from Aviva Systems Biology (San Diego, CA, USA). The anti-premelanosome protein 17 (Pmel17) antibody was purchased from Sigma-Aldrich Corp. and the anti-microphthalmia-associated transcription factor (Mitf) antibody (C5) was purchased from Abcam plc. (Cambridge, UK). The anti-dinitrophenyl-KLH, rabbit lgG antibody, Alexa Fluor 488 goat anti-rabbit lgG antibody and Alexa Fluor 546 goat anti-rat lgG antibody were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Other reagents used in experiments were purchased from Wako Pure Chemical Industries (Osaka, Japan).
2.2. Preparation of Melanosome-Fractions and Examination of Melanosomes
Cells were seeded (at 100,000 cells/mL) into ten 10-cm dishes and cultured for three days. B16 melanoma cells that had been cultured for three days were harvested with trypsin-EDTA solution, washed with a phosphate-buffered salt (PBS) solution, and pulverized with a homogenizer to prepare the cellular tissue. The cellular tissue was mixed with 0.25 mol/L sucrose solution to prepare a homogenate. A discontinuous sucrose density gradient was created with sucrose solutions at 1.8, 1.6, 1.4, 1.2, 1.0, 0.8, and 0.4 mol/L by overlaying lower concentrations on higher concentrations. The homogenate was subjected to density-gradient centrifugation at 700× g, and the supernatant was layered and further centrifuged with an ultracentrifugation device (21 h, 100,000× g, 4 °C). After centrifugation, fractions were collected by layer descending from the upper layer with a quantitative delivery liquid pump, to prepare melanosome fractions. The prepared fractions were numbered from 1 (fraction 1) for the highest layer (0.25 mol/L sucrose layer) to fraction 8 for the lowest layer (1.8 mol/L sucrose layer). Fractions were examined with an inverted microscope to confirm the presence of melanosomes.
2.3. Measurement of Melanosome-Fraction Tyrosinase Activity
Each fraction was added to wells on a 96-well plate (Corning Incorporated, Corning, NY, USA) at 10 µL/well (n = 3). Then, 80 µL of 1% Triton X-100 solution and 10 µL of 10 mmol/L DOPA were added to each well, and the absorbance was measured (wavelength: 475 nm) immediately (0 min) and two hours later using a microplate reader (Multi-Detection Microplate POWERSAN HT; BioTek, Winooski, VT, USA).
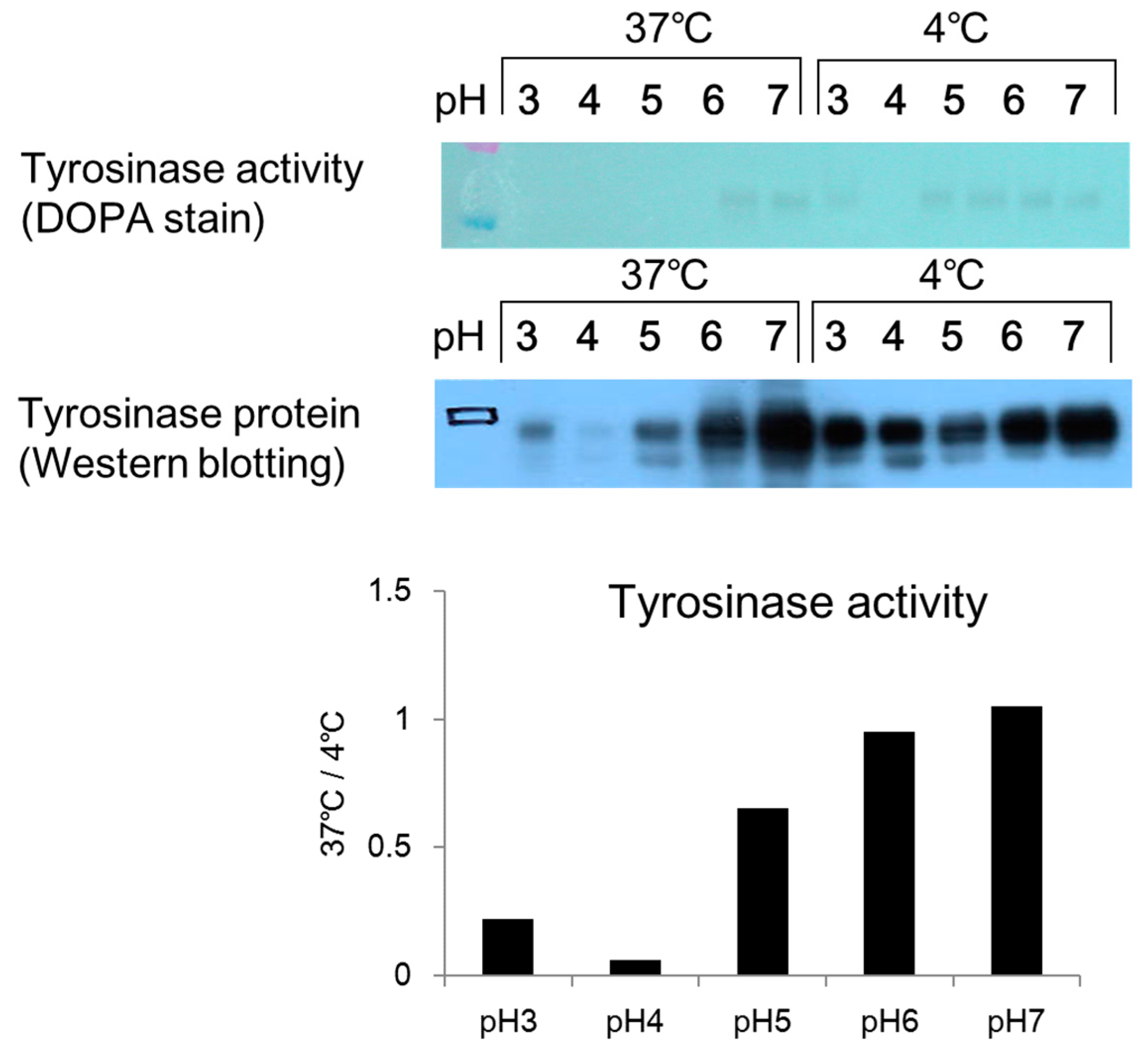
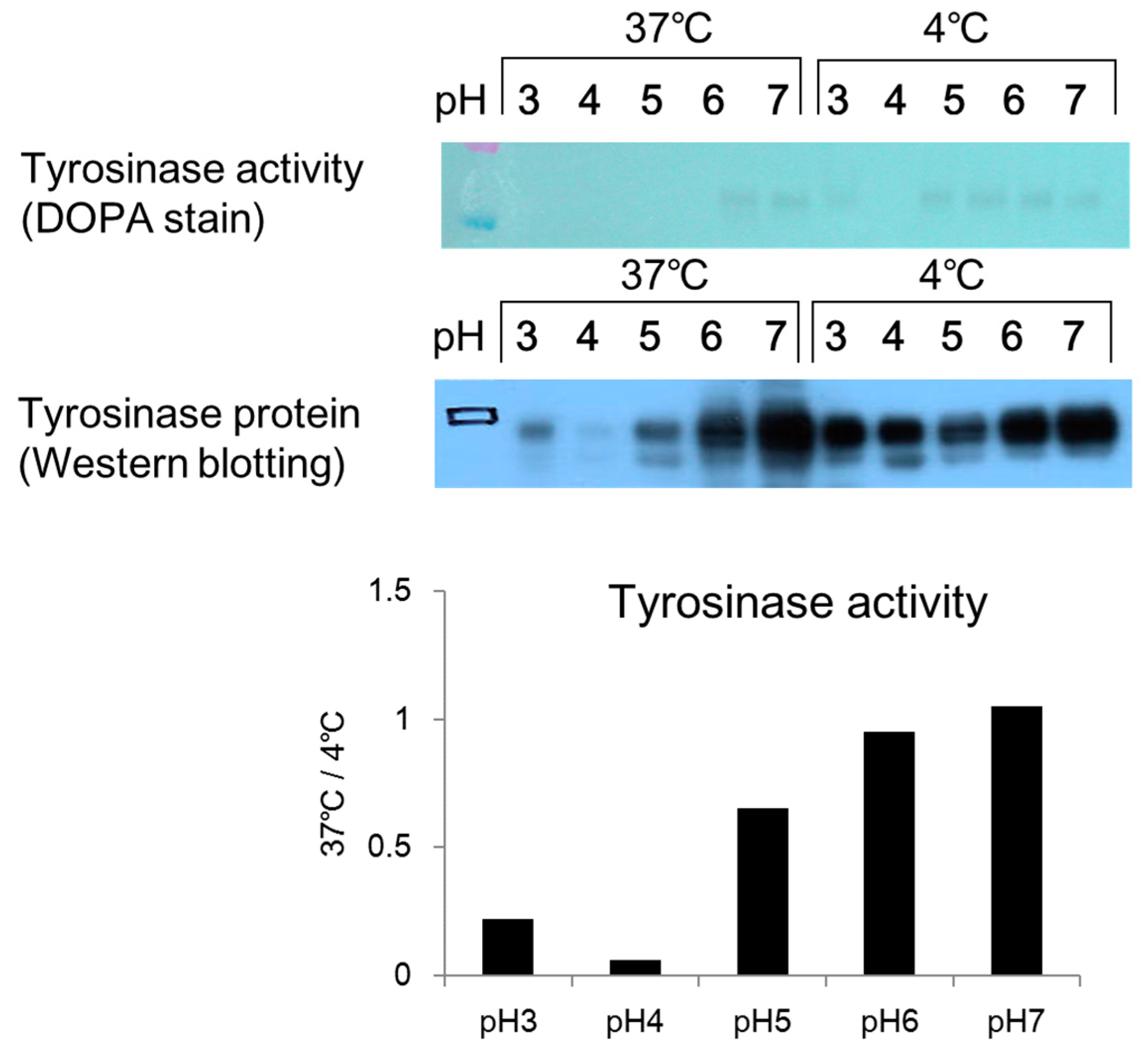
2.4. Effect of pH on Melanosome-Fraction Tyrosinase Activity
Citric acid–sodium citrate buffers (0.1 mol/L; pH 3, pH 4, and pH 5) and sodium phosphate buffers (0.1 mol/L; pH 6 and pH 7) were prepared. A 45 µL aliquot of each buffer was added to a 1.5 mL micro-tube, and 45 µL of a prepared melanosome fraction (fraction 8) were added to each tube. The cells were then left to stand overnight in an incubator (37 °C) and a freezer (−20 °C). After overnight incubation, 10 µL aliquots of the mixture in each tube were added to wells on a 96-well plate (n = 3). Then, 80 µL of PBS (pH 6.8) and 10 µL of 10 mmol/L DOPA solution were added to each well, and the absorbance was measured (wavelength: 475 nm) immediately (0 min) and 2 h later using the microplate reader. A 10% volume of glycerin (a 10% volume) was added to a sample of each mixture, which had been left to stand overnight, and 15 µL of each sample was applied to a 4–20% SDS-PAGE mini-gel and subjected to electrophoresis. After electrophoresis, the gel was removed by washing with PBS solution two to three times, and the sample was stained with a 2% DOPA solution. After two hours, the solution was exchanged for fresh DOPA solution and the stained sample was examined for a tyrosinase band.
2.5. Effect of pH on Melanosome-Fraction Tyrosinase Proteins
Citric acid–sodium citrate buffers (pH 3, pH 4, and pH 5) and sodium phosphate buffers (pH 6 and pH 7) were added to micro-tubes at 45 µL per tube. A 45 µL aliquot of a melanosome fraction prepared in each buffer was added to each micro-tube, and the tubes were left overnight in the incubator (37 °C) and a freezer (−20 °C). After being left overnight, a 10% volume of glycerin was added to each sample. A 15 µL aliquot of each sample was then added to a 4–20% SDS-PAGE gel and subjected to electrophoresis. After electrophoresis, each sample was submitted to a Western blotting assay. An anti-tyrosinase antibody was used as the primary antibody. The secondary antibody was a peroxidase-labeled anti-rabbit antibody, which had been prepared by dilution with a tris-buffered salts (TBS) solution.
2.6. Effect of Bafilomycin A1 on Melanin Content
B16 melanoma cells were seeded onto 10-cm dishes at 10 mL per dish (100,000 cells/mL) and cultured for one day. A 10 µL aliquot of 1 µmol/L bafilomycin A1 formulation was added to each dish, and the cells were then cultured for three days. Cultured cells were harvested with trypsin-EDTA and centrifuged. The culture medium was discarded, and the cells were repeatedly washed with PBS solution, and then counted with a hemocytometer. After being counted, the cells were centrifuged again, the supernatant was discarded, and the cellular sediment was dissolved in an amount of 1 mol/L NaOH according to the number of cells. Dissolved melanosome content was imaged with a digital camera.
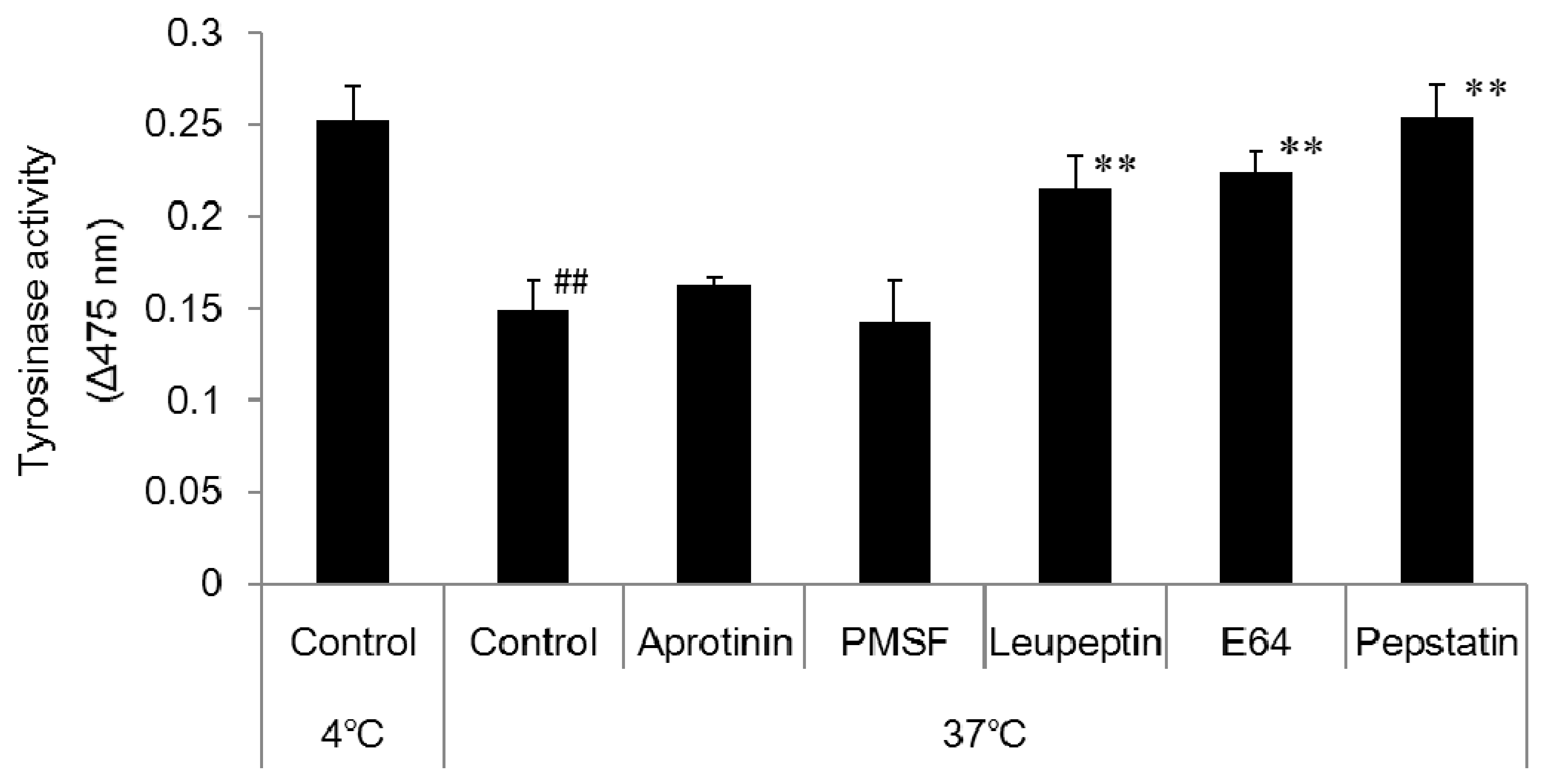
2.7. Effect of Enzyme Inhibitors on Tyrosinase Degradation
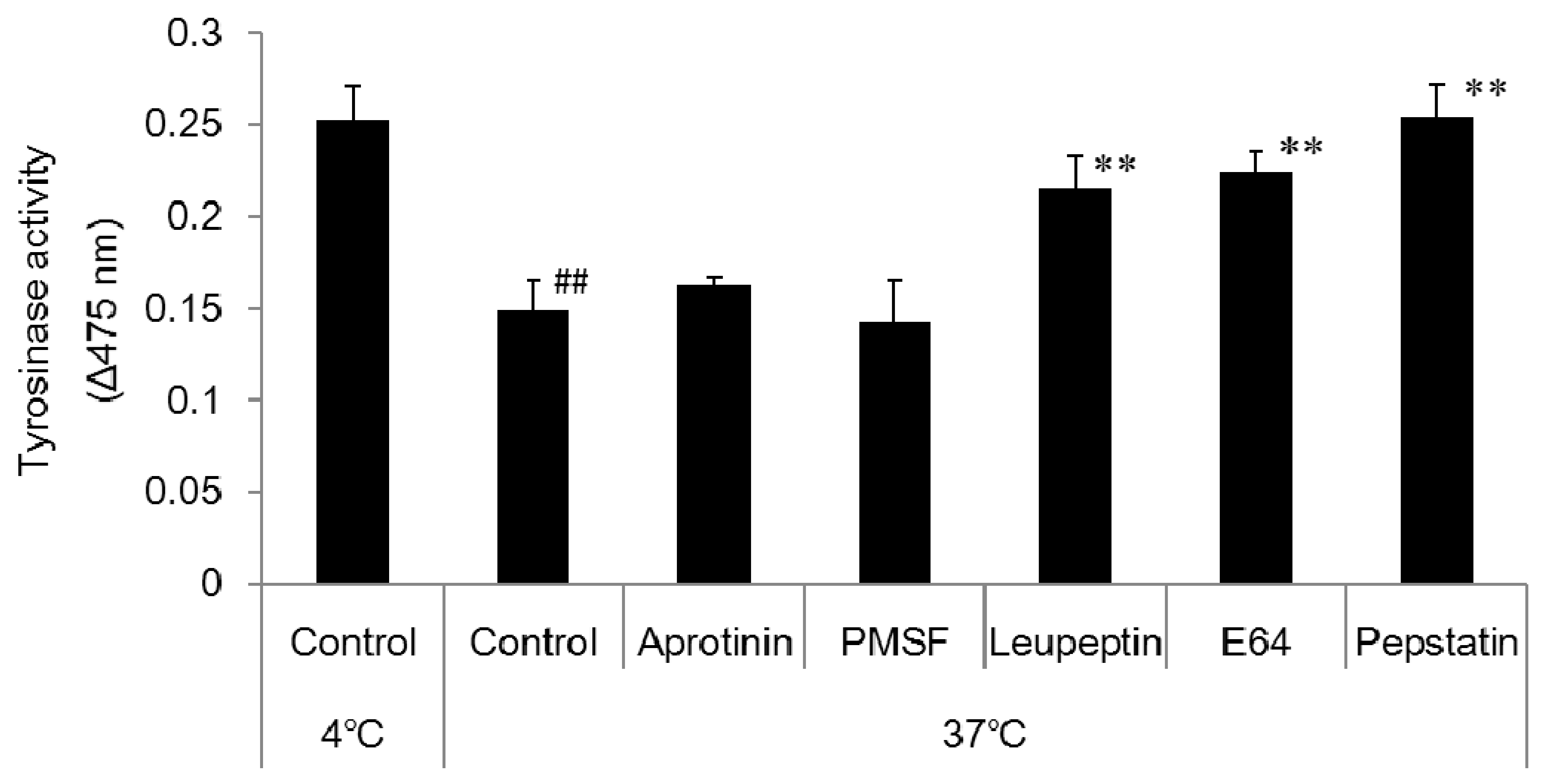
Citric acid-sodium citrate buffer (pH 4) was prepared and added to 1.5 mL micro-tubes at 50 µL per tube. A 50 µL aliquot of the prepared melanosome fraction was added to each tube followed by 1 µL of the relevant enzyme inhibitor. The cells were left overnight in the incubator (37 °C) and a freezer (−20 °C). The enzyme inhibitors were 1 mol/L phenylmethane sulfonyl fluoride (PMSF; a serine protease inhibitor), 2.2 mmol/L leupeptin (a serine/cysteine protease inhibitor), 1.4 mmol/L E64 (a cysteine protease inhibitor), and 10 mmol/L pepstatin A (an aspartic proteinase inhibitor). After being left overnight, 10 µL aliquots of each sample were added to wells on the 96-well plate (n = 3). An 80 µL aliquot of PBS solution and a 10 µL aliquot of 10 mmol/L DOPA solution were added to each well, and absorbance was measured (wavelength: 475 nm) with the microplate reader immediately (0 min), 2 h, and one day after to determine tyrosinase activity.
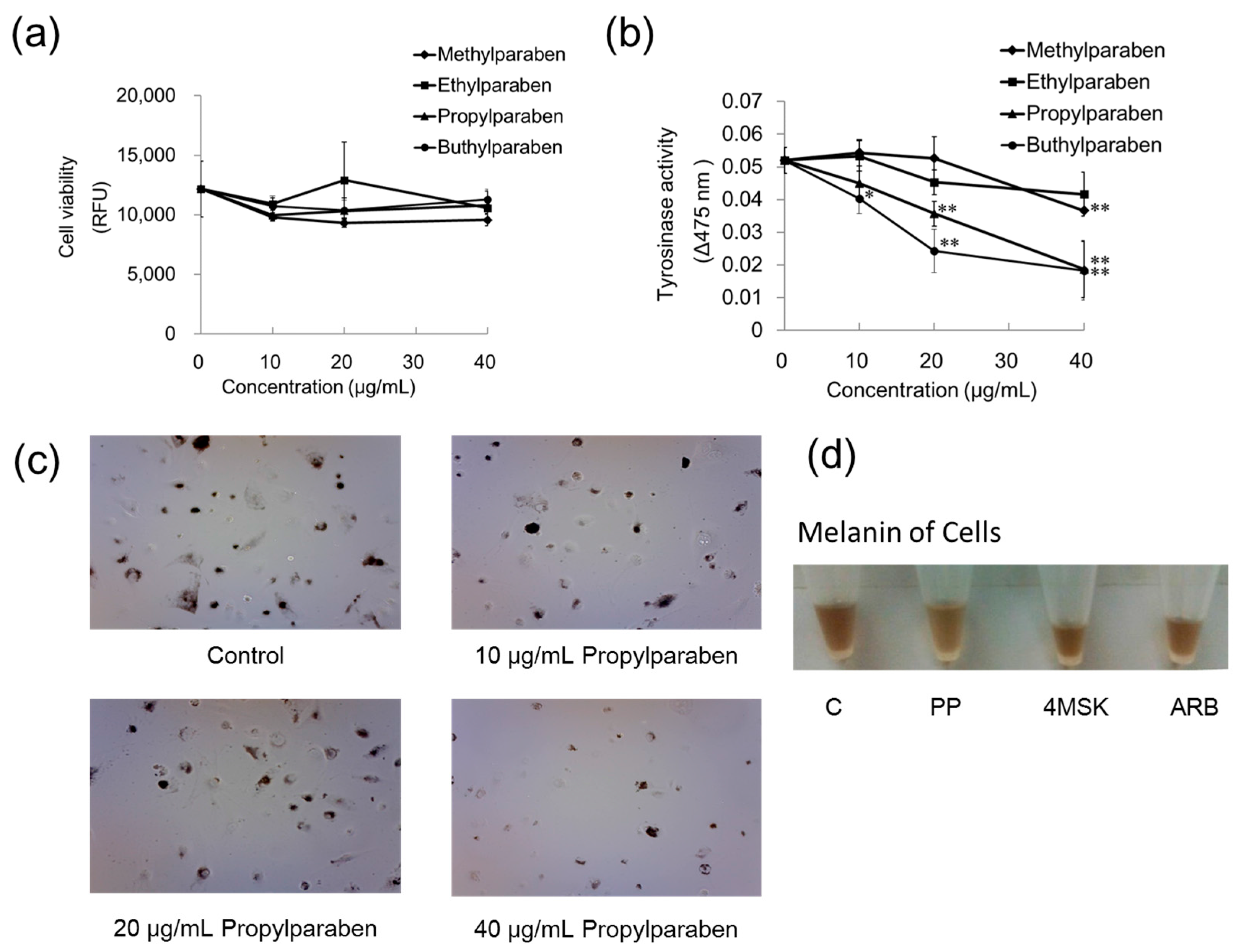
2.8. Effects of Parabens on Cell Proliferation
Parabens with methyl, ethyl, propyl, and butyl chains were used. B16 melanoma cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) and subjected to the experiments described below. Cells were seeded into wells of the 96-well plate at 99 µL per well (30,000 cells/mL) and cultured for one day. Methylparaben, ethylparaben, propylparaben, and butylparaben were dissolved in dimethyl sulfoxide (DMSO) solution to prepare 1, 2, and 4 mg/mL, and these solutions were added to wells at 1 µL per well (n = 3). The samples were cultured for two days, after which each well was washed with PBS solution and the cells were then cultured with DMEM diluted 100-fold with Hoechst 33258 (100 µg/mL DMSO) for 30 min. The relative fluorescence unit (RFU) was then measured with the microplate reader (excitation, 360 nm; emission, 465 nm).
2.9. Comparison of Paraben Inhibitory Effects on Intracellular Anti-Tyrosinase Activity
B16 melanoma cells were seeded into wells on a 96-well plate at 99 µL per well (30,000 cells/mL), and cultured for one day. Methylparaben, ethylparaben, propylparaben, and butylparaben were dissolved in dimethyl sulfoxide (DMSO) solution to prepare 1, 2, and 4 mg/mL, and these solutions were added to wells at 1 µL per well (n = 3). The samples were cultured for two days, after which 10 µL of 10 mmol/L DOPA solution and 90 µL of 1 mmol/L Triton X-100 solution were added to each well, and the absorbance was measured (wavelength: 475 nm) immediately (0 min) and 1 h later with the microplate reader.
2.10. Comparison of Anti-Melanogenic Effects of Propylparaben, 4-Methoxy Salicylic Acid Potassium Salt, and Arbutin
B16 melanoma cells were seeded onto 10-cm dishes at 9.99 mL per dish (100,000 cells/mL) and cultured for one day. Propylparaben, 4-methoxy salicylic acid potassium salt and arbutin were dissolved in DMSO/H2O solution to prepare 40 mg/mL solutions. Then 10 µL aliquots of these solutions were added to the dishes, and the dishes were cultured for two days. The cells were imaged with a microscope, and the cultured cells were then harvested with trypsin-EDTA. The cells were centrifuged, the culture medium was discarded, and the cells were repeatedly washed with PBS solution, then counted. After being counted, the cells were centrifuged again, the liquid was discarded, and the cellular sediment was dissolved in 1 mol/L NaOH and imaged with a digital camera.
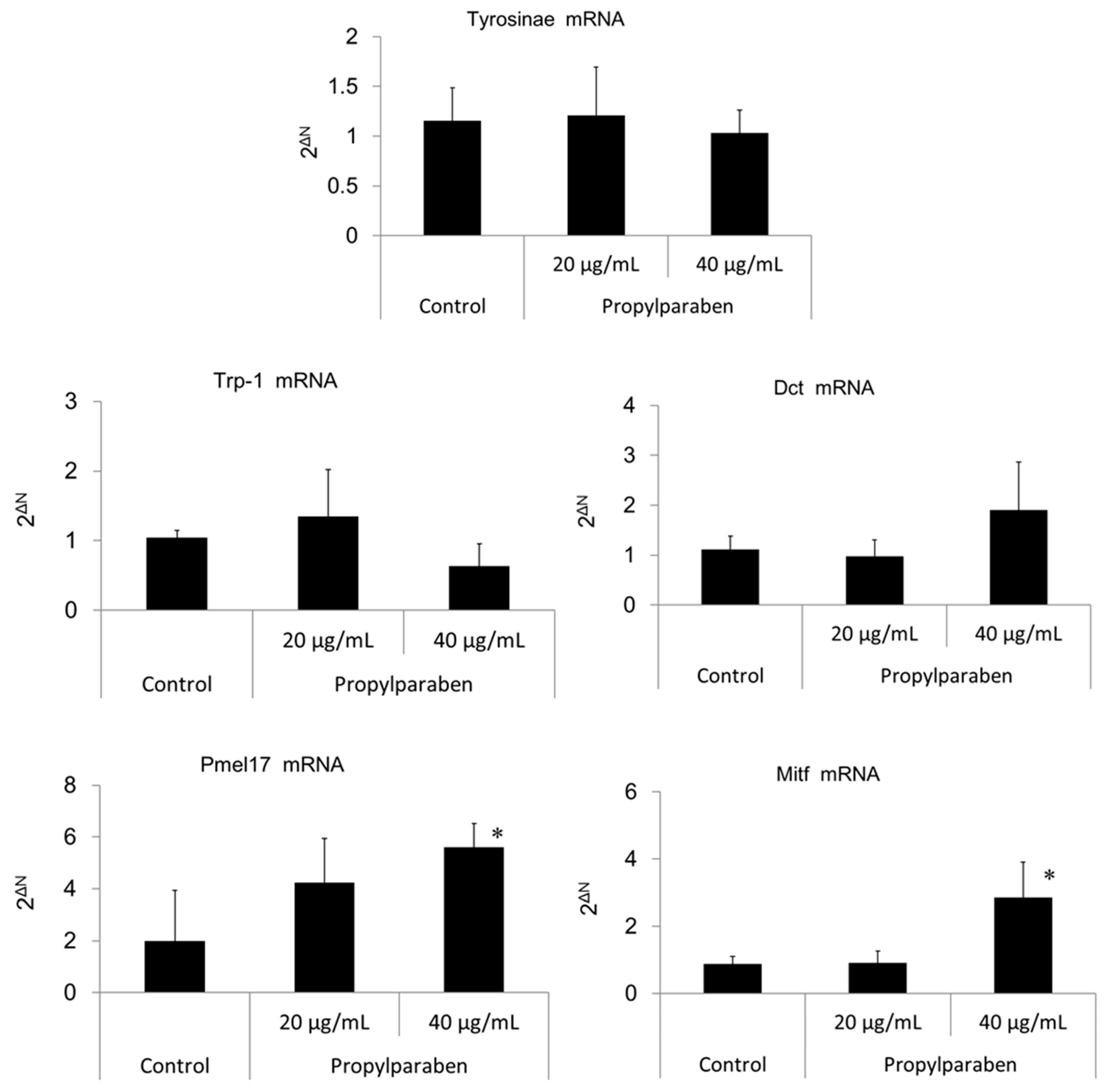
2.11. Effect of Propylparaben on mRNA Expression of Melanogenesis-Related Proteins
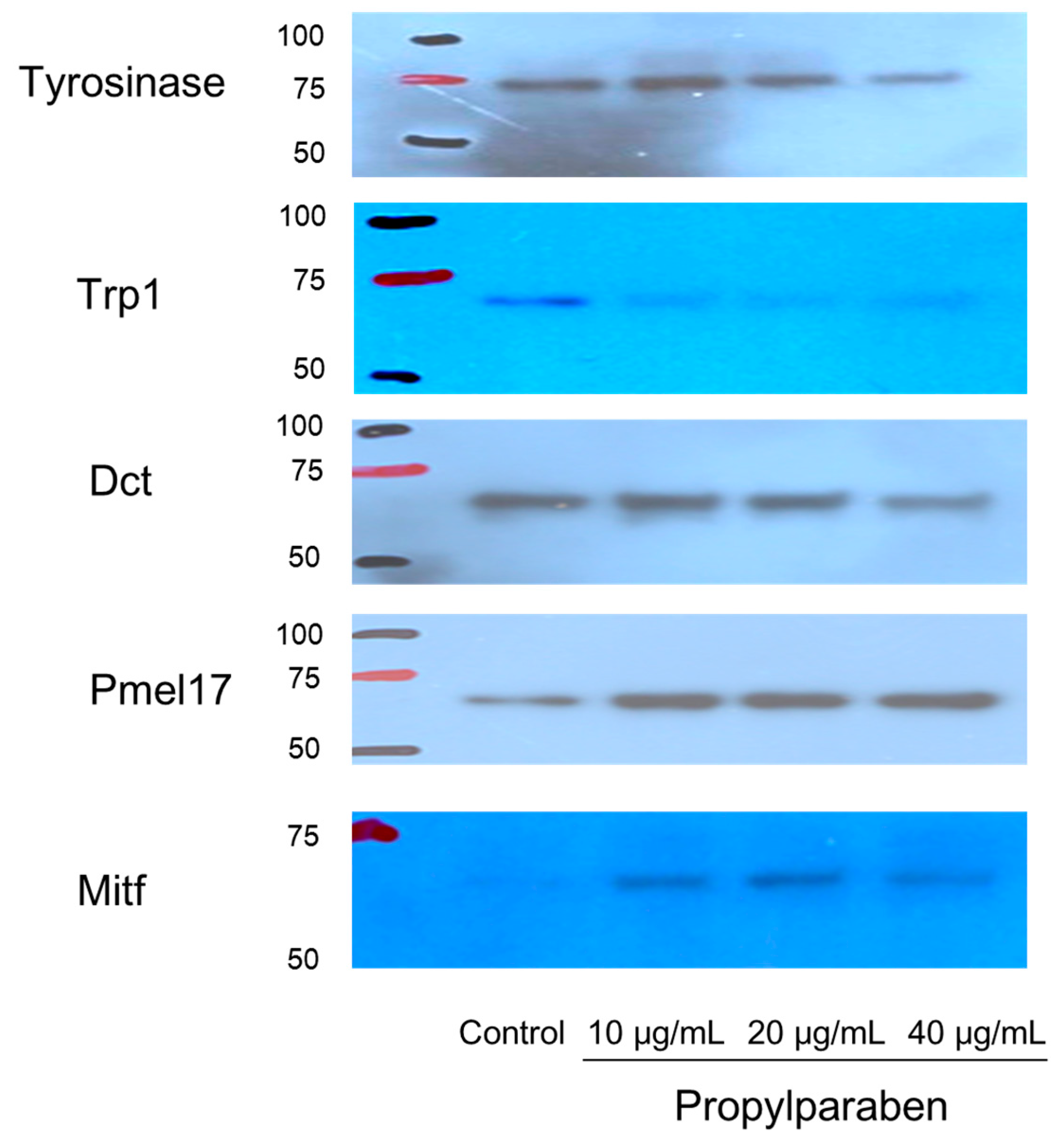
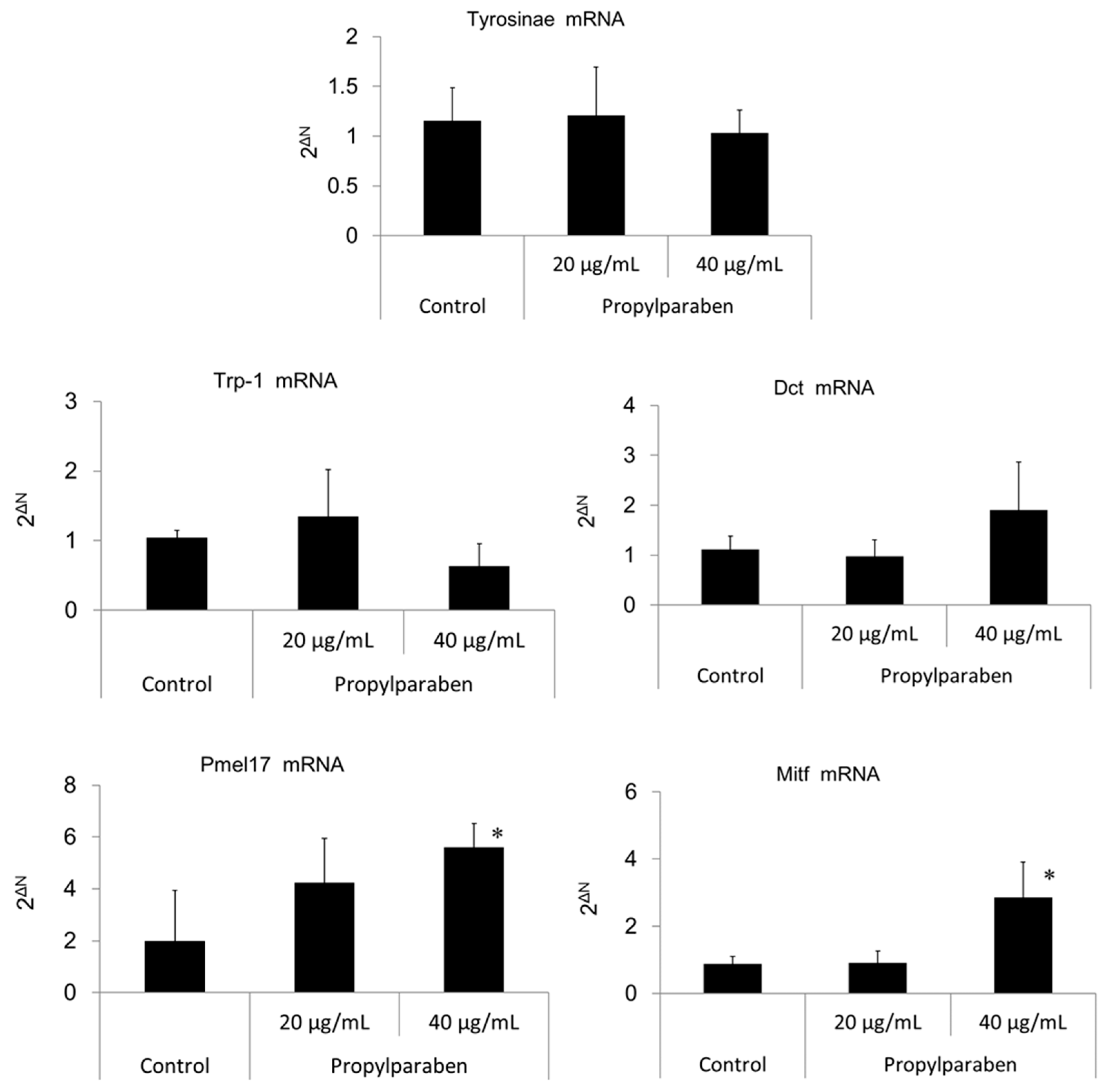
B16 melanoma cells were seeded onto 35-mm dishes at 1 mL per dish (100,000 cells/mL) and cultured for one day. Propylparaben was dissolved in DMSO solution to prepare 2 and 4 mg/mL solutions, 1 µL aliquots of these solutions were added to the dishes, and the cells were cultured for two days. The mRNA was extracted from cultured cells with an mRNA extraction kit (RNeasy Mini-kit, Qiagen K.K., Tokyo, Japan). The mRNA expression was measured for tyrosinase, Trp-1, Dct, Pmel17, Mitf, and glycerol-3-phosphate dehydrogenase (G3pdh) using a real-time polymerase chain reaction (PCR) system (ABI PRISM 7900HT, Applied Biosystems, Foster City, CA, USA).
2.12. Effects of Propylparaben on mRNA Expression of Melanogenesis-Related Proteins in Western Blotting
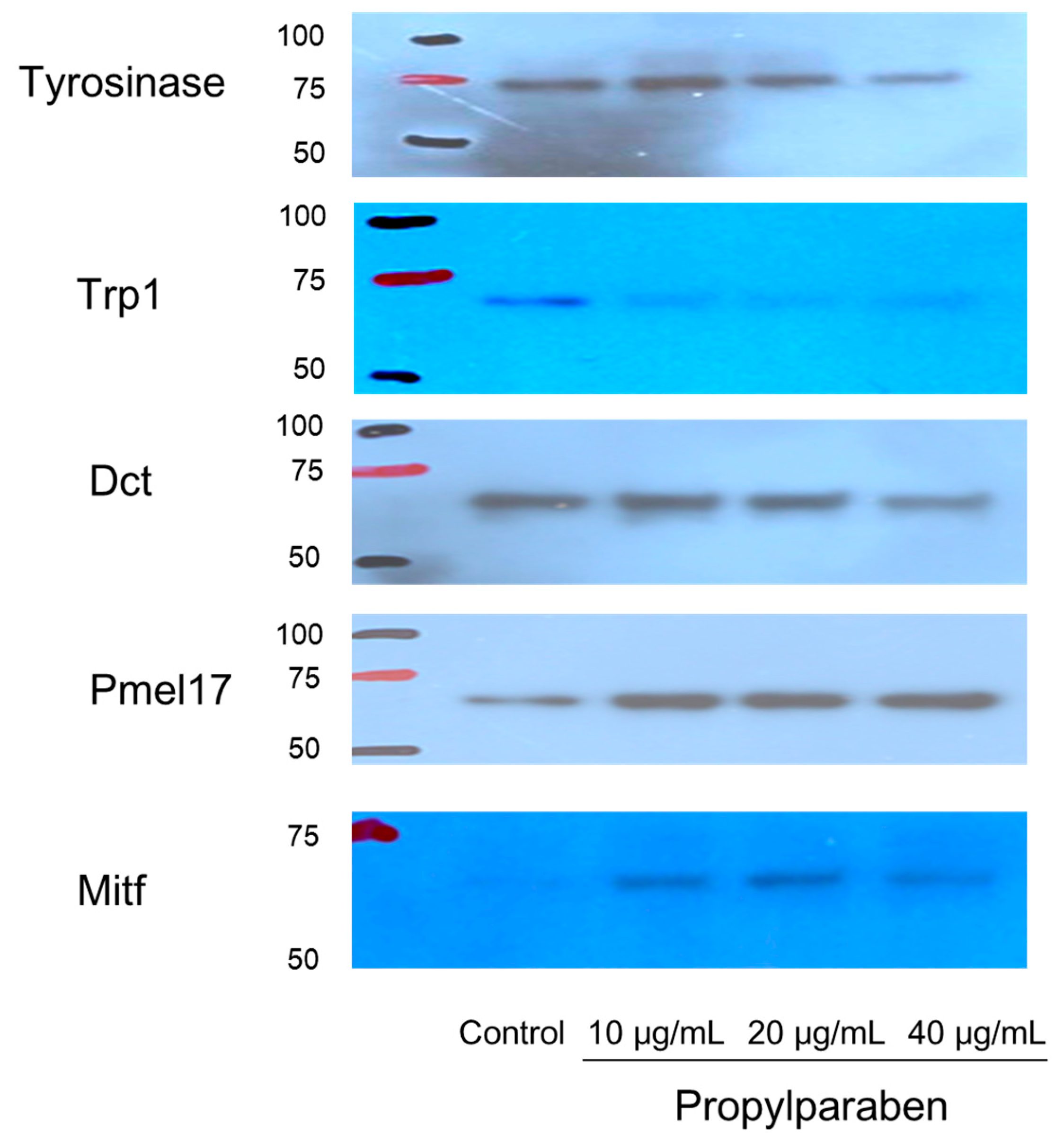
B16 melanoma cells were seeded into 10-cm dishes at 9.99 mL per dish (100,000 cells/mL) and cultured for one day. Propylparaben was dissolved in DMSO to prepare 1, 2, and 4 mg/mL solutions, 10 µL aliquots of these solutions were added to the dishes, and the cells were cultured for two days. The cells were harvested, and the proteins were quantified by the microplate reader measurement (at 560 nm) with a Pierce BCA Protein Assay kit (Thermo Fisher Scientific). After quantification, the proteins were separated with electrophoresis and subjected to a Western blot assay. The primary antibodies were anti-tyrosinase, anti-Trp1, anti-Dct, anti-Pmel17, and anti-Mitf antibodies, which were diluted 1000-fold. The secondary antibody was a peroxidase-labelled anti-rabbit IgG antibody, which was diluted 5000-fold in Tris-buffered saline (TBS) solution.
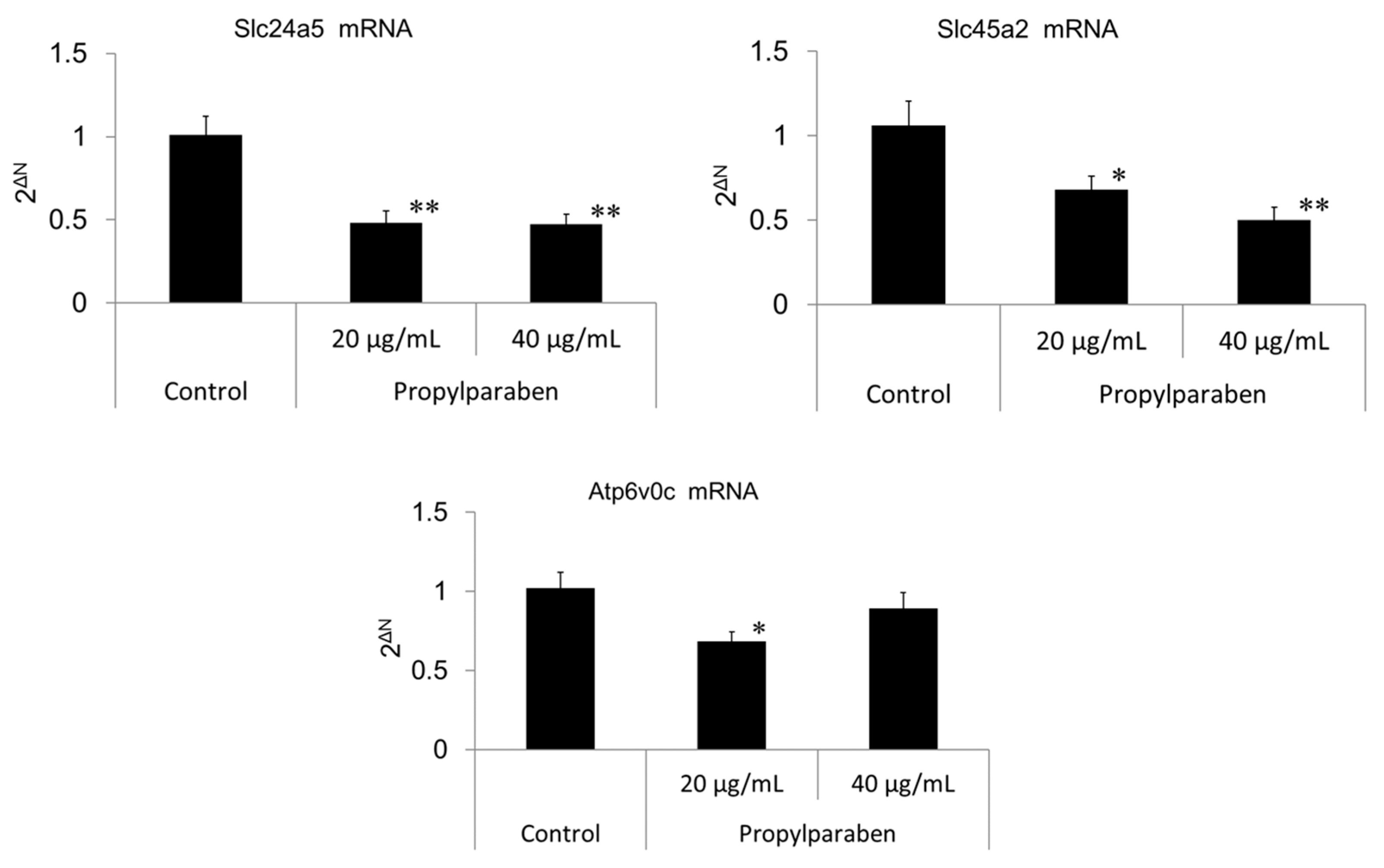
2.13. Effect of Propylparaben on mRNA Expression of Melanosomal pH Regulators (Slc24a5, Slc45a2, Atp6v0c-Encoded V-ATPase Sub-Unit, and G3pdh)
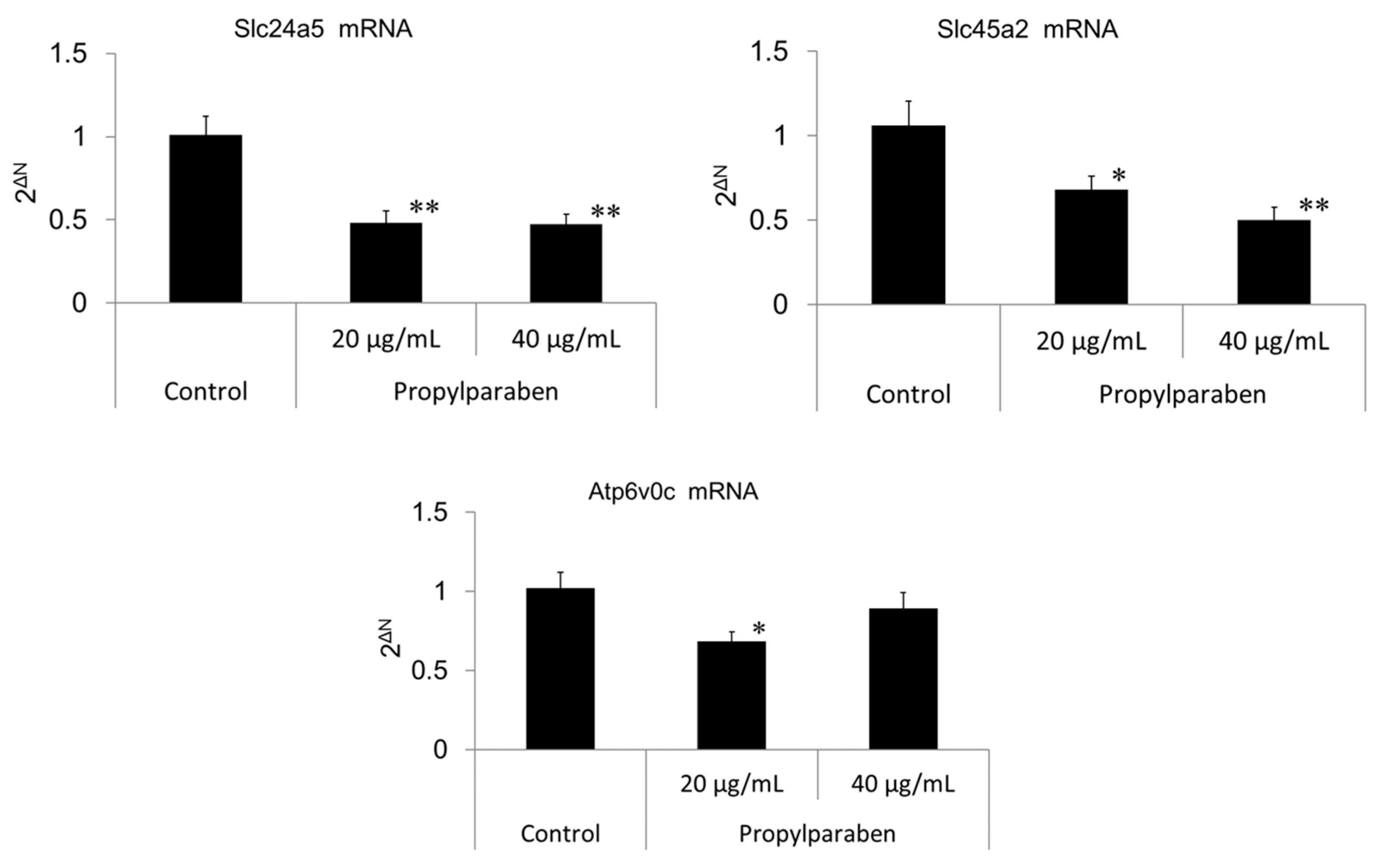
Cells were seeded into 35-mm dishes at 0.999 mL per dish (30,000 cells/mL) and cultured for one day. Propylparaben was dissolved in DMSO to prepare 2 and 4 mg/mL solutions, which were added to the dishes at 1 µL per dish, and the cells were cultured for three days (n = 3). mRNA was extracted from the cultured cells with the mRNA extraction kit (RNeasy Mini-kit) mRNA expression was measured for Slc24a5, Slc45a2, Atp6v0c-encoded sub-unit protein, and G3pdh using the real-time PCR system. The primers for Slc24a5, Slc45a2, Atp6v0c, and G3pdh were acquired from Qiagen K.K.
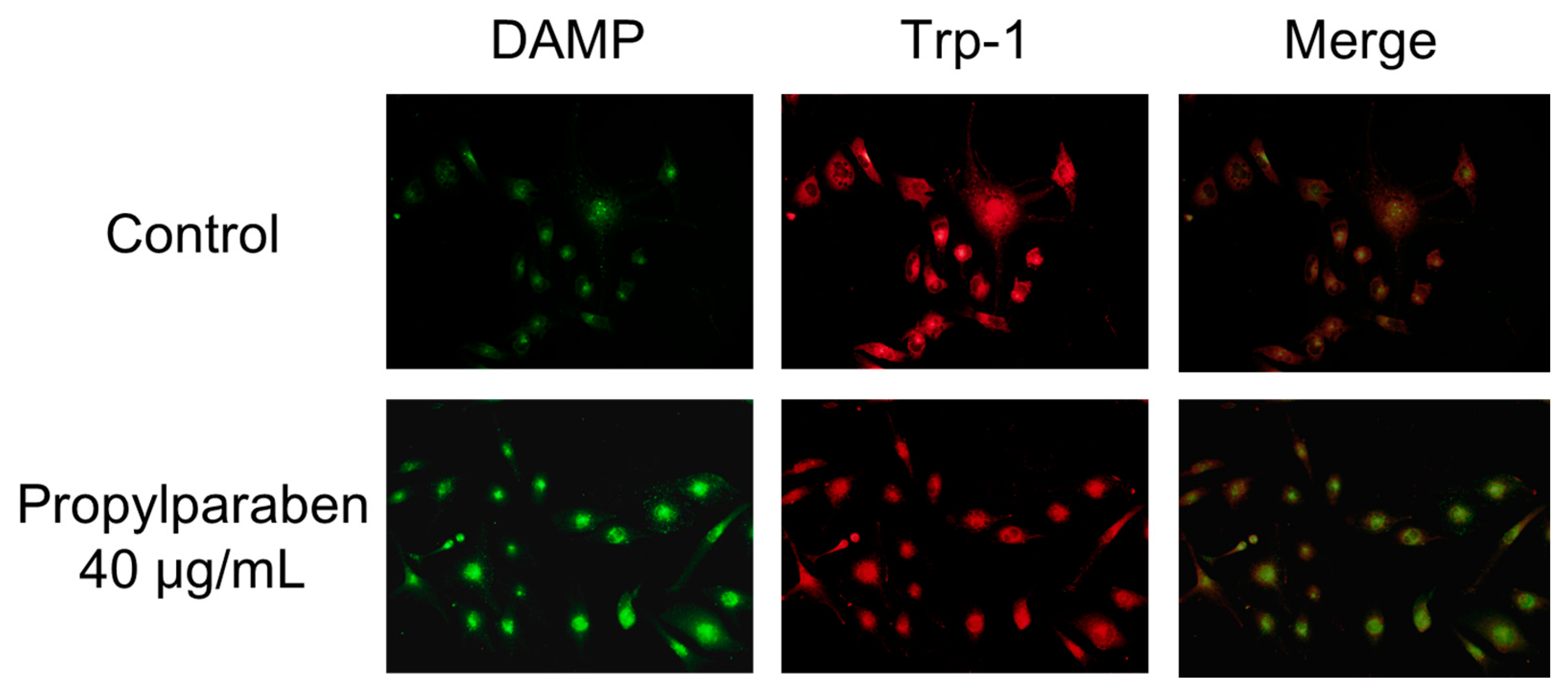
2.14. Effect of Propylparaben on Melanosome pH
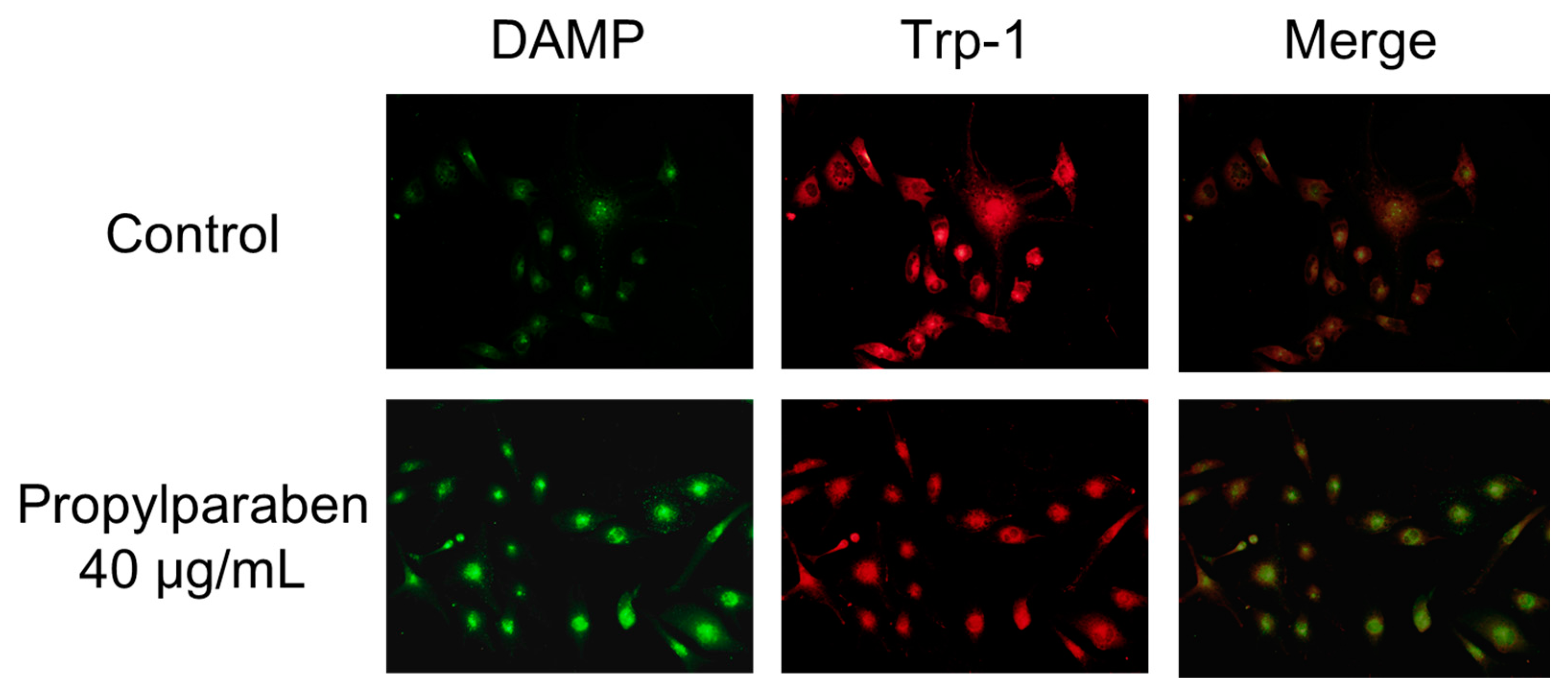
Cells were added to chamber slides at 0.999 mL per slide (10,000 cells/mL) and cultured for one day. Propylparaben was dissolved in DMSO to prepare 2.5 and 5 mg/mL solutions, which were added to the slides at 1 µL per slide, and the cells were cultured for three days. The chamber slides were washed with serum-free DMEM, and a 30 µmol/L DAMP solution was prepared with serum-free DMEM and added to the slides at 1 mL per slide. The cells were incubated for 40 min and then washed with PBS solution, 1 mL of 4% paraformaldehyde was added, and the slides were left to stand for 20 min to fix the cells in paraformaldehyde. After fixation, the cells were washed with PBS, 1 mL of 50 mmol/L NH4Cl was added, and the cells were incubated for 10 min. When DAMP is incubated with live cells, its intake is dependent on the intracellular organelle acidity pH. Any acidic granules with DAMP accumulation in fixed cells can be detected with an anti-dinitrophenol (DNP) antibody and a secondary antibody. DAMP is an acidotropic reagent and fluorescence will be stronger when the interior of the cell is acidic. The cells were permeabilized with 0.1% Triton X-100, isothiocyanate Alexa Fluor 488 goat anti-rabbit IgG antibody and an anti-2,4-dinitrophenol antibody diluted 2000-fold with 1% bovine serum albumin (BSA) was added. The cells were incubated for two hours. The cells were washed four times with PBS solution. The primary antibody (rat anti-Trp-1 antibody) was then added, and the cells incubated for two hours at room temperature. The cells were washed with PBS solution three times, then 2000-fold diluted Alexa Fluor 546 goat anti-rat IgG antibody was added and the cells incubated for one hour at room temperature. The cells were stained and examined under a fluorescence microscope (BX51; Olympus, Tokyo, Japan).
2.15. Statistical Analysis
Measured values were analyzed for significant differences using an unpaired t-test function (two-tailed) in Microsoft Excel. p < 0.05 was considered statistically significant.
4. Discussion
DOPA staining of the collected melanosome fraction revealed that tyrosinase activity decreased when the intramelanosomal pH was acidic. Western blotting revealed a decrease in tyrosinase protein content with acidification of intramelanosomal pH. In other words, we found that acidification of intramelanosomal pH enables tyrosinase degradation, as well as decreases tyrosinase activity.
Melanosomal pH is regulated by V-ATPase, which mediates H
+ influx and Na
+/H
+ exchangers, which mediate H
+ extrusion. We investigated the fluctuation in melanin content when acidification of pH was blocked with bafilomycin A1, a V-ATPase inhibitor. We found that both melanin and tyrosinase contents increased in bafilomycin A1-treated cells. These results suggested enhanced melanogenesis occurred after blocking intramelanosomal pH acidification. Consequentially, we considered that suppression of melanogenesis is facilitated by an acidic pH, and we, thus, investigated this mechanism with a variety of enzyme inhibitors. We found that both inhibitors of aspartic protease and cysteine protease decreased the degradation of tyrosinase. Accordingly, we consider that aspartic protease and cysteine protease are involved in accelerating tyrosinase degradation in acidic melanosomes. Aspartic proteases are one of the acidic proteases, and cathepsin D and cathepsin E are considered to be aspartic proteases in the degradation of tyrosinase in melanosomes. Cysteine proteases are another type of acidic proteases, and cathepsin L2 is considered to be a cysteine protease in the degradation of tyrosinase in melanosomes. In studies on racial or skin color differences, it has been reported that melanosomes in keratinocytes from light skin are more rapidly degraded than those from dark skin [
14] and that the activity of cathepsin L2, which is a lysosomal enzyme, is higher in keratinocytes from lightly-pigmented skin relative to darkly-pigmented skins [
15].
Slc45a2 is a known Na
+/H
+ exchanger, and increased Slc45a2 expression intensifies H
+ extrusion. This increase results in greater melanosomal H
+ extrusion and pH-neutralization of the intramelanosomal environment. Decreased Slc45a2 expression weakens melanosomal H
+ extrusion, which leads to greater melanosomal H
+ retention and the resulting acidification of the intramelanosomal environment. A mutation in the Slc45a2 gene induces oculocutaneous albinism type 4 [
16]. A comparison of mutant Slc45a2-gene-transfected melanocytes with normal melanocytes showed a lower melanin content in the latter cells, and Slc45a2 mutation abolishes or reduces melanogenesis [
17]. Slc24a5 plays a role in potassium-dependent Na
+/H
+ exchangers. H
+ is transported into the melanosome through V-ATPase, and Na
+ intake is mediated through Na
+/H
+ exchangers through an electrochemical gradient. Na
+ extrusion is conjugated with Ca
2+ influx through Slc24a5. In other words, decreased Slc24a5 expression results in intramelanosomal accumulation of Na
+ and a cessation of Na
+/H
+ exchange, which results in melanosomal retention of acidity [
18,
19]. Reduction of melanogenesis from normal levels has been reported to occur in B16 melanoma cells with Slc24a5 knockdown [
20].
Parabens are used in cosmetic formulations, and the C1 to C4 (side chain) parabens are indicated to reduce tyrosinase activity as the chain length increases. Parabens reported to have a wide antibacterial spectrum and high safety were investigated for anti-microbial action and were found to have a drug effect increasing in intensity in the order of methylparaben < ethylparaben < propylparaben < butylparaben. Accordingly, a very good linear relationship between the number of carbons on the alkyl chain and affinity was established. The carbon chain was determined to be the major player in differences in anti-microbial effects. Based on this good linear relationship between affinity and the number of carbons on the alkyl chain, proliferation of cells with a high number of carbons, and the relationships between differences in the number of carbons and differences in anti-microbial effects, we surmise that decreases in tyrosinase activity may be related to increases in paraben chain length. Propylparaben has an anti-melanogenic effect, and we consider that the mechanism of its anti-melanogenic effect encompasses intramelanosomal degradation of the protein tyrosinase, Trp-1, and Dct because these protein contents were reduced without any corresponding decrease in mRNA expression. Propylparaben also increased Mitf, but Mitf serves as a survival factor of melanocytes [
21], and so we believe that propylparaben will not induce cytotoxicity to melanocytes. On the other hand, we observed acidic changes in the melanosomes due to propylparaben. To determine the mechanism for this phenomenon, we investigated mRNA expression for Slc24a5 and Slc45a2, which are proteins involved in intramelanosomal pH regulation. We found that mRNA expression for Slc45a2, which plays a role in H
+ extrusion, was reduced in a concentration-dependent manner. Expression of Slc24a5, which is an Na
+/Ca
2+ exchanger, was also reduced. Based on these results, we determined a mechanism for the anti-melanogenic action of propylparaben, as follows: H
+ influx is reduced with decreased expression of the Slc45a2 gene, and intramelanosomal pH becomes acidic. Based on our findings, we consider that propylparaben-induced melanosomal acidification and the resulting tyrosinase degradation represents a promising new cosmetic mechanism.
Pmel17 exists in immature melanosome (stage II, III), and Trp-1 exists mainly in mature melanosome (stage IV) [
22,
23]. Dct exists mainly in stage I melanosome/coated endosome [
24]. B16 melanoma cells treated with propylparaben were thought to consist of many stage II and III melanosomes. The Pmel17 level was thought to be increased by propylparaben because the maturity of melanosome was controlled by promoting the acidification of the pH of melanosome. Moreover, as Mitf participates in the survival of pigment cells, Mitf level was increased not only by propylparaben, but also by 4-n-butylresorcinol, which inhibits melanin production at non-cytotoxic concentrations in cultured B16 melanoma cells (data not shown). Increased melanin inhibition showed a relationship with increased Mitf level in B16 melanoma cells treated with propylparaben, and further research is required on this.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}