
The Role of Oxidative Stress in TB Meningitis and Therapeutic Options

 ,
, 



Abstract
:1. Introduction: Meningitis Disease Process
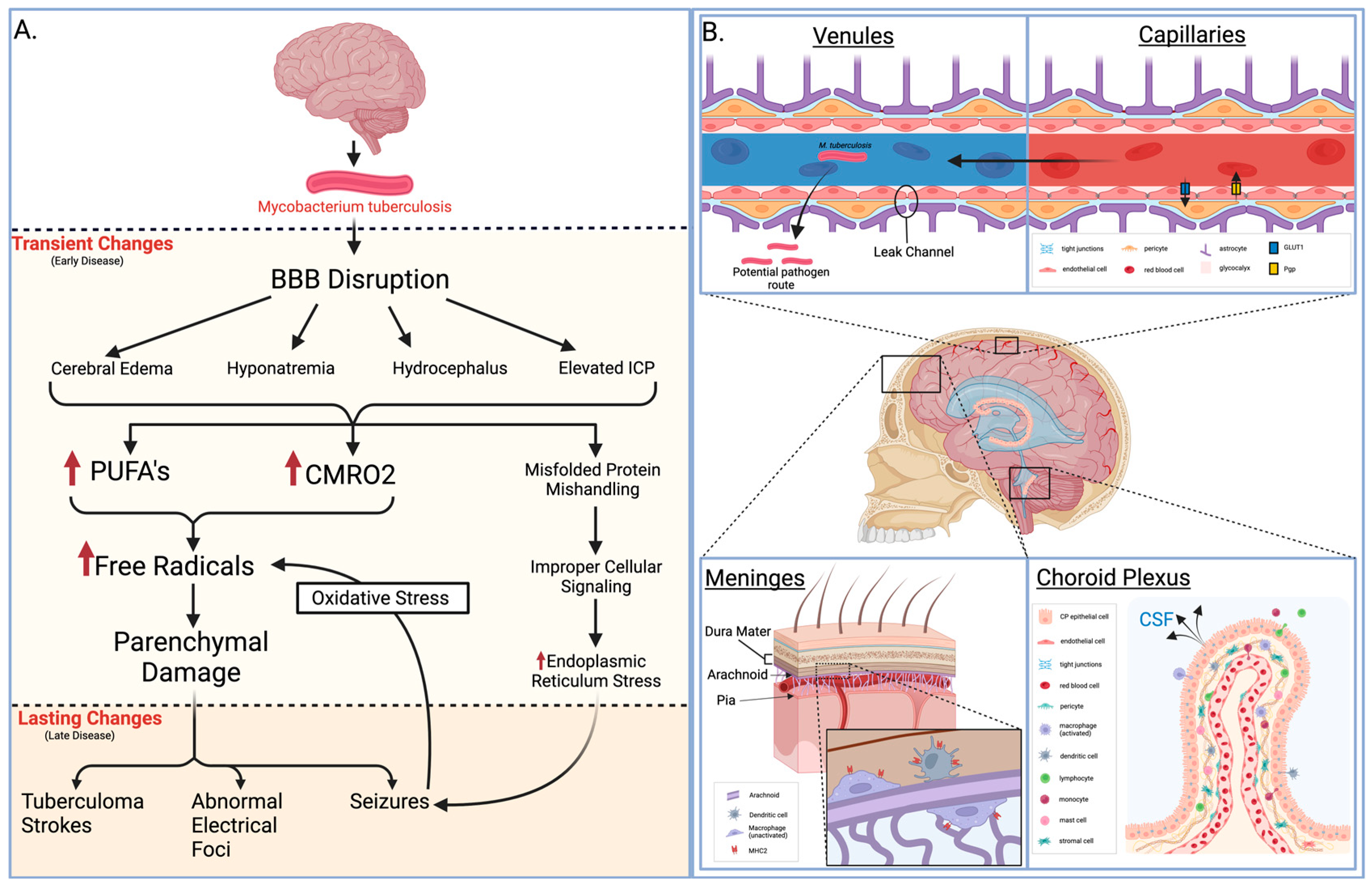
1.1. Molecular Bases of Meningitis Pathophysiology
1.2. Pathology of Meningitis
1.3. US Statistics of Meningitis
1.4. Global Statistics of Meningitis
2. Tuberculous Meningitis and Non-Tuberculous Meningitis
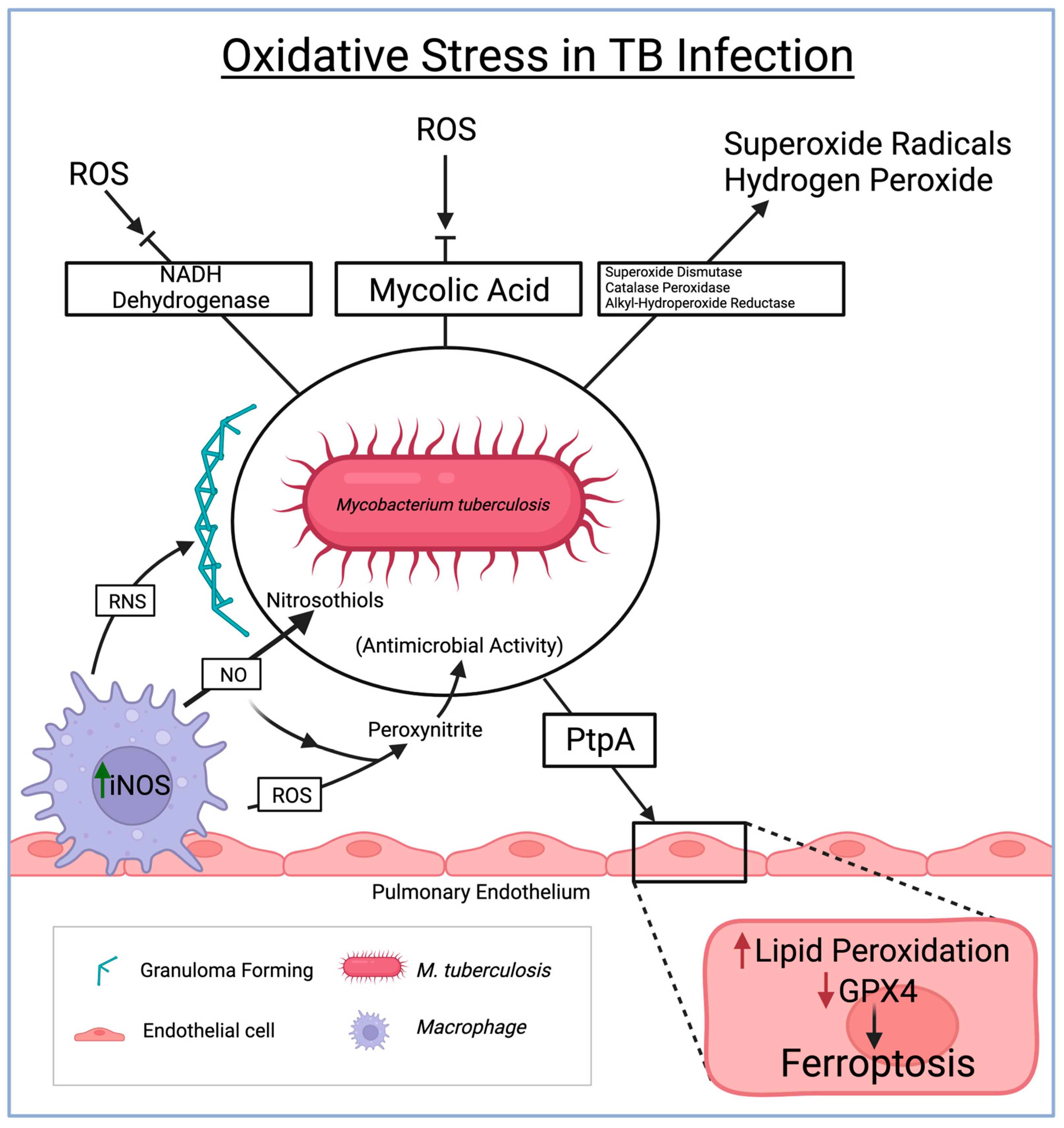
3. Oxidative Stress in Tuberculosis
4. TB Meningitis Treatment Options
5. Alternative Therapy for Oxidative Stress-Induced TB Meningitis
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Griffiths, M.J.; McGill, F.; Solomon, T. Management of acute meningitis. Clin. Med. 2018, 18, 164–169. [Google Scholar] [CrossRef]
- Erickson, M.A.; Banks, W.A. Neuroimmune axes of the Blood–Brain barriers and Blood–Brain interfaces: Bases for physiological regulation, disease states, and pharmacological interventions. Pharmacol. Rev. 2018, 70, 278–314. [Google Scholar] [CrossRef]
- Dias, M.C.; Mapunda, J.A.; Vladymyrov, M.; Engelhardt, B. Structure and junctional complexes of endothelial, epithelial and glial brain barriers. Int. J. Mol. Sci. 2019, 20, 5372. [Google Scholar] [CrossRef]
- Butt, A.M.; Jones, H.C.; Abbott, N.J. Electrical resistance across the blood-brain barrier in anaesthetized rats: A developmental study. J. Physiol. 1990, 429, 47–62. [Google Scholar] [CrossRef]
- Join-Lambert, O.; Morand, P.; Carbonnelle, É.; Coureuil, M.; Bille, E.; Bourdoulous, S.; Nassif, X. Mechanisms of meningeal invasion by a bacterial extracellular pathogen, the example of Neisseria meningitidis. Prog. Neurobiol. 2010, 91, 130–139. [Google Scholar] [CrossRef]
- Ghersi-Egea, J.; Strazielle, N.; Catala, M.; Silva-Vargas, V.; Doetsch, F.; Engelhardt, B. Molecular anatomy and functions of the choroidal blood-cerebrospinal fluid barrier in health and disease. Acta Neuropathol. 2018, 135, 337–361. [Google Scholar] [CrossRef]
- Weller, R.O.; Sharp, M.M.; Christodoulides, M.; Carare, R.O.; Møllgård, K. The meninges as barriers and facilitators for the movement of fluid, cells and pathogens related to the rodent and human CNS. Acta Neuropathol. 2018, 135, 363–385. [Google Scholar] [CrossRef]
- Christodoulides, M.; Makepeace, B.L.; Partridge, K.; Kaur, D.; Fowler, M.; Weller, R.O.; Heckels, J.E. Interaction of Neisseria meningitidis with Human Meningeal Cells Induces the Secretion of a Distinct Group of Chemotactic, Proinflammatory, and Growth-Factor Cytokines. Infect. Immun. 2002, 70, 4035–4044. [Google Scholar] [CrossRef]
- Rua, R.; McGavern, D.B. Advances in meningeal immunity. Trends Mol. Med. 2018, 24, 542–559. [Google Scholar] [CrossRef]
- Misra, U.K.; Tripathi, A.; Kumar, M. Oxidative and endoplasmic reticulum stress in tuberculous meningitis related seizures. Epilepsy Res. 2019, 156, 106160. [Google Scholar] [CrossRef]
- Charalambous, L.T.; Premji, A.; Tybout, C.; Hunt, A.; Cutshaw, D.; Elsamadicy, A.A.; Yang, S.; Xie, J.; Giamberardino, C.; Pagadala, P.; et al. Prevalence, healthcare resource utilization and overall burden of fungal meningitis in the United States. J. Med. Microbiol. 2018, 67, 215–227. [Google Scholar] [CrossRef]
- Bhimraj, A. Acute community-acquired bacterial meningitis in adults: An evidence-based review. Clevel. Clin. J. Med. 2012, 79, 393–400. [Google Scholar] [CrossRef]
- Mount, H.R.; Boyle, S.D. Aseptic and bacterial meningitis: Evaluation, treatment, and prevention. PubMed 2017, 96, 314–322. [Google Scholar]
- Amidu, N.; Antuamwine, B.B.; Addai-Mensah, O.; Abdul-Karim, A.; Azure, S.; Abubakari, B.B.; Abenyeri, J.; Opoku, A.S.; Nkukah, J.E.; Najibullah, A.S. Diagnosis of bacterial meningitis in Ghana: Polymerase chain reaction versus latex agglutination methods. PLoS ONE 2019, 14, e0210812. [Google Scholar] [CrossRef]
- Houri, H.; Pormohammad, A.; Riahi, S.M.; Nasiri, M.J.; Fallah, F.; Dabiri, H.; Pouriran, R. Acute bacterial meningitis in Iran: Systematic review and meta-analysis. PLoS ONE 2017, 12, e0169617. [Google Scholar] [CrossRef]
- Abassi, M.; Boulware, D.R.; Rhein, J. Cryptococcal meningitis: Diagnosis and Management Update. Curr. Trop. Med. Rep. 2015, 2, 90–99. [Google Scholar] [CrossRef]
- Bahr, N.C.; Nuwagira, E.; Evans, E.E.; Cresswell, F.V.; Bystrom, P.V.; Byamukama, A.; Bridge, S.; Bangdiwala, A.S.; Meya, D.B.; Denkinger, C.M.; et al. Diagnostic accuracy of Xpert MTB/RIF Ultra for tuberculous meningitis in HIV-infected adults: A prospective cohort study. Lancet Infect. Dis. 2018, 18, 68–75. [Google Scholar] [CrossRef]
- Rajasingham, R.; Smith, R.; Park, B.J.; Jarvis, J.N.; Govender, N.P.; Chiller, T.; Denning, D.W.; Loyse, A.; Boulware, D.R. Global burden of disease of HIV-associated cryptococcal meningitis: An updated analysis. Lancet Infect. Dis. 2017, 17, 873–881. [Google Scholar] [CrossRef]
- Antinori, S.; Corbellino, M.; Meroni, L.; Resta, F.; Sollima, S.; Tonolini, M.; Tortorano, A.M.; Milazzo, L.; Bello, L.; Furfaro, E.; et al. Aspergillus meningitis: A rare clinical manifestation of central nervous system aspergillosis. Case report and review of 92 cases. J. Infect. 2013, 66, 218–238. [Google Scholar] [CrossRef]
- Stevens, D.A.; Zhang, Y.; Finkelman, M.; Pappagianis, D.; Clemons, K.V.; Martinez, M. Cerebrospinal Fluid (1,3)-Beta-d-Glucan Testing Is Useful in Diagnosis of Coccidioidal Meningitis. J. Clin. Microbiol. 2016, 54, 2707–2710. [Google Scholar] [CrossRef]
- Wheat, L.J.; Batteiger, B.E.; Sathapatayavongs, B. Histoplasma capsulatum Infections of the Central Nervous System. Medicine 1990, 69, 244. [Google Scholar] [CrossRef]
- Wunrow, H.Y.; Bender, R.G.; Vongpradith, A.; Sirota, S.B.; Swetschinski, L.R.; Novotney, A.; Gray, A.P.; Ikuta, K.S.; Sharara, F.; Wool, E.E.; et al. Global, regional, and national burden of meningitis and its aetiologies, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet Neurol. 2023, 22, 685–711. [Google Scholar] [CrossRef]
- Kwambana-Adams, B. Global burden of meningitis and implications for strategy. Lancet Neurol. 2023, 22, 646–648. [Google Scholar] [CrossRef]
- Qu, C.; Wang, Y.; Wang, X.; He, R.; Cao, H.; Liu, B.; Zhang, H.; Zhang, N.; Lai, Z.; Dai, Z.; et al. Global Burden and Its Association with Socioeconomic Development Status of Meningitis Caused by Specific Pathogens over the Past 30 years: A Population-Based Study. Neuroepidemiology 2023, 57, 316–335. [Google Scholar] [CrossRef]
- Kumar, R.; Singh, S.; Kohli, N. A diagnostic rule for tuberculous meningitis. Arch. Dis. Child. 1999, 81, 221–224. [Google Scholar] [CrossRef]
- Nudelman, Y.; Tunkel, A.R. Bacterial meningitis. Drugs 2009, 69, 2577–2596. [Google Scholar] [CrossRef]
- Zwijnenburg, P.J.; Van Der Poll, T.; Roord, J.J.; Van Furth, A.M. Chemotactic Factors in Cerebrospinal Fluid during Bacterial Meningitis. Infect. Immun. 2006, 74, 1445–1451. [Google Scholar] [CrossRef]
- Van Furth, A.M.; Seijmonsbergen, E.M.; Langermans, J.A.M.; Groeneveld, P.H.P.; De Bel, C.E.; Van Furth, R. High levels of interleukin 10 and tumor necrosis factor in cerebrospinal fluid during the onset of bacterial meningitis. Clin. Infect. Dis. 1995, 21, 220–222. [Google Scholar] [CrossRef]
- Naghavi, M.; Abajobir, A.A.; Abbafati, C.; Abbas, K.; Abd-Allah, F.; Abera, S.F.; Aboyans, V.; Adetokunboh, O.; Afshin, A.; Agrawal, A.; et al. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1151–1210. [Google Scholar] [CrossRef]
- Van De Beek, D.; Brouwer, M.C.; Koedel, U.; Wall, E.C. Community-acquired bacterial meningitis. Lancet 2021, 398, 1171–1183. [Google Scholar] [CrossRef]
- Hasbun, R.; Rosenthal, N.; Balada-Llasat, J.; Chung, J.; Duff, S.; Bozzette, S.A.; Zimmer, L.; Ginocchio, C.C. Epidemiology of Meningitis and Encephalitis in the United States, 2011–2014. Clin. Infect. Dis. 2017, 65, 359–363. [Google Scholar] [CrossRef]
- Hasbun, R.; Wootton, S.H.; Rosenthal, N.; Balada-Llasat, J.M.; Chung, J.; Duff, S.; Bozzette, S.A.; Zimmer, L.; Ginocchio, C.C. Epidemiology of Meningitis and encephalitis in infants and Children in the United States, 2011–2014. Pediatr. Infect. Dis. J. 2019, 38, 37–41. [Google Scholar] [CrossRef]
- Wright, W.F.; Pinto, C.N.; Palisoc, K.; Baghli, S. Viral (aseptic) meningitis: A review. J. Neurol. Sci. 2019, 398, 176–183. [Google Scholar] [CrossRef]
- Williamson, P.R.; Jarvis, J.N.; Panackal, A.A.; Fisher, M.C.; Molloy, S.F.; Loyse, A.; Harrison, T.S. Cryptococcal meningitis: Epidemiology, immunology, diagnosis and therapy. Nat. Rev. Neurol. 2016, 13, 13–24. [Google Scholar] [CrossRef]
- Seddon, J.A.; Tugume, L.; Solomons, R.; Prasad, K.; Bahr, N.C. The current global situation for tuberculous meningitis: Epidemiology, diagnostics, treatment and outcomes. Wellcome Open Res. 2019, 4, 167. [Google Scholar] [CrossRef]
- Dodd, P.J.; Osman, M.; Cresswell, F.V.; Stadelman, A.; Lân, N.H.; Thuong, N.T.T.; Muzyamba, M.; Glaser, L.; Dlamini, S.N.; Seddon, J.A. The global burden of tuberculous meningitis in adults: A modelling study. PLoS Glob. Public Health 2021, 1, e0000069. [Google Scholar] [CrossRef]
- Jung, J.; Shin, I.; Choi, Y. A Rare Case of Nontuberculous Mycobacterial Abscess Mimicking Brain Tumor in an Immunocompetent Patient. Brain Tumor Res. Treat. 2023, 11, 219–222. [Google Scholar] [CrossRef]
- Banks, H.S.; Broom, J.C.; Crawford, J.V. Treatment of non-tuberculous meningitis. Proc. R. Soc. Med. 1953, 46, 149–154. [Google Scholar]
- Arshad, A.; Dayal, S.; Gadhe, R.; Mawley, A.; Shin, K.; Tellez, D.; Phan, P.; Venketaraman, V. Analysis of tuberculosis meningitis Pathogenesis, diagnosis, and treatment. J. Clin. Med. 2020, 9, 2962. [Google Scholar] [CrossRef]
- Donald, P.R.; Schaaf, H.S.; Schoeman, J. Tuberculous meningitis and miliary tuberculosis: The Rich focus revisited. J. Infect. 2005, 50, 193–195. [Google Scholar] [CrossRef]
- Davis, A.; Rohlwink, U.K.; Proust, A.; Figaji, A.; Wilkinson, R.J. The pathogenesis of tuberculous meningitis. J. Leukoc. Biol. 2019, 105, 267–280. [Google Scholar] [CrossRef]
- Shastri, M.D.; Shukla, S.D.; Chong, W.C.; Dua, K.; Peterson, G.; Patel, R.; Hansbro, P.M.; Eri, R.; O’Toole, R. Role of oxidative stress in the pathology and management of human tuberculosis. Oxidative Med. Cell. Longev. 2018, 2018, 7695364. [Google Scholar] [CrossRef]
- Tyagi, P.; Dharmaraja, A.T.; Bhaskar, A.; Chakrapani, H.; Singh, A. Mycobacterium tuberculosis has diminished capacity to counteract redox stress induced by elevated levels of endogenous superoxide. Free Radic. Biol. Med. 2015, 84, 344–354. [Google Scholar] [CrossRef]
- Xiao, L.; Huang, H.; Fan, S.; Zheng, B.; Wu, J.; Zhang, J.; Pi, J.; Xu, J. Ferroptosis: A mixed blessing for infectious diseases. Front. Pharmacol. 2022, 13, 992734. [Google Scholar] [CrossRef]
- Imai, H.; Matsuoka, M.; Kumagai, T.; Sakamoto, T.; Koumura, T. Lipid Peroxidation-Dependent Cell Death Regulated by GPx4 and Ferroptosis. In Apoptotic and Non-Apoptotic Cell Death; Nagata, S., Nakano, H., Eds.; Current Topics in Microbiology and Immunology; Springer: Cham, Switzerland, 2016; Volume 403. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Liang, T.; Chen, J.; Xu, G.; Zhang, Z.; Xue, J.; Zeng, H.; Jiang, J.; Chen, T.; Qin, Z.; Li, H.; et al. Ferroptosis-related gene SOCS1, a marker for tuberculosis diagnosis and treatment, involves in macrophage polarization and facilitates bone destruction in tuberculosis. Tuberculosis 2022, 132, 102140. [Google Scholar] [CrossRef]
- Gan, B. Ferroptosis hijacking by Mycobacterium tuberculosis. Nat. Commun. 2023, 14, 1431. [Google Scholar] [CrossRef]
- Yang, C.; Yuk, J.; Jo, E. The role of nitric oxide in mycobacterial infections. Immune Netw. 2009, 9, 46. [Google Scholar] [CrossRef]
- Nahid, P.; Dorman, S.E.; Alipanah, N.; Barry, P.M.; Brożek, J.; Cattamanchi, A.; Chaisson, L.H.; Chaisson, R.E.; Daley, C.L.; Grzemska, M.; et al. Official American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America Clinical Practice Guidelines: Treatment of Drug-Susceptible Tuberculosis. Clin. Infect. Dis. 2016, 63, e147–e195. [Google Scholar] [CrossRef]
- Marx, G.E.; Chan, E.D. Tuberculous meningitis: Diagnosis and treatment Overview. Tuberc. Res. Treat. 2011, 2011, 798764. [Google Scholar] [CrossRef]
- Davis, A.; Meintjes, G.; Wilkinson, R.J. Treatment of tuberculous meningitis and its complications in adults. Curr. Treat. Options Neurol. 2018, 20, 5. [Google Scholar] [CrossRef]
- Thwaites, G.; Hien, T.T. Tuberculous meningitis: Many questions, too few answers. Lancet Neurol. 2005, 4, 160–170. [Google Scholar] [CrossRef]
- Kalita, J.; Misra, U.K.; Dubey, A. Role of Oxidative Stress in Tuberculous Meningitis: A Clinico-Radiological Correlation. J. Mol. Neurosci. 2019, 68, 287–294. [Google Scholar] [CrossRef]
- Kumar, R.; Kolloli, A.; Singh, P.; Vinnard, C.; Kaplan, G.; Subbian, S. Thalidomide and phosphodiesterase 4 inhibitors as host directed therapeutics for tuberculous Meningitis: Insights from the Rabbit Model. Front. Cell. Infect. Microbiol. 2020, 9, 450. [Google Scholar] [CrossRef]
- Thwaites, G.; Van Toorn, R.; Schoeman, J. Tuberculous meningitis: More questions, still too few answers. Lancet Neurol. 2013, 12, 999–1010. [Google Scholar] [CrossRef]
- Török, M.E. Tuberculous meningitis: Advances in diagnosis and treatment. Br. Med. Bull. 2015, 113, 117–131. [Google Scholar] [CrossRef]
- Thwaites, G.; Bang, N.D.; Dung, N.H.; Quy, H.T.; Oanh, D.T.T.; Thoa, N.T.C.; Hien, N.Q.; Thuc, N.T.; Hải, N.N.; Lan, N.T.N.; et al. Dexamethasone for the treatment of tuberculous meningitis in adolescents and adults. N. Engl. J. Med. 2004, 351, 1741–1751. [Google Scholar] [CrossRef]
- Daniel, B.; Gathungu, G.; Natrajan, M. Tuberculous meningitis in children: Clinical management & outcome. Indian J. Med. Res. 2019, 150, 117. [Google Scholar] [CrossRef]
- Davis, A.; Donovan, J.; Bremer, M.; Van Toorn, R.; Schoeman, J.; Dadabhoy, A.; Lai, R.; Cresswell, F.; Boulware, D.R.; Wilkinson, R.J.; et al. Host directed therapies for tuberculous meningitis. Wellcome Open Res. 2021, 5, 292. [Google Scholar] [CrossRef]
- Ren, J.; Li, C.; Yan, X.; Qu, Y.; Yang, Y.; Guo, Z. Crosstalk between Oxidative Stress and Ferroptosis/Oxytosis in Ischemic Stroke: Possible Targets and Molecular Mechanisms. Oxidative Med. Cell. Longev. 2021, 2021, 6643382. [Google Scholar] [CrossRef]
- Entezari, S.; Haghi, S.M.; Norouzkhani, N.; Sahebnazar, B.; Vosoughian, F.; Akbarzadeh, D.; Islampanah, M.; Naghsh, N.; Abbasalizadeh, M.; Deravi, N. Iron chelators in treatment of iron overload. J. Toxicol. 2022, 2022, 4911205. [Google Scholar] [CrossRef]
- Zhang, S.; Wei, X.; Anderson, G.J.; Li, R.; Gao, L.; Chen, S.; Zhao, J.; Liu, S. Double-edge sword roles of iron in driving energy production versus instigating ferroptosis. Cell Death Dis. 2022, 13, 40. [Google Scholar] [CrossRef]
- Feng, W.; Xiao, Y.; Zhao, C.; Zhang, Z.; Liu, W.; Ma, J.; Ganz, T.; Zhang, J.; Liu, S. New deferric amine compounds efficiently chelate excess iron to treat iron overload disorders and to prevent ferroptosis. Adv. Sci. 2022, 9, 2202679. [Google Scholar] [CrossRef]
- Cronje, L.; Bornman, L. Iron overload and tuberculosis: A case for iron chelation therapy. Int. J. Tuberc. Lung Dis. Off. J. Int. Union Against Tuberc. Lung Dis. 2005, 9, 2–9. [Google Scholar]
- Adeoye, O.; Johnson, O.; Adeleke, O.; Christiania, O. Review on the role of glutathione on oxidative stress and infertility. JBRA Assist. Reprod. 2018, 22, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.; Mishra, P.C. Modeling the activity of glutathione as a hydroxyl radical scavenger considering its neutral non-zwitterionic form. J. Mol. Model. 2012, 19, 767–777. [Google Scholar] [CrossRef]
- Kwon, D.H.; Cha, H.; Lee, H.; Hong, S.; Park, C.; Park, S.; Kim, G.; Kim, S.; Kim, H.; Hwang, H.; et al. Protective Effect of Glutathione against Oxidative Stress-induced Cytotoxicity in RAW 264.7 Macrophages through Activating the Nuclear Factor Erythroid 2-Related Factor-2/Heme Oxygenase-1 Pathway. Antioxidants 2019, 8, 82. [Google Scholar] [CrossRef]
- Rushworth, G.F.; Megson, I.L. Existing and potential therapeutic uses for N-acetylcysteine: The need for conversion to intracellular glutathione for antioxidant benefits. Pharmacol. Ther. 2014, 141, 150–159. [Google Scholar] [CrossRef]
- Tan, M.; Yin, Y.; Ma, X.; Zhang, J.; Pan, W.; Tan, M.; Zhao, Y.; Yang, T.; Jiang, T.; Li, H. Glutathione system enhancement for cardiac protection: Pharmacological options against oxidative stress and ferroptosis. Cell Death Dis. 2023, 14, 131. [Google Scholar] [CrossRef]
- Morvaridzadeh, M.; Agah, S.; Estêvão, M.D.; Hosseini, A.S.; Heydari, H.; Toupchian, O.; Abdollahi, S.; Persad, E.; Abu-Zaid, A.; Rezamand, G.; et al. Effect of saffron supplementation on oxidative stress parameters: A systematic review and meta-analysis of randomized placebo-controlled trials. Food Sci. Nutr. 2021, 9, 5809–5819. [Google Scholar] [CrossRef]
- Abedi, A.; Ghobadi, H.; Sharghi, A.; Iranpour, S.; Fazlzadeh, M.; Aslani, M.R. Effect of saffron supplementation on oxidative stress markers (MDA, TAC, TOS, GPx, SOD, and pro-oxidant/antioxidant balance): An updated systematic review and meta-analysis of randomized placebo-controlled trials. Front. Med. 2023, 10, 1071514. [Google Scholar] [CrossRef]

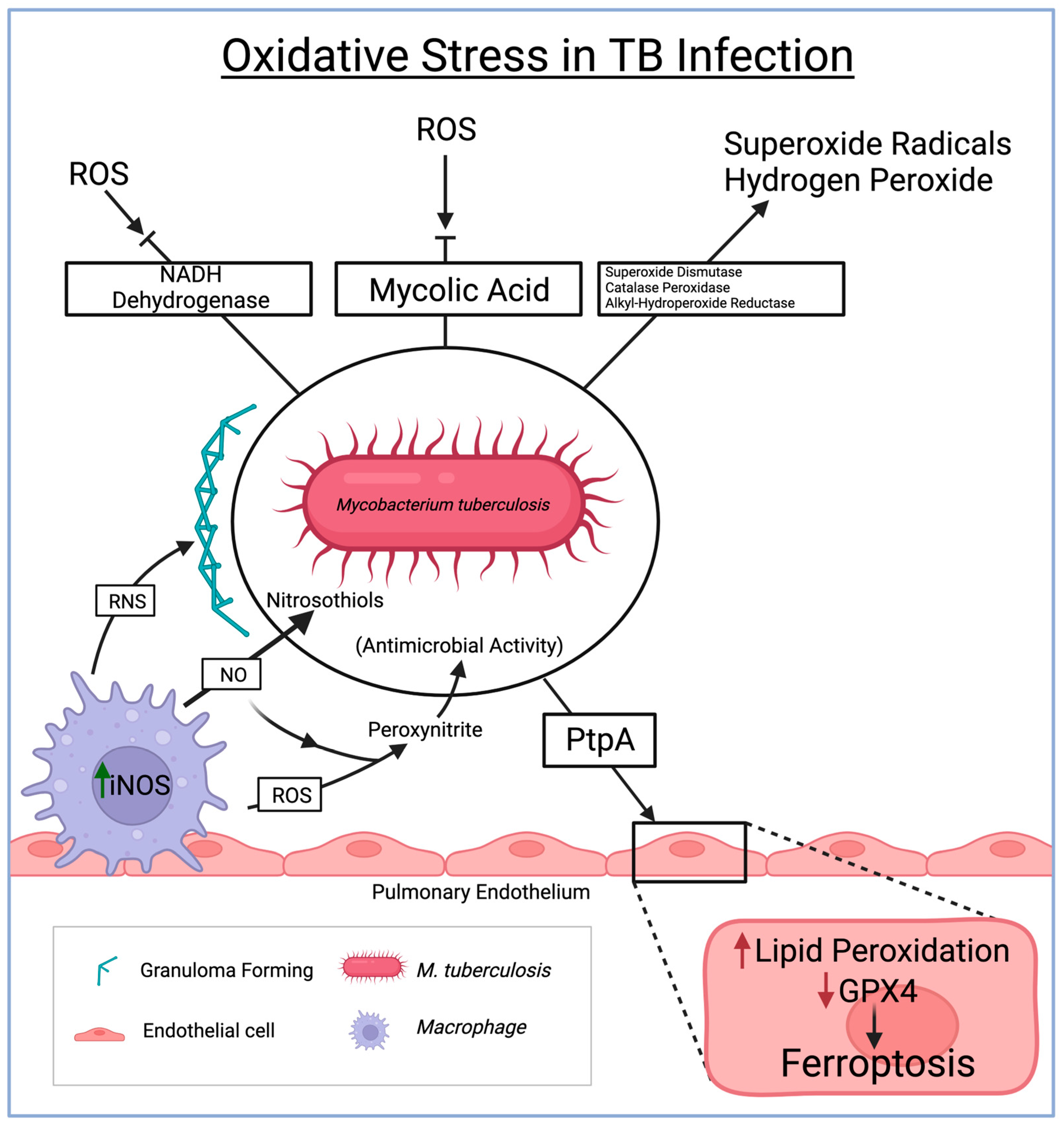

{kind=link}
{kind=link}
| Treatment | Dosage (Experimental) | Effect/Key Features |
|---|---|---|
| Antibiotics | ||
| Isoniazid (INH) | 5 mg/kg/day (~300 mg/day) | High CSF penetration |
| Rifampicin (RMP) | 10 mg/kg/day (~450 mg/day) | High/Moderate CSF penetration |
| Pyrazinamide (PZE) | 25 mg/kg/day (~1500 mg/day) | Moderate CSF penetration |
| Streptomycin (SM) | 15 mg/kg/day (~800 mg/day) | Low CSF penetration |
| Ethambutol (ETB) | 15 mg/kg/day (~800 mg/day) | Low CSF penetration |
| Fluoroquinolones | ||
| Levofloxacin | 500 mg every 12 h | High potent activity High CSF penetration |
| Ciprofloxacin | 750 mg every 12 h | Increased survival rates Reduced disability burden Lower incidence of disease relapse |
| Gatifloxacin | 400 mg every 12 h | Increased survival rates Reduced disability burden Lower incidence of disease relapse |
| Moxifloxacin | 400 mg every 12 h | Highly potent activity High CSF penetration |
| Corticosteroids | ||
| Dexamethasone | Varies based on disease severity | Reduced risk of adverse effects Suppresses inflammation Decreased brain-stem encephalopathy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dawi, J.; Mohan, A.S.; Misakyan, Y.; Affa, S.; Gonzalez, E.; Hajjar, K.; Nikoghosyan, D.; Fardeheb, S.; Tuohino, C.; Venketaraman, V. The Role of Oxidative Stress in TB Meningitis and Therapeutic Options. Diseases 2024, 12, 50. https://doi.org/10.3390/diseases12030050
Dawi J, Mohan AS, Misakyan Y, Affa S, Gonzalez E, Hajjar K, Nikoghosyan D, Fardeheb S, Tuohino C, Venketaraman V. The Role of Oxidative Stress in TB Meningitis and Therapeutic Options. Diseases. 2024; 12(3):50. https://doi.org/10.3390/diseases12030050
Chicago/Turabian StyleDawi, John, Aishvaryaa Shree Mohan, Yura Misakyan, Scarlet Affa, Edgar Gonzalez, Karim Hajjar, David Nikoghosyan, Sabrina Fardeheb, Christopher Tuohino, and Vishwanath Venketaraman. 2024. "The Role of Oxidative Stress in TB Meningitis and Therapeutic Options" Diseases 12, no. 3: 50. https://doi.org/10.3390/diseases12030050