
Effects of High-Dose Cyclophosphamide on Ultrastructural Changes and Gene Expression Profiles in the Cardiomyocytes of C57BL/6J Mice
 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Animals
2.3. Plasma CY, HCY, and CEPM Concentrations after the Administration of High-Dose CY
2.4. Histological Examinations
2.5. Detection of Gene Expression Alterations
2.6. Total RNA Isolation
2.7. Microarray Analysis
2.8. Data Analysis and Filter Criteria
2.9. cDNA Synthesis and RT-PCR
2.10. Statistical Analysis
3. Results
3.1. Pharmacokinetics of High-Dose CY in Mice
3.2. Histopathological Examination
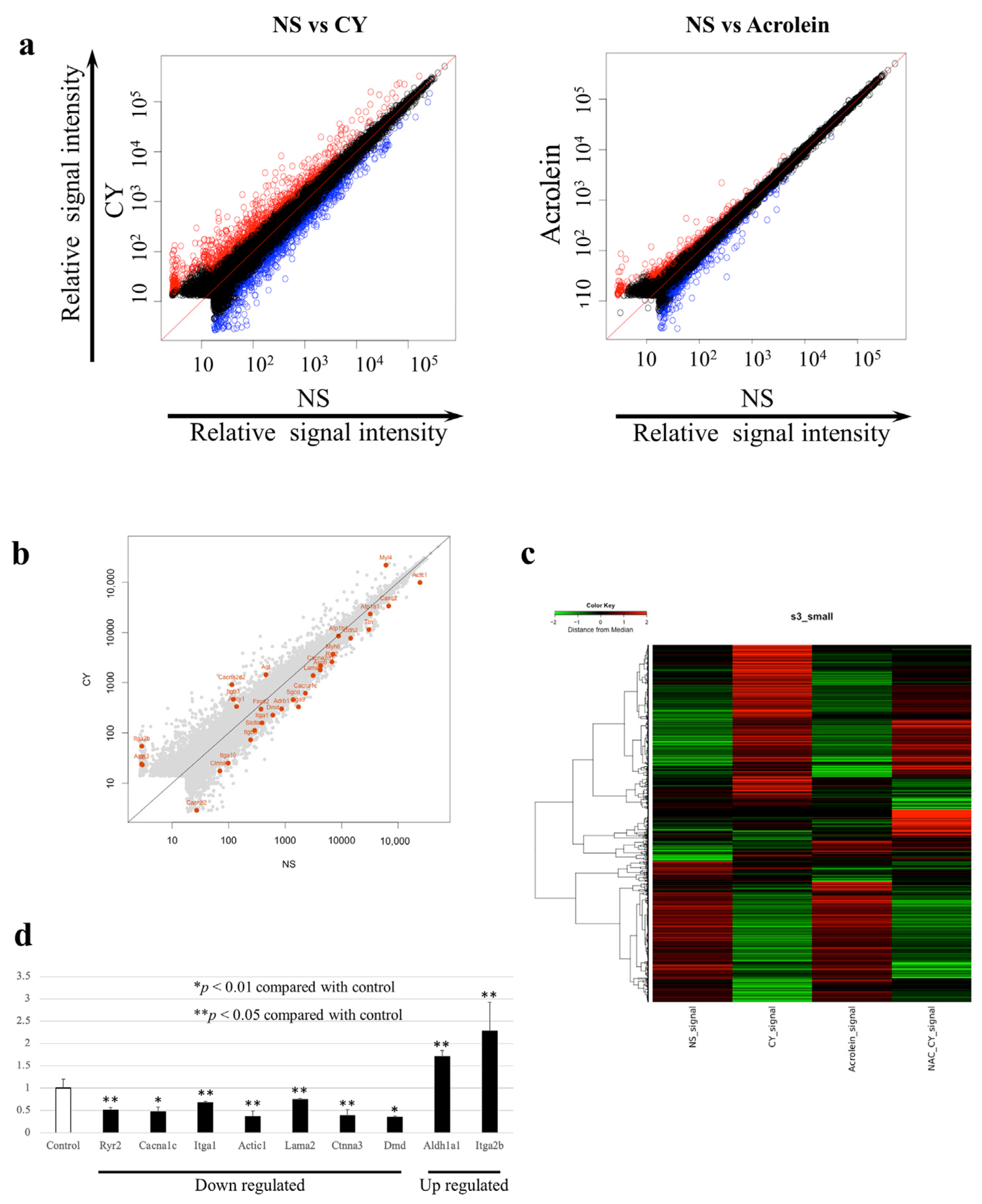
3.3. Identification of Gene Expression Profiles after the Administration of High-Dose CY
3.4. Gene-Enrichment and Functional Annotation Analyses
3.5. RT-PCR Validation of the Microarray Analysis Findings
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Abbreviation | Full term or phrase |
| Actβ | actin beta |
| Akt | protein kinase B |
| AldoCY | aldocyclophosphamide |
| ALDH1 | aldehyde dehydrogenase 1 |
| AUC | area under the concentration-time curve |
| CEPM | o-carboxyethyl-phosphoramide mustard |
| CY | cyclophosphamide |
| CYP | cytochrome P-450 |
| Ct | cycle time |
| DAVID | Database for Annotation, Visualization, and Integrated Discovery |
| EDTA | ethylenediaminetetraacetic acid |
| GO | gene ontology |
| GSK-3β | glycogen synthase kinase 3β |
| HE | haematoxylin-Eosin |
| HCL | hierarchical clustering |
| 4-HCY | 4-hydroxy-cyclophosphamide |
| HSCT | hematopoietic stem cell transplantation |
| i.p. | Intraperitoneally |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LC/MS/MS | liquid chromatography/tandem mass spectrometry |
| NAC | N-acetylcysteine |
| NS | normal saline |
| p38MAPK | P38 mitogen-activated protein kinase |
| PI3K | Phosphoinositide 3-kinase |
| PM | phosphoramide mustard |
| RT-PCR | reverse transcription PCR |
| TnC | troponin C |
References
- Santos, G.W.; Sensenbrenner, L.L.; Burke, P.J.; Mullins, G.M.; Blas, W.B.; Tutschka, P.J.; Slavin, R.E. The use of cyclophosphamide for clinical marrow transplantation. Transplant. Proc. 1972, 4, 559–564. [Google Scholar] [PubMed]
- Emadi, A.; Jones, R.J.; Brodsky, R.A. Cyclophosphamide and cancer: Golden anniversary. Nat. Rev. Clin. Oncol. 2009, 6, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Vivarelli, M.; Gibson, K.; Sinha, A.; Boyer, O. Childhood nephrotic syndrome. Lancet 2023, 2, 809–824. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, P.V.; Luznik, L.; Jones, R.J.; Vogelsang, G.B.; Leffell, M.S.; Phelps, M.; Rhubart, P.; Cowan, K.; Piantados, S.; Fuchs, E.J. Nonmyeloablative bone marrow transplantation from partially HLA-mismatched related donors using posttransplantation cyclophosphamide. Biol. Blood Marrow Transplant. 2002, 8, 377–386. [Google Scholar] [CrossRef]
- Nishikawa, T. Human leukocyte antigen-haploidentical haematopoietic stem cell transplantation using post-transplant cyclophosphamide for paediatric haematological malignancies. Cancers 2024, 16, 600. [Google Scholar] [CrossRef] [PubMed]
- Atalay, F.; Gulmez, O.; Ozsancak Ugurlu, A. Cardiotoxicity following cyclophosphamide therapy: A case report. J. Med. Case Rep. 2014, 8, 252. [Google Scholar] [CrossRef] [PubMed]
- Dhesi, S.; Chu, M.P.; Blevins, G.; Paterson, I.; Larratt, L.; Oudit, G.Y.; Kim, D.H. Cyclophosphamide-induced cardiomyopathy: A case report, review, and recommendations for management. J. Investig. Med. High Impact Case Rep. 2013, 1, 2324709613480346. [Google Scholar] [CrossRef] [PubMed]
- Krüger-Genge, A.; Köhler, S.; Laube, M.; Haileka, V.; Lemm, S.; Majchrzak, K.; Kammerer, S.; Schulz, C.; Storsberg, J.; Pietzsch, J.; et al. Anti-Cancer Prodrug Cyclophosphamide Exerts Thrombogenic Effects on Human Venous Endothelial Cells Independent of CYP450 Activation-Relevance to Thrombosis. Cells 2023, 12, 1965. [Google Scholar] [CrossRef]
- Gottdiener, J.S.; Appelbaum, F.R.; Ferrans, V.J.; Deisseroth, A.; Ziegler, J. Cardiotoxicity associated with high-dose cyclophosphamide therapy. Arch. Intern. Med. 1981, 141, 758–763. [Google Scholar] [CrossRef]
- McDonald, G.B.; Slattery, J.T.; Bouvier, M.E.; Ren, S.; Batchelder, A.L.; Kalhorn, T.F.; Schoch, H.G.; Anasetti, C.; Gooley, T. Cyclophosphamide metabolism, liver toxicity, and mortality following hematopoietic stem cell transplantation. Blood 2003, 101, 2043–2048. [Google Scholar] [CrossRef]
- Nishikawa, T.; Miyahara, E.; Kurauchi, K.; Watanabe, E.; Ikawa, K.; Asaba, K.; Tanabe, T.; Okamoto, Y.; Kawano, Y. Mechanisms of fatal cardiotoxicity following high-dose cyclophosphamide therapy and a method for its prevention. PLoS ONE 2015, 10, e0131394. [Google Scholar] [CrossRef] [PubMed]
- Kurauchi, K.; Nishikawa, T.; Miyahara, E.; Okamoto, Y.; Kawano, Y. Role of metabolites of cyclophosphamide in cardiotoxicity. BMC Res. Notes 2017, 10, 406. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Tomitori, H.; Machi, Y.; Hagihara, M.; Higashi, K.; Goda, H.; Ohya, T.; Niitsu, M.; Kashiwagi, K.; Igarashi, K. Acrolein toxicity: Comparison with reactive oxygen species. Biochem. Biophys. Res. Commun. 2009, 378, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.; Rangarajan, R.; Philidet, C.; Krüger-Genge, A.; Braune, S.; Kammerer, S.; Küpper, J.H.; Lendlein, A.; Jung, F. Effects of acrolein in comparison to its prodrug cyclophosphamide on human primary endothelial cells in vitro. Toxicol. In Vitro 2020, 62, 104685. [Google Scholar] [CrossRef] [PubMed]
- Dionísio, F.; Araújo, A.M.; Duarte-Araújo, M.; Bastos, M.L.; Guedes de Pinho, P.; Carvalho, F.; Costa, V.M. Cardiotoxicity of cyclophosphamide’s metabolites: An in vitro metabolomics approach in AC16 human cardiomyocytes. Arch. Toxicol. 2022, 96, 653–671. [Google Scholar] [CrossRef] [PubMed]
- Podgurskaya, A.D.; Slotvitsky, M.M.; Tsvelaya, V.A.; Frolova, S.R.; Romanova, S.G.; Balashov, V.A.; Agladze, K.I. Cyclophosphamide arrhythmogenicitytesting using human-induced pluripotent stem cell-derived cardiomyocytes. Sci. Rep. 2021, 11, 2336. [Google Scholar] [CrossRef]
- Liu, W.; Zhai, X.; Wang, W.; Zheng, B.; Zhang, Z.; Fan, X.; Chen, Y.; Wang, J. Aldehyde dehydrogenase 2 activation ameliorates cyclophosphamide-induced acute cardiotoxicity via detoxification of toxic aldehydes and suppression of cardiac cell death. J. Mol. Cell. Cardiol. 2018, 121, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Muguruma, K.; Pradipta, A.R.; Ode, Y.; Terashima, K.; Michiba, H.; Fujii, M.; Tanaka, K. Disease-associated acrolein: A possible diagnostic and therapeutic substrate for in vivo synthetic chemistry. Bioorg. Med. Chem. 2020, 28, 115831. [Google Scholar] [CrossRef]
- Kehrer, J.P.; Biswal, S.S. The molecular effects of acrolein. Toxicol. Sci. 2000, 57, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef]
- Quackenbush, J. Microarray data normalization and transformation. Nat. Genet. 2002, 32, S496–S501. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes And Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.I.; Sharov, V.; White, J.; Li, J.; Liang, W.; Bhagabati, N.; Braisted, J.; Klapa, M.; Currier, T.; Thiagarajan, M.; et al. TM4: A free, open-source system for microarray data management and analysis. BioTechniques 2003, 34, 374–378. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Iqubal, A.; Iqubal, M.K.; Sharma, S.; Ansari, M.A.; Najmi, A.K.; Ali, S.M.; Ali, J.; Haque, S.E. Molecular mechanism involved in cyclophosphamide-induced cardiotoxicity: Old drug with a new vision. Life Sci. 2019, 218, 112–131. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zhang, C.; Wang, C.; Zhao, L.; Wang, Z.; Dai, Z.; Lin, S.; Kang, H.; Ma, X. Ferulic acid against cyclophosphamide-induced heart toxicity in mice by inhibiting NF-κB pathway. Evid. Based Complement. Altern. Med. 2016, 2016, 1261270. [Google Scholar] [CrossRef]
- Fatani, A.G.; Darweesh, A.Q.; Rizwan, L.; Aleisa, A.M.; Al-Shabanah, O.A.; Sayed-Ahmed, M.M. Carnitine deficiency aggravates cyclophosphamide-induced cardiotoxicity in rats. Chemotherapy 2010, 56, 71–81. [Google Scholar] [CrossRef]
- Asiri, Y.A. Probucol attenuates cyclophosphamide-induced oxidative apoptosis, p53 and Bax signal expression in rat cardiac tissues. Oxid. Med. Cell. Longev. 2010, 3, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Park, E.S.; Kang, J.C.; Jang, Y.C.; Park, J.S.; Jang, S.Y.; Kim, D.E.; Kim, B.; Shin, H.S. Cardioprotective effects of rhamnetin in H9c2 cardiomyoblast cells under H2O2-induced apoptosis. J. Ethnopharmacol. 2014, 153, 552–560. [Google Scholar] [CrossRef] [PubMed]
- El-Agamy, D.S.; Elkablawy, M.A.; Abo-Haded, H.M. Modulation of cyclophosphamide-induced cardiotoxicity by methyl palmitate. Cancer Chemother. Pharmacol. 2017, 79, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Excitation-Contraction Coupling and Cardiac Contractile Force; Springer: Dordrecht, The Netherlands, 2001. [Google Scholar]
- Avila, G.; de la Rosa, J.A.; Monsalvo-Villegas, A.; Montiel-Jaen, M.G. Ca2+ channels mediate bidirectional signaling between sarcolemma and sarcoplasmic reticulum in muscle cells. Cells 2019, 9, 55. [Google Scholar] [CrossRef] [PubMed]
- Protasi, F. Structural interaction between RYRs and DHPRs in calcium release units of cardiac and skeletal muscle cells. Front. Biosci. 2002, 7, d650–d658. [Google Scholar] [CrossRef]
- Ríos, E.; Figueroa, L.; Manno, C.; Kraeva, N.; Riazi, S. The couplonopathies: A comparative approach to a class of diseases of skeletal and cardiac muscle. J. Gen. Physiol. 2015, 145, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.E.; Dirksen, R.T. Sarcoplasmic reticulum: The dynamic calcium governor of muscle. Muscle Nerve 2006, 33, 715–731. [Google Scholar] [CrossRef]
- Tadini-Buoninsegni, F.; Smeazzetto, S.; Gualdani, R.; Moncelli, M.R. Drug interactions with the Ca2+-ATPase from sarco (Endo) plasmic reticulum (SERCA). Front. Mol. Biosci. 2018, 5, 36. [Google Scholar] [CrossRef]
- Endoh, M. Cardiac Ca2+ signaling and Ca2+ sensitizers. Circ. J. 2008, 72, 1915–1925. [Google Scholar] [CrossRef]
- Pascual-Figal, D.A.; Januzzi, J.L. The Biology of ST2: The International ST2 Consensus Panel. Am. J. Cardiol. 2015, 115, 3B–7B. [Google Scholar] [CrossRef]
- Riccardi, M.; Myhre, P.L.; Zelniker, T.A.; Metra, M.; Januzzi, J.L.; Inciardi, R.M. Soluble ST2 in Heart Failure: A Clinical Role beyond B-Type Natriuretic Peptide. J. Cardiovasc. Dev. Dis. 2023, 17, 468. [Google Scholar] [CrossRef] [PubMed]
- Aula, H.; Skyttä, T.; Tuohinen, S.; Luukkaala, T.; Hämäläinen, M.; Virtanen, V.; Raatikainen, P.; Moilanen, E.; Kellokumpu-Lehtinen, P.L. ST2 levels increased and were associated with changes in left ventricular systolic function during a three-year follow-up after adjuvant radiotherapy for breast cancer. Breast 2020, 49, 183–186. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Upregulated genes: | |||
| Gene Symbol | Acc# | Z score | Gene name |
| Hmox1 | NM_010442 | 9.075 | Haem oxygenase 1 |
| Hamp | NM_032541 | 8.900 | Hepcidin antimicrobial peptide |
| Mt2 | NM_008630 | 8.579 | Metallothionein 2 |
| Serpina3n | NM_009252 | 8.301 | Serine (or cysteine) peptidase inhibitor clade A, member 3N |
| Dmkn | NM_001166173 | 7.662 | Dermokine, transcript variant 3 |
| Cd207 | NM_144943 | 7.595 | CD207 antigen |
| Dkk3 | NM_015814 | 7.498 | Dickkopf WNT signalling pathway inhibitor 3, transcript variant 2 |
| Mlana | XM_011247400 | 7.345 | Melan-A |
| Sfrp5 | NM_018780 | 7.028 | Secreted frizzled-related sequence protein 5 |
| Pip5k1a | AK167816 | 6.856 | Phosphatidylinositol-4-phosphate 5-kinase, type 1 alpha |
| Clec1b | NM_019985 | 6.784 | C-type lectin domain family 1, member b, transcript variant 1 |
| Anxa8 | NM_013473 | 6.760 | Annexin A8, transcript variant 1 |
| Fam32a | NM_026455 | 6.494 | Family with sequence similarity 32, member A |
| Gp9 | NM_018762 | 6.430 | Glycoprotein 9 (platelet) |
| Lgals3 | NM_001145953 | 6.298 | Lectin, galactose binding, soluble 3, transcript variant 1 |
| Fam183b | NM_029283 | 6.237 | Family with sequence similarity 183, member B transcript variant 1 |
| Clec4d | NM_010819 | 6.155 | C-type lectin domain family 4, member d, transcript variant 1 |
| Cntf | NM_170786 | 6.133 | Ciliary neurotrophic factor |
| Ppbp | NM_023785 | 6.065 | Pro-platelet basic protein |
| Gata2 | NM_008182 | 5.986 | Glutathione S-transferase, alpha 2 |
| Downregulated genes: | |||
| Gene Symbol | Acc# | Z score | Gene name |
| Cx3cr1 | NM_009987 | −6.630 | Chemokine (C-X3-C motif) receptor 1 |
| Nckap5 | NM_172484 | −5.346 | NCK-associated protein 5, transcript variant 2 |
| Aplnr | NM_011784 | −5.227 | Apelin receptor |
| Apln | NM_013912 | −5.118 | Apelin |
| Itga9 | NM_133721 | −4.791 | Integrin alpha 9, transcript variant 1 |
| Tfrc | NM_011638 | −4.554 | Transferrin receptor, transcript variant 1 |
| Ppargc1b | NM_133249 | −4.504 | Peroxisome proliferative activated receptor, gamma, coactivator 1 beta, transcript variant 1 |
| Akap7 | XM_006512789 | −4.497 | A kinase (PRKA) anchor protein 7 |
| Cops8 | NM_133805 | −4.479 | COP9 signalosome subunit 8 |
| Plekhh1 | NM_181073 | −4.462 | Pleckstrin homology domain containing, family H (with MyTH4 domain) member 1 |
| Galnt17 | NM_145218 | −4.418 | Polypeptide N-acetylgalactosaminyltransferase 17 |
| Large1 | NM_010687 | −4.360 | LARGE xylosyl- and glucuronyltransferase 1, transcript variant 2 |
| Klhdc8a | NM_144810 | −4.358 | Kelch domain containing 8A |
| Kcne1 | NM_008424 | −4.307 | Potassium voltage-gated channel, lsk-related subfamily, member 1 |
| Nrn1 | NM_001374754 | −4.275 | Neuritin 1, transcript variant 2 |
| Klra8 | NM_010650 | −4.228 | Killer cell lectin-like receptor, subfamily A, member 8, transcript variant 2 |
| Ccl21a | NM_011124 | −4.226 | Chemokine (C-C motif) ligand 21A (serine) |
| Dbp | NM_016974 | −4.233 | D site albumin promoter binding protein |
| Fhl2 | NM_010212 | −4.201 | Four and a half LIM domains 2, transcript variant 1 |
| Tuba4a | NM_009447 | −4.169 | Tubulin, alpha 4A, transcript variant 1 |
| Annotation Cluster | Count | p |
|---|---|---|
| 1 (Enrichment Score: 20.72) | ||
| Extracellular region | 210 | 2.8 × 10−29 |
| Extracellular space | 196 | 1.3 × 10−20 |
| Signal | 349 | 7.0 × 10−12 |
| 2 (Enrichment Score: 13.13) | ||
| Signal | 349 | 7.0 × 10−12 |
| Disulphide bond | 249 | 2.3 × 10−11 |
| Glycoprotein | 345 | 4.9 × 10−11 |
| 3 (Enrichment Score: 7.52) | ||
| Cell junction | 89 | 2.3 × 10−9 |
| Synapse | 55 | 4.0 × 10−7 |
| 4 (Enrichment Score: 6.14) | ||
| Dilated cardiomyopathy | 21 | 3.1 × 10−7 |
| Hypertrophic cardiomyopathy | 20 | 8.5 × 10−7 |
| Arrhythmogenic right ventricular cardiomyopathy | 18 | 1.4 × 10−6 |
| 5 (Enrichment Score: 4.00) | ||
| Membrane | 451 | 1.9 × 10−12 |
| Plasma membrane | 361 | 1.2 × 10−6 |
| Cell membrane | 237 | 4.8 × 10−5 |
| Term | Count | p |
|---|---|---|
| Dilated cardiomyopathy | 21 | 3.1 × 10−7 |
| Hypertrophic cardiomyopathy (HCM) | 20 | 8.5 × 10−7 |
| Arrhythmogenic right ventricular | ||
| Cardiomyopathy (ARVC) | 18 | 1.4 × 10−6 |
| Viral protein interaction with cytokine | ||
| and cytokine receptor | 20 | 1.7 × 10−6 |
| ECM-receptor interaction | 19 | 2.2 × 10−6 |
| Focal adhesion | 29 | 1.3 × 10−5 |
| Metabolism of xenobiotics by cytochrome P450 | 16 | 1.5 × 10−5 |
| Hematopoietic cell lineage | 18 | 2.4 × 10−5 |
| Fluid shear stress and atherosclerosis | 23 | 4.1 × 10−5 |
| Platinum drug resistance | 16 | 4.6 × 10−5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishikawa, T.; Miyahara, E.; Yamazaki, I.; Ikawa, K.; Nakagawa, S.; Kodama, Y.; Kawano, Y.; Okamoto, Y. Effects of High-Dose Cyclophosphamide on Ultrastructural Changes and Gene Expression Profiles in the Cardiomyocytes of C57BL/6J Mice. Diseases 2024, 12, 85. https://doi.org/10.3390/diseases12050085
Nishikawa T, Miyahara E, Yamazaki I, Ikawa K, Nakagawa S, Kodama Y, Kawano Y, Okamoto Y. Effects of High-Dose Cyclophosphamide on Ultrastructural Changes and Gene Expression Profiles in the Cardiomyocytes of C57BL/6J Mice. Diseases. 2024; 12(5):85. https://doi.org/10.3390/diseases12050085
Chicago/Turabian StyleNishikawa, Takuro, Emiko Miyahara, Ieharu Yamazaki, Kazuro Ikawa, Shunsuke Nakagawa, Yuichi Kodama, Yoshifumi Kawano, and Yasuhiro Okamoto. 2024. "Effects of High-Dose Cyclophosphamide on Ultrastructural Changes and Gene Expression Profiles in the Cardiomyocytes of C57BL/6J Mice" Diseases 12, no. 5: 85. https://doi.org/10.3390/diseases12050085
APA StyleNishikawa, T., Miyahara, E., Yamazaki, I., Ikawa, K., Nakagawa, S., Kodama, Y., Kawano, Y., & Okamoto, Y. (2024). Effects of High-Dose Cyclophosphamide on Ultrastructural Changes and Gene Expression Profiles in the Cardiomyocytes of C57BL/6J Mice. Diseases, 12(5), 85. https://doi.org/10.3390/diseases12050085