TRAIL Deficient Mice Are Protected from Sugen/Hypoxia Induced Pulmonary Arterial Hypertension


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Animals
2.2. Echocardiography
2.3. Cardiac Catheterisation
2.4. Right Ventricular Hypertrophy
2.5 Immunohistochemistry
2.6. Quantification of Pulmonary Vascular Remodelling
2.7. Western Immunoblotting
2.8. Statistical Analysis
3. Results and Discussion
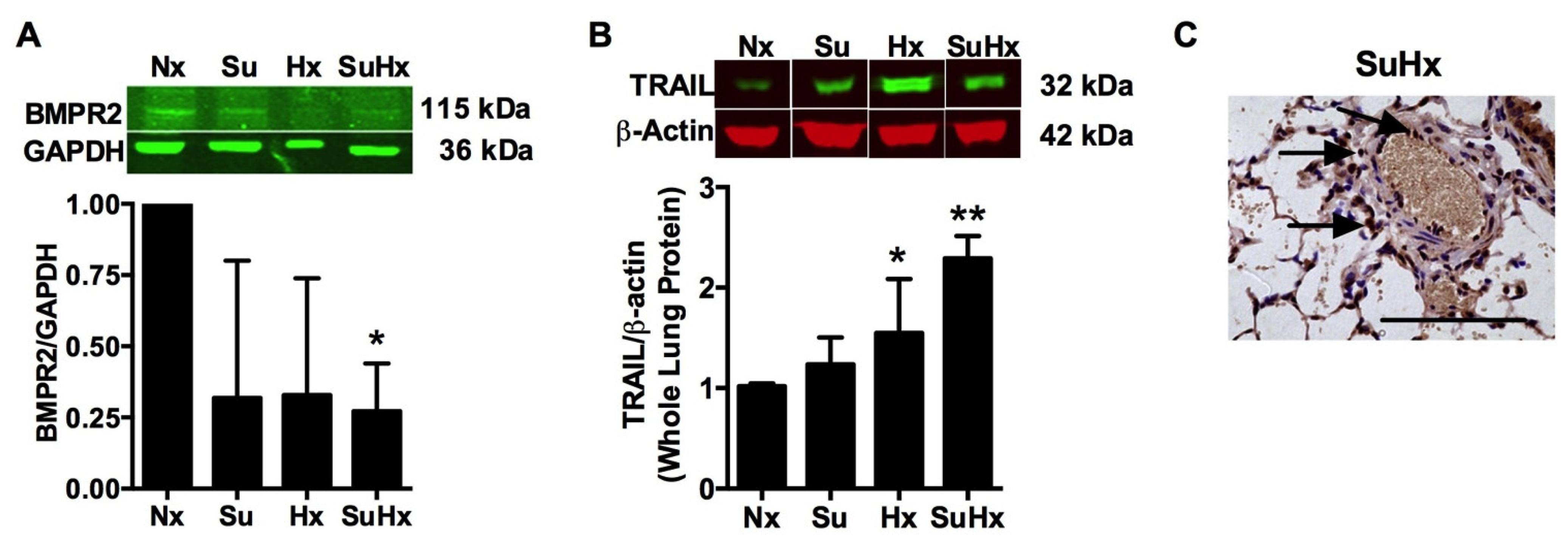
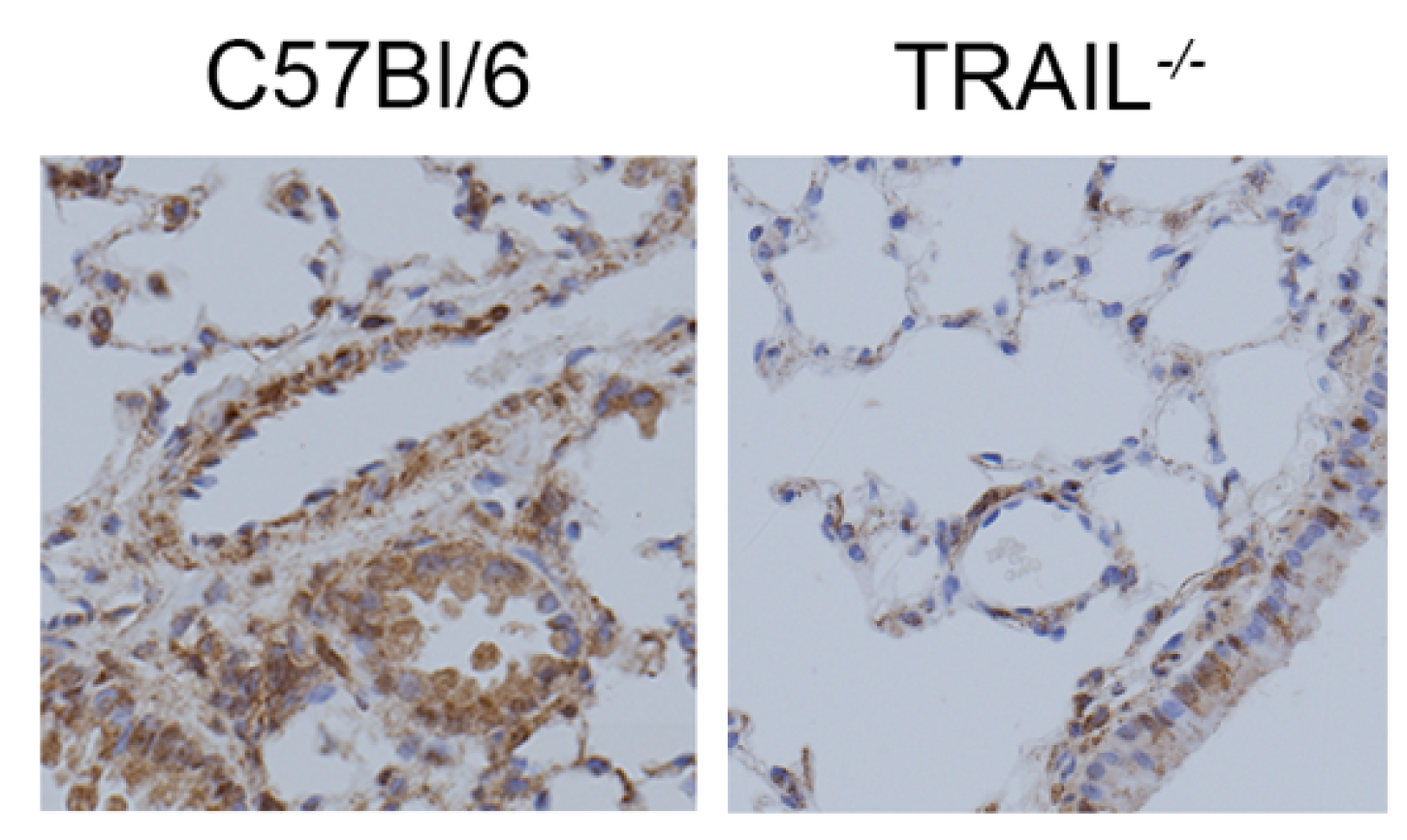
3.1. BMPR2 Expression Is Decreased and TRAIL Expression Is Increased in the SuHx Model of PAH

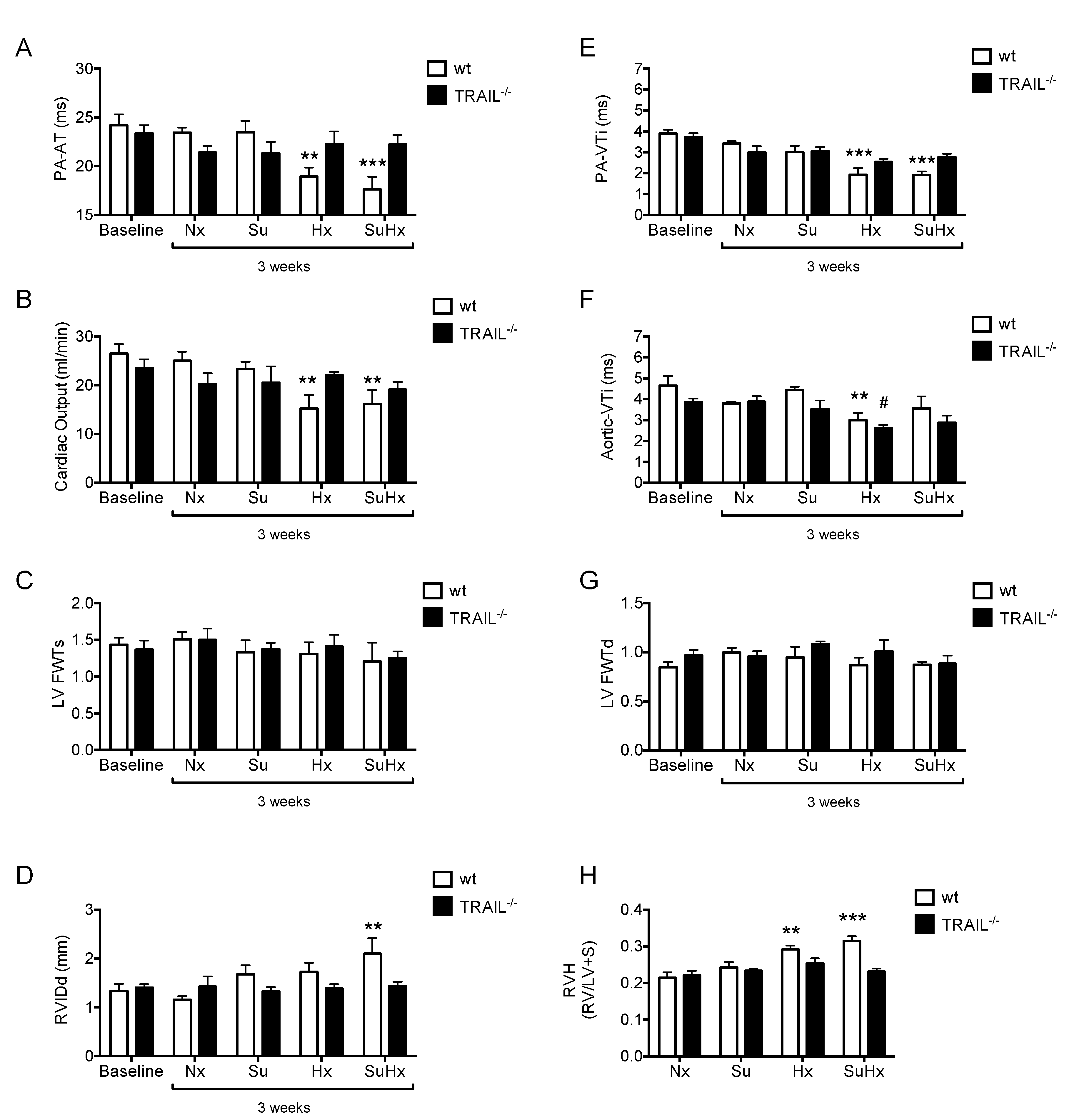
3.2. TRAIL−/−Mice Show no Echocardiographic Signs of PAH in Response to SuHx

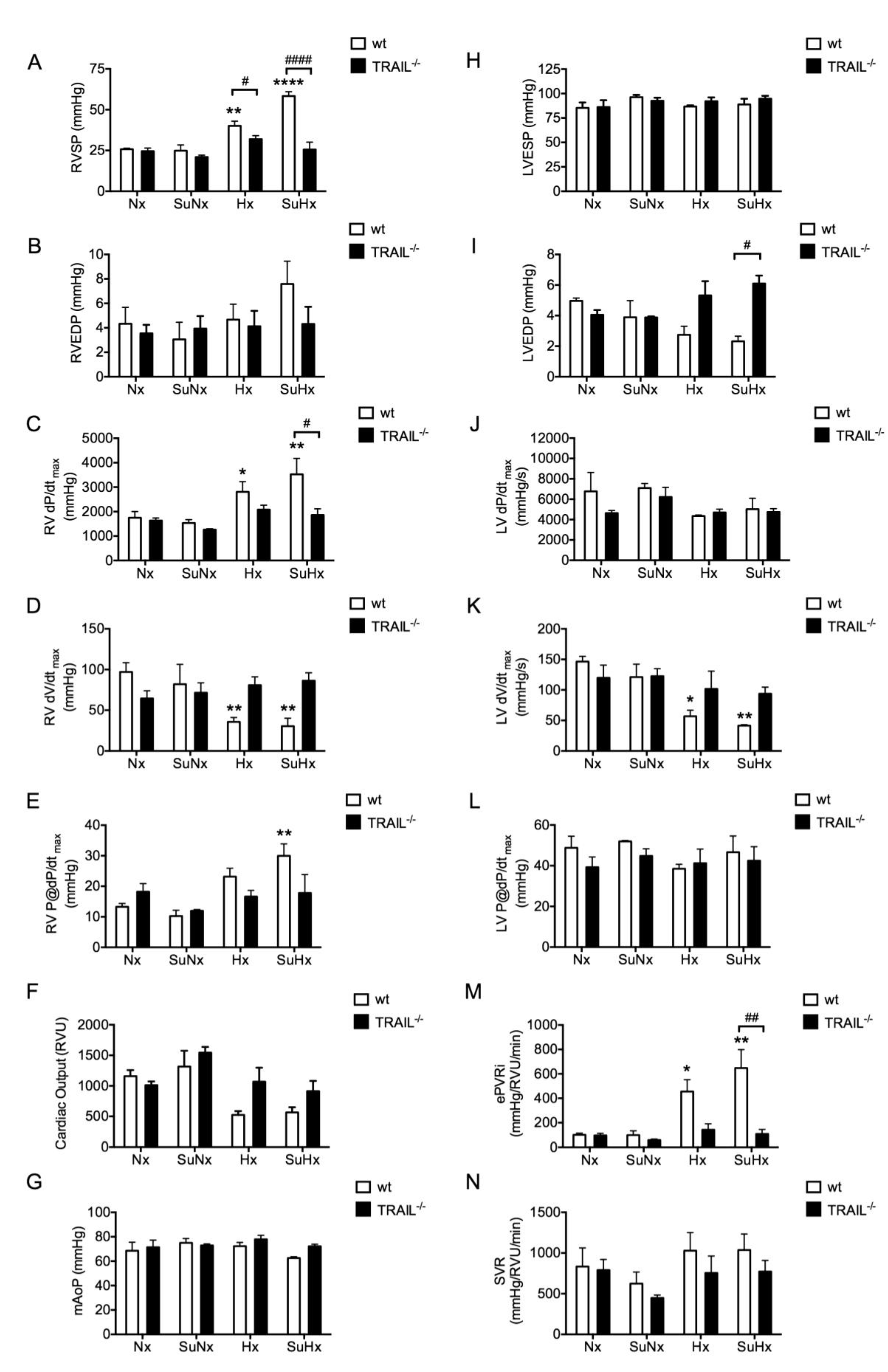
3.3. TRAIL−/− Mice Show no Significant Haemodynamic Signs of PAH in Response to SuHx
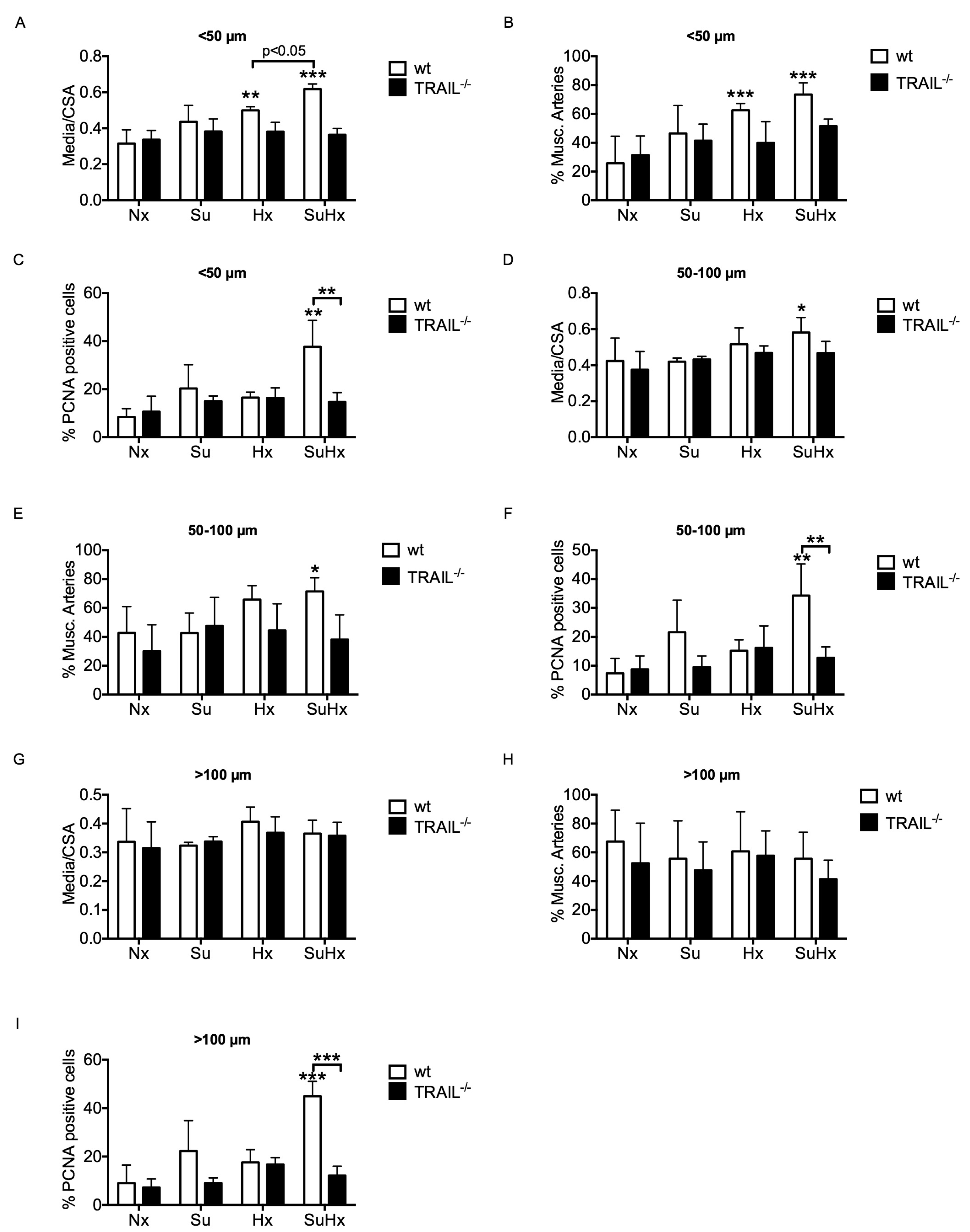
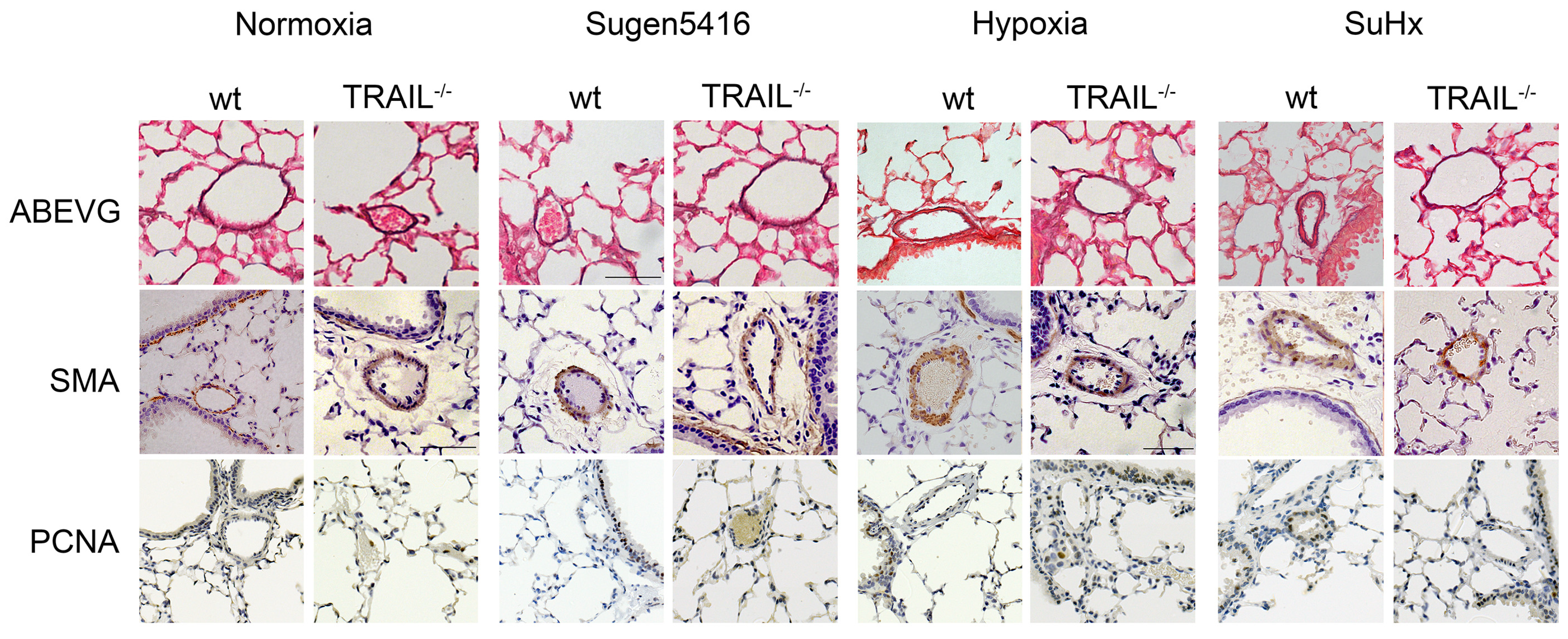
3.4. TRAIL−/− Mice Show Reduced Pulmonary Vascular Remodelling in Response to SuHx

3.5. The Protection from Pulmonary Vascular Remodelling in TRAIL−/− Mice in the SuHx Model Is Associated with Reduced Recruitment of Inflammatory Cells



4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kiely, D.G.; Elliot, C.A.; Sabroe, I.; Condliffe, R. Pulmonary hypertension: Diagnosis and management. BMJ 2013, 346, f2028. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Abman, S.H.; Braun, T.; Capron, F.; Stevens, T.; Thistlethwaite, P.A.; Haworth, S.G. Development and pathology of pulmonary hypertension. J. Am. Coll. Cardiol. 2009, 54, S3–S9. [Google Scholar] [CrossRef] [PubMed]
- Vonk-Noordegraaf, A.; Haddad, F.; Chin, K.M.; Forfia, P.R.; Kawut, S.M.; Lumens, J.; Naeije, R.; Newman, J.; Oudiz, R.J.; Provencher, S.; et al. Right Heart Adaptation to Pulmonary Arterial Hypertension. J. Am. Coll. Cardiol. 2013, 62, D22–D33. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitch, M. Molecular pathogenesis of pulmonary arterial hypertension. J. Clin. Invest. 2008, 118, 2372–2379. [Google Scholar] [CrossRef] [PubMed]
- Schermuly, R.T.; Ghofrani, H.A.; Wilkins, M.R.; Grimminger, F. Mechanisms of disease: pulmonary arterial hypertension. Nat. Rev. 2011, 8, 443–455. [Google Scholar]
- Tuder, R.M.; Groves, B.; Badesch, D.B.; Voelkel, N.F. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am. J. Pathol. 1994, 144, 275–285. [Google Scholar] [PubMed]
- Perros, F.; Montani, D.; Dorfmuller, P.; Durand-Gasselin, I.; Tcherakian, C.; Le Pavec, J.; Mazmanian, M.; Fadel, E.; Mussot, S.; Mercier, O.; et al. Platelet-derived growth factor expression and function in idiopathic pulmonary arterial hypertension. Am. J. Resp. Crit. Care Med. 2008, 178, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Schermuly, R.T.; Dony, E.; Ghofrani, H.A.; Pullamsetti, S.; Savai, R.; Roth, M.; Sydykov, A.; Lai, Y.J.; Weissmann, N.; Seeger, W.; et al. Reversal of experimental pulmonary hypertension by PDGF inhibition. J. Clin. Invest. 2005, 115, 2811–2821. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Monti, G.; Brenot, F.; Sitbon, O.; Portier, A.; Grangeot-Keros, L.; Duroux, P.; Galanaud, P.; Simonneau, G.; Emilie, D. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am. J. Resp. Crit. Care Med. 1995, 151, 1628–1631. [Google Scholar] [CrossRef] [PubMed]
- Voelkel, N.; Tuder, R.; Bridges, J.; Arend, W. Interleukin-1 receptor antagonist treatment reduces pulmonary hypertension generated in rats by monocrotaline. Am. J. Resp. Cell Mol. Biol. 1994, 11, 664–675. [Google Scholar] [CrossRef]
- Lawrie, A.; Hameed, A.G.; Chamberlain, J.; Arnold, N.; Kennerley, A.; Hopkinson, K.; Pickworth, J.; Kiely, D.G.; Crossman, D.C.; Francis, S.E. Paigen diet-fed apolipoprotein E knockout mice develop severe pulmonary hypertension in an interleukin-1-dependent manner. Am. J. Pathol. 2011, 179, 1693–1705. [Google Scholar] [CrossRef] [PubMed]
- Soon, E.; Holmes, A.M.; Treacy, C.M.; Doughty, N.J.; Southgate, L.; Machado, R.D.; Trembath, R.C.; Jennings, S.; Barker, L.; Nicklin, P.; et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 2010, 122, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Steiner, M.K.; Syrkina, O.L.; Kolliputi, N.; Mark, E.J.; Hales, C.A.; Waxman, A.B. Interleukin-6 overexpression induces pulmonary hypertension. Circ. Res. 2009, 104, 236–244, 228. [Google Scholar]
- Hameed, A.G.; Arnold, N.D.; Chamberlain, J.; Pickworth, J.A.; Paiva, C.; Dawson, S.; Cross, S.; Long, L.; Zhao, L.; Morrell, N.W.; et al. Inhibition of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) reverses experimental pulmonary hypertension. J. Exp. Med. 2012, 209, 1919–1935. [Google Scholar] [CrossRef] [PubMed]
- Wiley, S.R.; Schooley, K.; Smolak, P.J.; Din, W.S.; Huang, C.P.; Nicholl, J.K.; Sutherland, G.R.; Smith, T.D.; Rauch, C.; Smith, C.A. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 1995, 3, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Pan, G. An Antagonist Decoy Receptor and a Death Domain-Containing Receptor for TRAIL. Science 1997, 277, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Walczak, H.; DegliEsposti, M.; Johnson, R.; Smolak, P.; Waugh, J.; Boiani, N.; Timour, M.; Gerhart, M.; Schooley, K.; Smith, C.; et al. TRAIL-R2: A novel apoptosis-mediating receptor for TRAIL. EMBO J. 1997, 16, 5386–5397. [Google Scholar] [CrossRef] [PubMed]
- Degli-Esposti, M.; Smolak, P.; Walczak, H.; Waugh, J.; Huang, C.; DuBose, R.; Goodwin, R.; Smith, C. Cloning and characterization of TRAIL-R3,a novel member of the emerging TRAIL receptor family. J. Exp. Med. 1997, 186, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Degli-Esposti, M.A.; Dougall, W.C.; Smolak, P.J.; Waugh, J.Y.; Smith, C.A.; Goodwin, R.G. The novel receptor TRAIL-R4 induces NF-kappaB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity 1997, 7, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Pan, G.; Ni, J.; Yu, G.; Wei, Y.; Dixit, V. TRUNDD, a new member of the TRAIL receptor family that antagonizes TRAIL signalling. FEBS Lett. 1998, 424, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Emery, J.; McDonnell, P.; Burke, M.; Deen, K.; Lyn, S.; Silverman, C.; Dul, E.; Appelbaum, E.; Eichman, C.; DiPrinzio, R.; et al. Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J. Biol. Chem. 1998, 273, 14363–14367. [Google Scholar]
- Condliffe, R.; Pickworth, J.A.; Hopkinson, K.; Walker, S.J.; Hameed, A.G.; Suntharaligam, J.; Soon, E.; Treacy, C.; Pepke-Zaba, J.; Francis, S.E.; et al. Serum osteoprotegerin is increased and predicts survival in idiopathic pulmonary arterial hypertension. Pulmonary Circulation 2012, 2, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Kavurma, M.M.; Schoppet, M.; Bobryshev, Y.V.; Khachigian, L.M.; Bennett, M.R. TRAIL stimulates proliferation of vascular smooth muscle cells via activation of NF-kappaB and induction of insulin-like growth factor-1 receptor. J. Biol. Chem. 2008, 283, 7754–7762. [Google Scholar] [CrossRef] [PubMed]
- Lawrie, A.; Waterman, E.; Southwood, M.; Evans, D.; Suntharalingam, J.; Francis, S.; Crossman, D.; Croucher, P.; Morrell, N.; Newman, C. Evidence of a role for osteoprotegerin in the pathogenesis of pulmonary arterial hypertension. Am. J. Pathol. 2008, 172, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Lawrie, A. A report on the use of animal models and phenotyping methods in pulmonary hypertension research. Pulmonary Circulation 2014. [Google Scholar] [CrossRef]
- Stenmark, K.R.; Meyrick, B.; Galie, N.; Mooi, W.J.; McMurtry, I.F. Animal models of pulmonary arterial hypertension: the hope for etiological discovery and pharmacological cure. Am. J. Physiol. 2009, 297, L1013–L1032. [Google Scholar]
- Taraseviciene-Stewart, L.; Kasahara, Y.; Alger, L.; Hirth, P.; Mc Mahon, G.; Waltenberger, J.; Voelkel, N.F.; Tuder, R.M. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 2001, 15, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Ciuclan, L.; Bonneau, O.; Hussey, M.; Duggan, N.; Holmes, A.M.; Good, R.; Stringer, R.; Jones, P.; Morrell, N.W.; Jarai, G.; et al. A novel murine model of severe pulmonary arterial hypertension. Am. J. Resp. Crit. Care Med. 2011, 184, 1171–1182. [Google Scholar]
- Cretney, E.; Takeda, K.; Yagita, H.; Glaccum, M.; Peschon, J.J.; Smyth, M.J. Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing ligand-deficient mice. J. Immunol. 2002, 168, 1356–1361. [Google Scholar] [CrossRef] [PubMed]
- Watt, V.; Chamberlain, J.; Steiner, T.; Francis, S.; Crossman, D. TRAIL attenuates the development of atherosclerosis in apolipoprotein E deficient mice. Atherosclerosis 2011, 215, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Simmons, G.H.; Minson, C.T.; Cracowski, J.L.; Halliwill, J.R. Systemic hypoxia causes cutaneous vasodilation in healthy humans. J. Appl. Physiol. 2007, 103, 608–615. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dawson, S.H.; Arnold, N.D.; Pickworth, J.A.; Francis, S.E.; Lawrie, A. TRAIL Deficient Mice Are Protected from Sugen/Hypoxia Induced Pulmonary Arterial Hypertension. Diseases 2014, 2, 260-273. https://doi.org/10.3390/diseases2030260
Dawson SH, Arnold ND, Pickworth JA, Francis SE, Lawrie A. TRAIL Deficient Mice Are Protected from Sugen/Hypoxia Induced Pulmonary Arterial Hypertension. Diseases. 2014; 2(3):260-273. https://doi.org/10.3390/diseases2030260
Chicago/Turabian StyleDawson, Sarah H., Nadine D. Arnold, Josephine A. Pickworth, Sheila E. Francis, and Allan Lawrie. 2014. "TRAIL Deficient Mice Are Protected from Sugen/Hypoxia Induced Pulmonary Arterial Hypertension" Diseases 2, no. 3: 260-273. https://doi.org/10.3390/diseases2030260
APA StyleDawson, S. H., Arnold, N. D., Pickworth, J. A., Francis, S. E., & Lawrie, A. (2014). TRAIL Deficient Mice Are Protected from Sugen/Hypoxia Induced Pulmonary Arterial Hypertension. Diseases, 2(3), 260-273. https://doi.org/10.3390/diseases2030260