
Hyperosmolarity Triggers the Warburg Effect in Chinese Hamster Ovary Cells and Reveals a Reduced Mitochondria Horsepower

 ,
,
Abstract
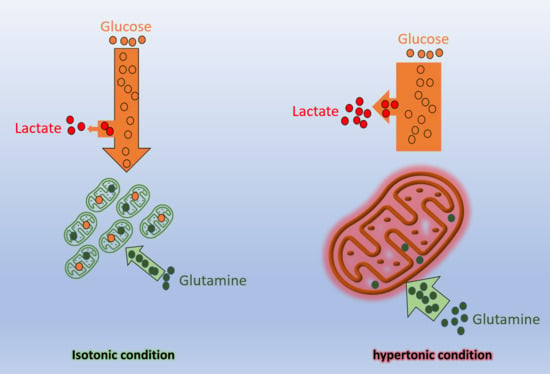
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. High Osmolarity Condition Affects CHO Cell Growth Rate but Maintains Viability
2.2. High Osmolarity Condition Favors Glycolytic and Glutaminolytic Phenotypes
2.3. Cell Cycle Distribution and Early Effect of Hyperosmolarity and Drugs Combination
2.3.1. Cell Cycle Distribution
2.3.2. Early Events Following Drugs and Mannitol Addition
2.3.3. Long-Term Effects on Cell Mitochondrial Activity
Membrane Potential
Cell Mitochondrial Activity
3. Discussion
4. Materials and Methods
4.1. Cell Line and Medium
4.2. Cultures and Drug Treatments
4.3. Analytical Methods
4.3.1. Cell Count
4.3.2. Extracellular Metabolites Measurements
4.3.3. Flow Cytometer and Microscopy Analysis
Sample Preparation
Flow Cytometer
Mitochondrial Membrane Potential Measurement
Fluorescence Microscopy
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schwartz, L.; Guais, A.; Pooya, M.; Abolhassani, M. Is inflammation a consequence of extracellular hyperosmolarity? J. Inflamm. 2009, 6, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemeth, Z.H.; Deitch, E.A.; Szabo, C.; Hasko, G. Hyperosmotic stress induces nuclear factor-kappaB activation and interleukin-8 production in human intestinal epithelial cells. Am. J. Pathol. 2002, 161, 987–996. [Google Scholar] [CrossRef]
- Moloney, E.D.; Griffin, S.; Burke, C.M.; Poulter, L.W.; O’Sullivan, S. Release of inflammatory mediators from eosinophils following a hyperosmolar stimulus. Respir. Med. 2003, 97, 928–932. [Google Scholar] [CrossRef] [Green Version]
- Hubert, A.; Cauliez, B.; Chedeville, A.; Husson, A.; Lavoinne, A. Osmotic stress, a proinflammatory signal in Caco-2 cells. Biochimie 2004, 86, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Li, D.Q.; Corrales, R.M.; Pflugfelder, S.C. Hyperosmolar saline is a proinflammatory stress on the mouse ocular surface. Eye Contact Lens 2005, 31, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Messmer, E.M. Osmoprotection as a new therapeutic principle. Ophthalmologe 2007, 104, 987–990. [Google Scholar] [CrossRef] [PubMed]
- Foulks, G.N. Pharmacological management of dry eye in the elderly patient. Drugs Aging 2008, 25, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Abolhassani, M.; Wertz, X.; Pooya, M.; Chaumet-Riffaud, P.; Guais, A.; Schwartz, L. Hyperosmolarity causes inflammation through the methylation of protein phosphatase 2A. Inflamm. Res. 2008, 57, 419–429. [Google Scholar] [CrossRef]
- Schwartz, L.; Abolhassani, M.; Pooya, M.; Steyaert, J.M.; Wertz, X.; Israel, M.; Guais, A.; Chaumet-Riffaud, P. Hyperosmotic stress contributes to mouse colonic inflammation through the methylation of protein phosphatase 2A. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G934–G941. [Google Scholar] [CrossRef] [Green Version]
- Light, R.W.; Macgregor, M.I.; Luchsinger, P.C.; Ball, W.C., Jr. Pleural effusions: The diagnostic separation of transudates and exudates. Ann. Intern. Med. 1972, 77, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Schilli, R.; Breuer, R.I.; Klein, F.; Dunn, K.; Gnaedinger, A.; Bernstein, J.; Paige, M.; Kaufman, M. Comparison of the composition of faecal fluid in Crohn’s disease and ulcerative colitis. Gut 1982, 23, 326–332. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Mantovani, A.; Pierotti, M.A. Cancer and inflammation: A complex relationship. Cancer Lett. 2008, 267, 180–181. [Google Scholar] [CrossRef]
- Schwartz, L.; da Veiga Moreira, J.; Jolicoeur, M. Physical forces modulate cell differentiation and proliferation processes. J. Cell. Mol. Med. 2018, 22, 738–745. [Google Scholar] [CrossRef] [Green Version]
- Kleihues, P.; Stewart, B.W. From host factors to genetic susceptibility. Prog. Clin. Biol. Res. 1996, 395, 201–209. [Google Scholar]
- Di Paolo, A.; Bocci, G. Drug distribution in tumors: Mechanisms, role in drug resistance, and methods for modification. Curr. Oncol. Rep. 2007, 9, 109–114. [Google Scholar] [CrossRef]
- Jain, R.K. Haemodynamic and transport barriers to the treatment of solid tumours. Int. J. Radiat. Biol. 1991, 60, 85–100. [Google Scholar] [CrossRef]
- Stohrer, M.; Boucher, Y.; Stangassinger, M.; Jain, R.K. Oncotic pressure in solid tumors is elevated. Cancer Res. 2000, 60, 4251–4255. [Google Scholar]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Schwartz, L.; Seyfried, T.; Alfarouk, K.O.; Da Veiga Moreira, J.; Fais, S. Out of Warburg effect: An effective cancer treatment targeting the tumor specific metabolism and dysregulated pH. Semin. Cancer Biol. 2017, 43, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Moreira, J.D.; Hamraz, M.; Abolhassani, M.; Bigan, E.; Peres, S.; Pauleve, L.; Nogueira, M.L.; Steyaert, J.M.; Schwartz, L. The Redox Status of Cancer Cells Supports Mechanisms behind the Warburg Effect. Metabolites 2016, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. Glutaminolysis: Supplying carbon or nitrogen or both for cancer cells? Cell Cycle 2010, 9, 3884–3886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herranz, D. Glutaminolysis gets the spotlight in cancer. Oncotarget 2017, 8, 10761–10762. [Google Scholar] [CrossRef]
- Villar, V.H.; Merhi, F.; Djavaheri-Mergny, M.; Duran, R.V. Glutaminolysis and autophagy in cancer. Autophagy 2015, 11, 1198–1208. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A Hallmark of Cancer Metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 163–194. [Google Scholar] [CrossRef]
- Akins, N.S.; Nielson, T.C.; Le, H.V. Inhibition of Glycolysis and Glutaminolysis: An Emerging Drug Discovery Approach to Combat Cancer. Curr. Top. Med. Chem. 2018, 18, 494–504. [Google Scholar] [CrossRef]
- Cervantes-Madrid, D.; Dominguez-Gomez, G.; Gonzalez-Fierro, A.; Perez-Cardenas, E.; Taja-Chayeb, L.; Trejo-Becerril, C.; Duenas-Gonzalez, A. Feasibility and antitumor efficacy in vivo, of simultaneously targeting glycolysis, glutaminolysis and fatty acid synthesis using lonidamine, 6-diazo-5-oxo-L-norleucine and orlistat in colon cancer. Oncol. Lett. 2017, 13, 1905–1910. [Google Scholar] [CrossRef] [Green Version]
- Cervantes-Madrid, D.; Duenas-Gonzalez, A. Antitumor effects of a drug combination targeting glycolysis, glutaminolysis and de novo synthesis of fatty acids. Oncol. Rep. 2015, 34, 1533–1542. [Google Scholar] [CrossRef]
- Dornier, E.; Rabas, N.; Mitchell, L.; Novo, D.; Dhayade, S.; Marco, S.; Mackay, G.; Sumpton, D.; Pallares, M.; Nixon, C.; et al. Glutaminolysis drives membrane trafficking to promote invasiveness of breast cancer cells. Nat. Commun 2017, 8, 2255. [Google Scholar] [CrossRef]
- Jin, L.; Alesi, G.N.; Kang, S. Glutaminolysis as a target for cancer therapy. Oncogene 2016, 35, 3619–3625. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Yin, Y.; Clark, L.H.; Sun, W.; Sullivan, S.A.; Tran, A.Q.; Han, J.; Zhang, L.; Guo, H.; Madugu, E.; et al. Dual inhibition of glycolysis and glutaminolysis as a therapeutic strategy in the treatment of ovarian cancer. Oncotarget 2017, 8, 63551–63561. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Yang, X.; Li, D.; Liang, Z.; Chen, Y.; Ma, G.; Wang, Y.; Li, Y.; Liang, Y.; Niu, H. The elevated glutaminolysis of bladder cancer and T cells in a simulated tumor microenvironment contributes to the up-regulation of PD-L1 expression by interferon-gamma. OncoTargets Ther. 2018, 11, 7229–7243. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Liu, L.; Yao, W. Activation of p53 by costunolide blocks glutaminolysis and inhibits proliferation in human colorectal cancer cells. Gene 2018, 678, 261–269. [Google Scholar] [CrossRef]
- Glutaminolysis Drives Lung Cancer Metastasis via the PLAG1-GDH1 Axis. Cancer Discov. 2018, 8, 135.
- Guais, A.; Baronzio, G.; Sanders, E.; Campion, F.; Mainini, C.; Fiorentini, G.; Montagnani, F.; Behzadi, M.; Schwartz, L.; Abolhassani, M. Adding a combination of hydroxycitrate and lipoic acid (METABLOC) to chemotherapy improves effectiveness against tumor development: Experimental results and case report. Investig. New Drugs 2012, 30, 200–211. [Google Scholar] [CrossRef]
- Ghorbaniaghdam, A.; Chen, J.; Henry, O.; Jolicoeur, M. Analyzing clonal variation of monoclonal antibody-producing CHO cell lines using an in silico metabolomic platform. PLoS ONE 2014, 9, e90832. [Google Scholar] [CrossRef] [Green Version]
- Zagari, F.; Jordan, M.; Stettler, M.; Broly, H.; Wurm, F.M. Lactate metabolism shift in CHO cell culture: The role of mitochondrial oxidative activity. New Biotechnol. 2013, 30, 238–245. [Google Scholar] [CrossRef]
- Dworacka, M.; Chukanova, G.; Iskakova, S.; Kurmambayev, Y.; Wesolowska, A.; Frycz, B.A.; Jagodzinski, P.P.; Dworacki, G. New arguments for beneficial effects of alpha-lipoic acid on the cardiovascular system in the course of type 2 diabetes. Eur. J. Pharm. Sci. 2018, 117, 41–47. [Google Scholar] [CrossRef]
- Gomes, M.B.; Negrato, C.A. Alpha-lipoic acid as a pleiotropic compound with potential therapeutic use in diabetes and other chronic diseases. Diabetol. Metab. Syndr. 2014, 6, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochette, L.; Ghibu, S.; Muresan, A.; Vergely, C. Alpha-lipoic acid: Molecular mechanisms and therapeutic potential in diabetes. Can. J. Physiol. Pharmacol. 2015, 93, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Udupa, A.; Nahar, P.; Shah, S.; Kshirsagar, M.; Ghongane, B. A comparative study of effects of omega-3 Fatty acids, alpha lipoic Acid and vitamin e in type 2 diabetes mellitus. Ann. Med. Health Sci Res. 2013, 3, 442–446. [Google Scholar] [PubMed]
- Bingham, P.M.; Stuart, S.D.; Zachar, Z. Lipoic acid and lipoic acid analogs in cancer metabolism and chemotherapy. Expert Rev. Clin. Pharmacol. 2014, 7, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Korotchkina, L.G.; Sidhu, S.; Patel, M.S. R-lipoic acid inhibits mammalian pyruvate dehydrogenase kinase. Free. Radic. Res. 2004, 38, 1083–1092. [Google Scholar] [CrossRef]
- Da Veiga Moreira, J.; Hamraz, M.; Abolhassani, M.; Schwartz, L.; Jolicoeur, M.; Peres, S. Metabolic therapies inhibit tumor growth in vivo and in silico. Sci. Rep. 2019, 9, 3153. [Google Scholar] [CrossRef] [Green Version]
- Uchendu, C.; Ambali, S.F.; Ayo, J.O.; Esievo, K.A.N.; Umosen, A.J. Erythrocyte osmotic fragility and lipid peroxidation following chronic co-exposure of rats to chlorpyrifos and deltamethrin, and the beneficial effect of alpha-lipoic acid. Toxicol. Rep. 2014, 1, 373–378. [Google Scholar] [CrossRef] [Green Version]
- Onakpoya, I.; Hung, S.K.; Perry, R.; Wider, B.; Ernst, E. The Use of Garcinia Extract (Hydroxycitric Acid) as a Weight loss Supplement: A Systematic Review and Meta-Analysis of Randomised Clinical Trials. J. Obes. 2011, 2011, 509038. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef]
- Pietrocola, F.; Pol, J.; Vacchelli, E.; Rao, S.; Enot, D.P.; Kroemer, G. Caloric Restriction Mimetics Enhance Anticancer Immunosurveillance. Cancer Cell 2016, 30, 147–160. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, L.; Abolhassani, M.; Guais, A.; Sanders, E.; Steyaert, J.M.; Campion, F.; Israel, M. A combination of alpha lipoic acid and calcium hydroxycitrate is efficient against mouse cancer models: Preliminary results. Oncol. Rep. 2010, 23, 1407–1416. [Google Scholar] [CrossRef] [Green Version]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Demaria, S.; Pikarsky, E.; Karin, M.; Coussens, L.M.; Chen, Y.C.; El-Omar, E.M.; Trinchieri, G.; Dubinett, S.M.; Mao, J.T.; Szabo, E.; et al. Cancer and inflammation: Promise for biologic therapy. J. Immunother. 2010, 33, 335–351. [Google Scholar] [CrossRef] [Green Version]
- Takagi, M.; Hayashi, H.; Yoshida, T. The effect of osmolarity on metabolism and morphology in adhesion and suspension chinese hamster ovary cells producing tissue plasminogen activator. Cytotechnology 2000, 32, 171–179. [Google Scholar] [CrossRef]
- Sheikholeslami, Z.; Jolicoeur, M.; Henry, O. Probing the metabolism of an inducible mammalian expression system using extracellular isotopomer analysis. J. Biotechnol. 2013, 164, 469–478. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [Green Version]
- Colombo, S.L.; Palacios-Callender, M.; Frakich, N.; De Leon, J.; Schmitt, C.A.; Boorn, L.; Davis, N.; Moncada, S. Anaphase-promoting complex/cyclosome-Cdh1 coordinates glycolysis and glutaminolysis with transition to S phase in human T lymphocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 18868–18873. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
da Veiga Moreira, J.; De Staercke, L.; César Martínez-Basilio, P.; Gauthier-Thibodeau, S.; Montégut, L.; Schwartz, L.; Jolicoeur, M. Hyperosmolarity Triggers the Warburg Effect in Chinese Hamster Ovary Cells and Reveals a Reduced Mitochondria Horsepower. Metabolites 2021, 11, 344. https://doi.org/10.3390/metabo11060344
da Veiga Moreira J, De Staercke L, César Martínez-Basilio P, Gauthier-Thibodeau S, Montégut L, Schwartz L, Jolicoeur M. Hyperosmolarity Triggers the Warburg Effect in Chinese Hamster Ovary Cells and Reveals a Reduced Mitochondria Horsepower. Metabolites. 2021; 11(6):344. https://doi.org/10.3390/metabo11060344
Chicago/Turabian Styleda Veiga Moreira, Jorgelindo, Lenny De Staercke, Pablo César Martínez-Basilio, Sandrine Gauthier-Thibodeau, Léa Montégut, Laurent Schwartz, and Mario Jolicoeur. 2021. "Hyperosmolarity Triggers the Warburg Effect in Chinese Hamster Ovary Cells and Reveals a Reduced Mitochondria Horsepower" Metabolites 11, no. 6: 344. https://doi.org/10.3390/metabo11060344