
Comprehensive Metabolic Profiling of Euphorbiasteroid in Rats by Integrating UPLC-Q/TOF-MS and NMR as Well as Microbial Biotransformation
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Instrumentation and Analysis Conditions
2.3. Animal and Drug Administration
2.4. Sample Collection of Blood, Urine, and Feces
2.5. Preparation of Blood, Urine, and Feces Samples
2.6. In Vitro Incubation of Euphorbiasteroid with Rat Liver Microsomes
2.7. Microbial Transformation of Euphorbiasteroid
2.8. Preparation of the Transformation Products of Euphorbiasteroid
2.9. Cell Culture and Cell Cytotoxicity Assay
3. Results and Discussion
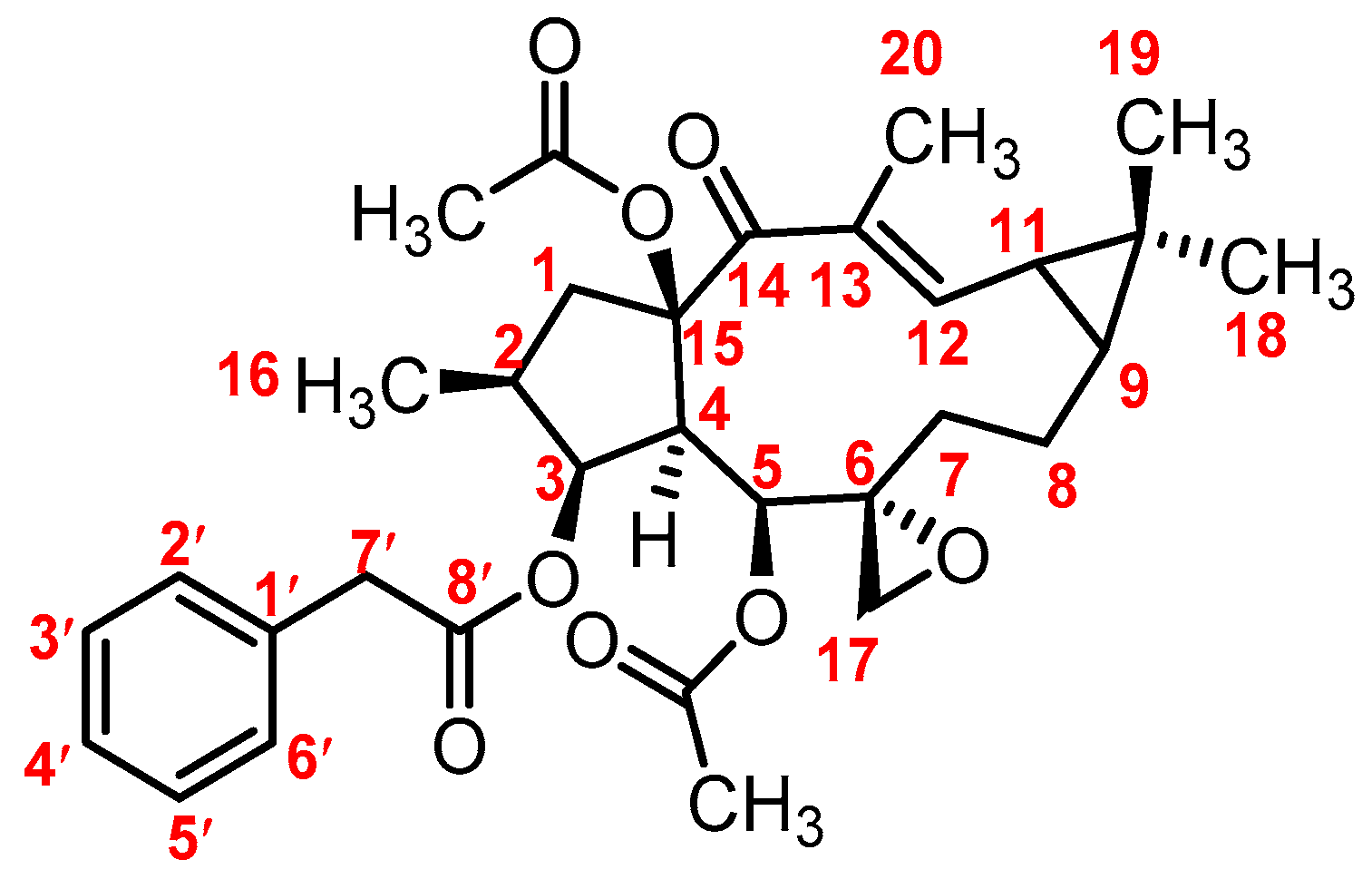
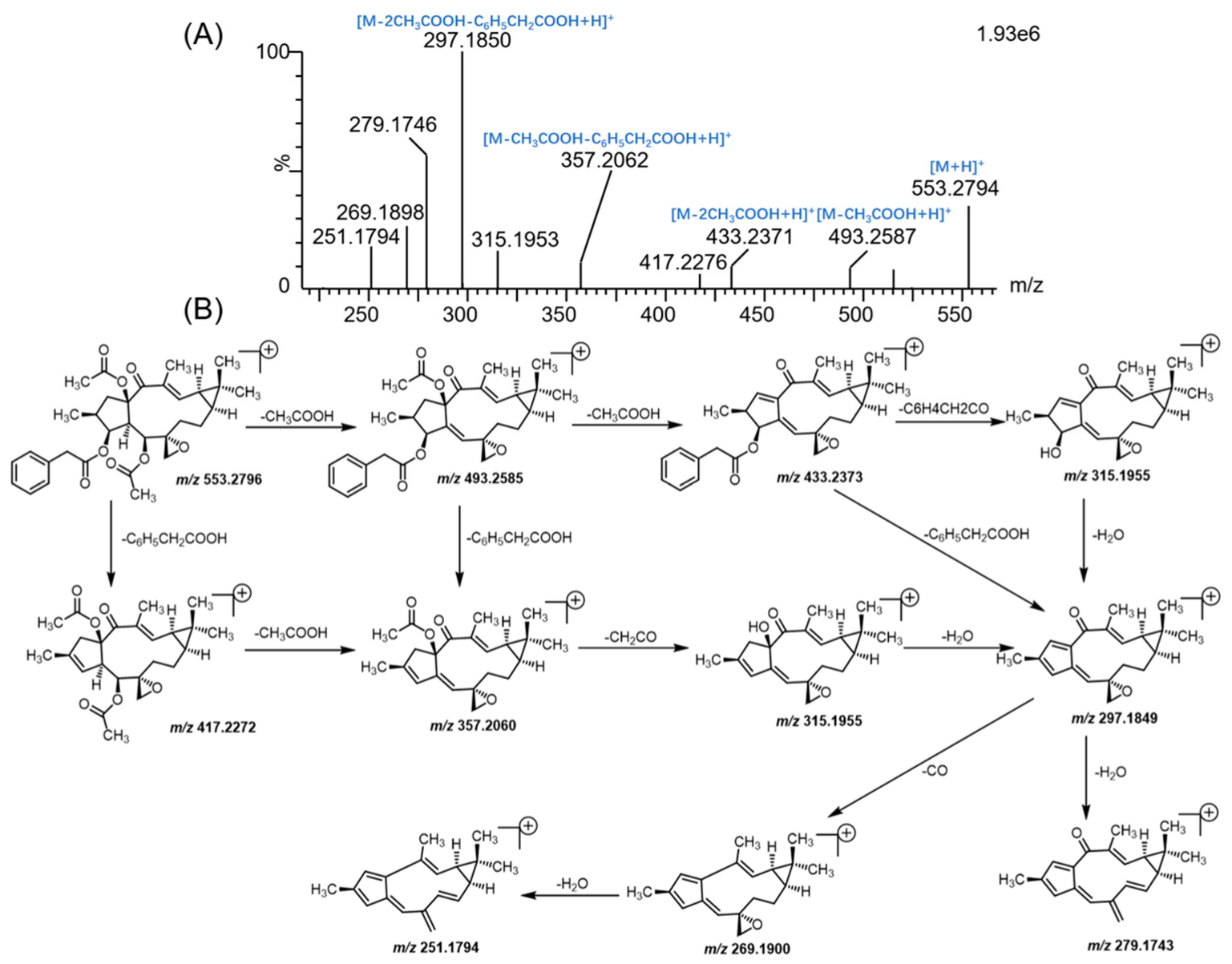
3.1. Mass Fragmentation Behavior Analyses of Euphorbiasteroid
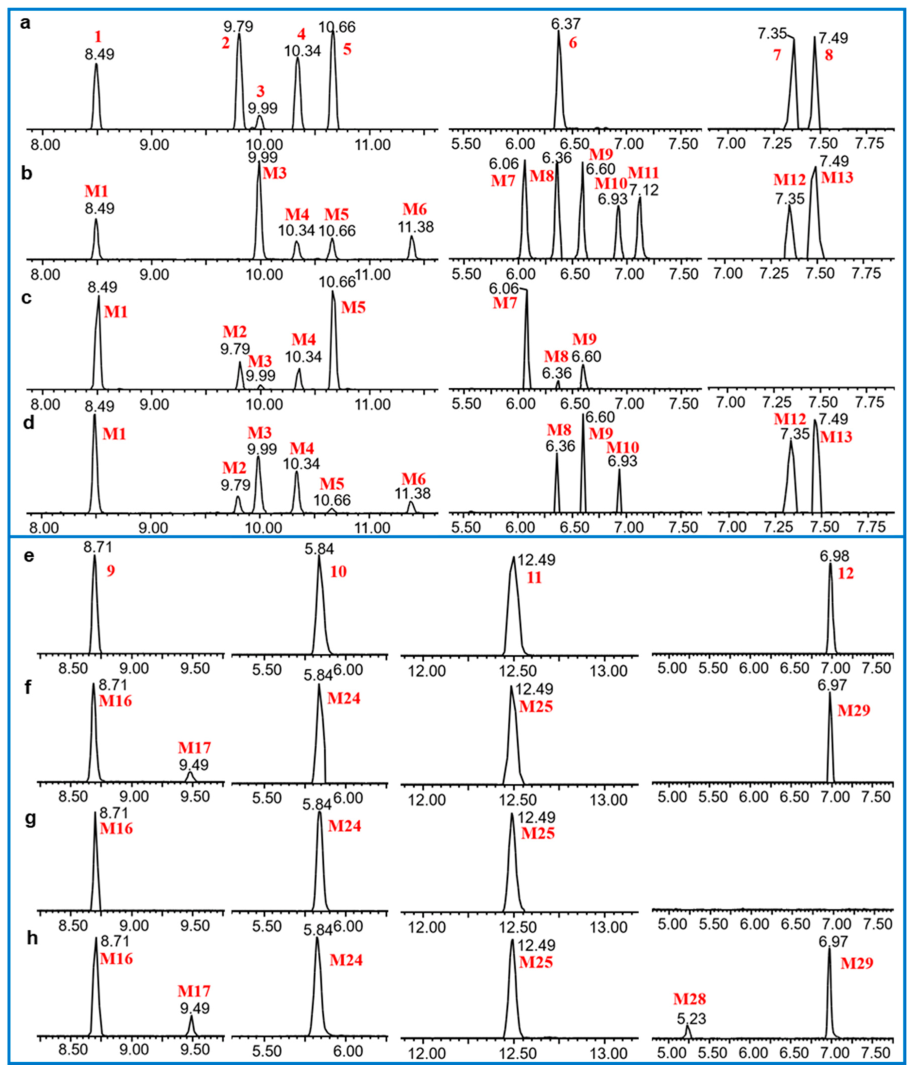
3.2. Identification of Metabolites of Euphorbiasteroid In Vitro and In Vivo
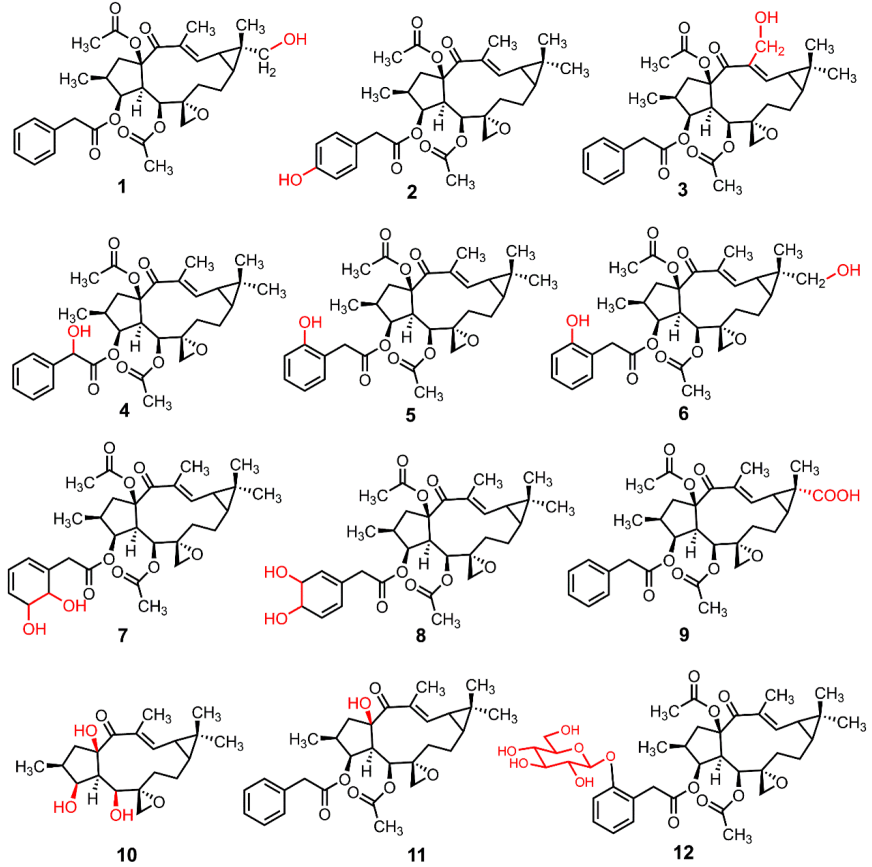
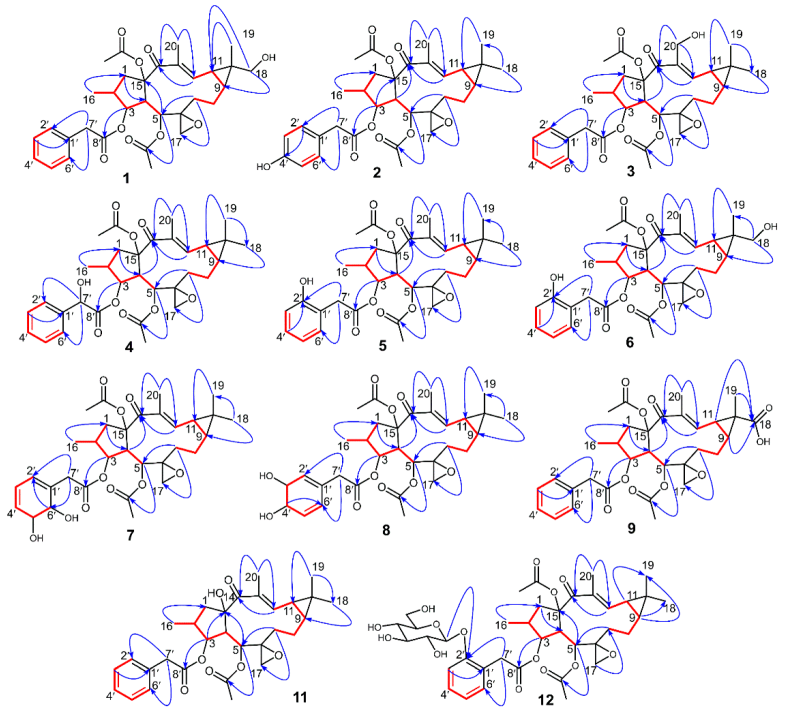
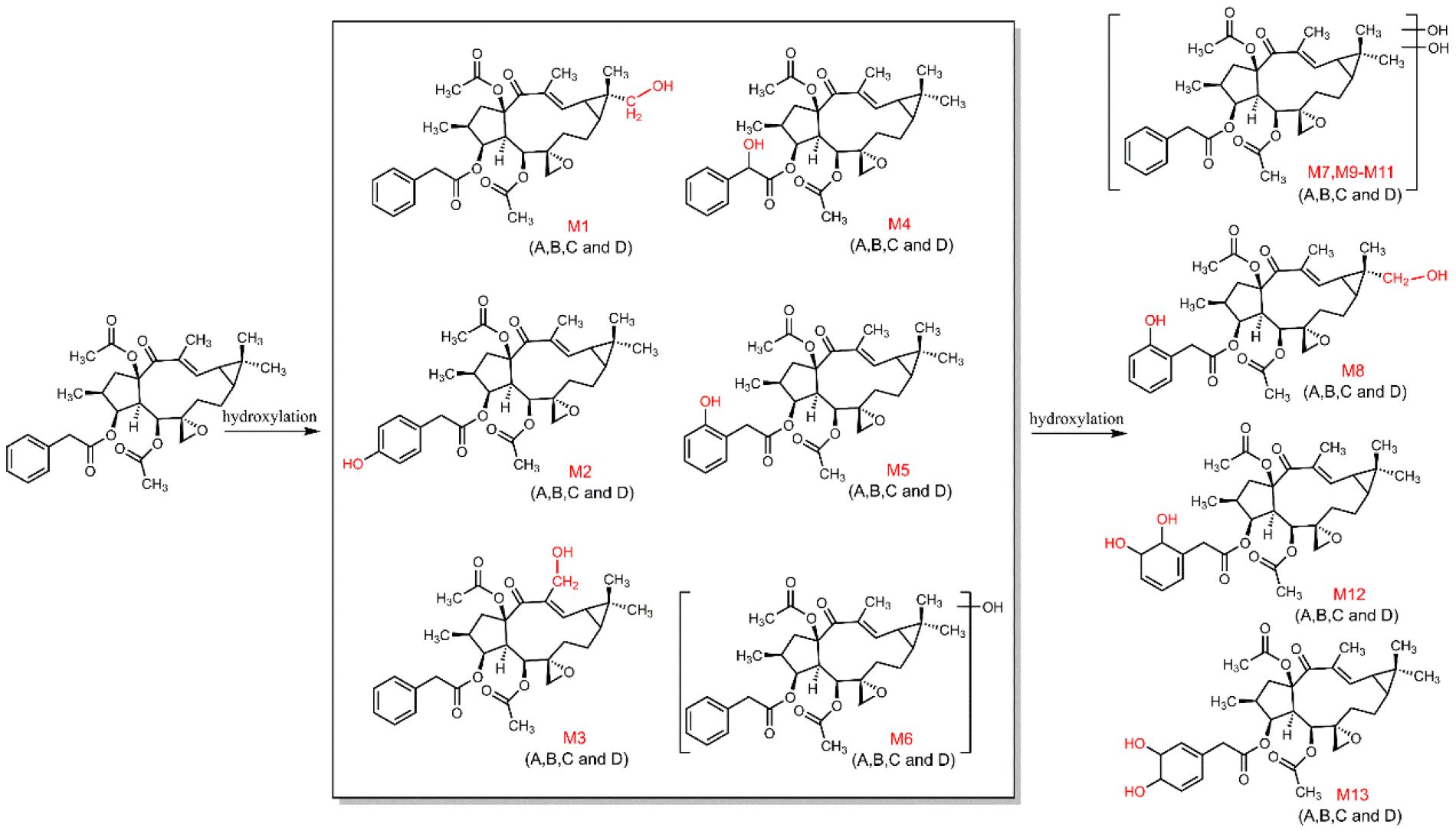
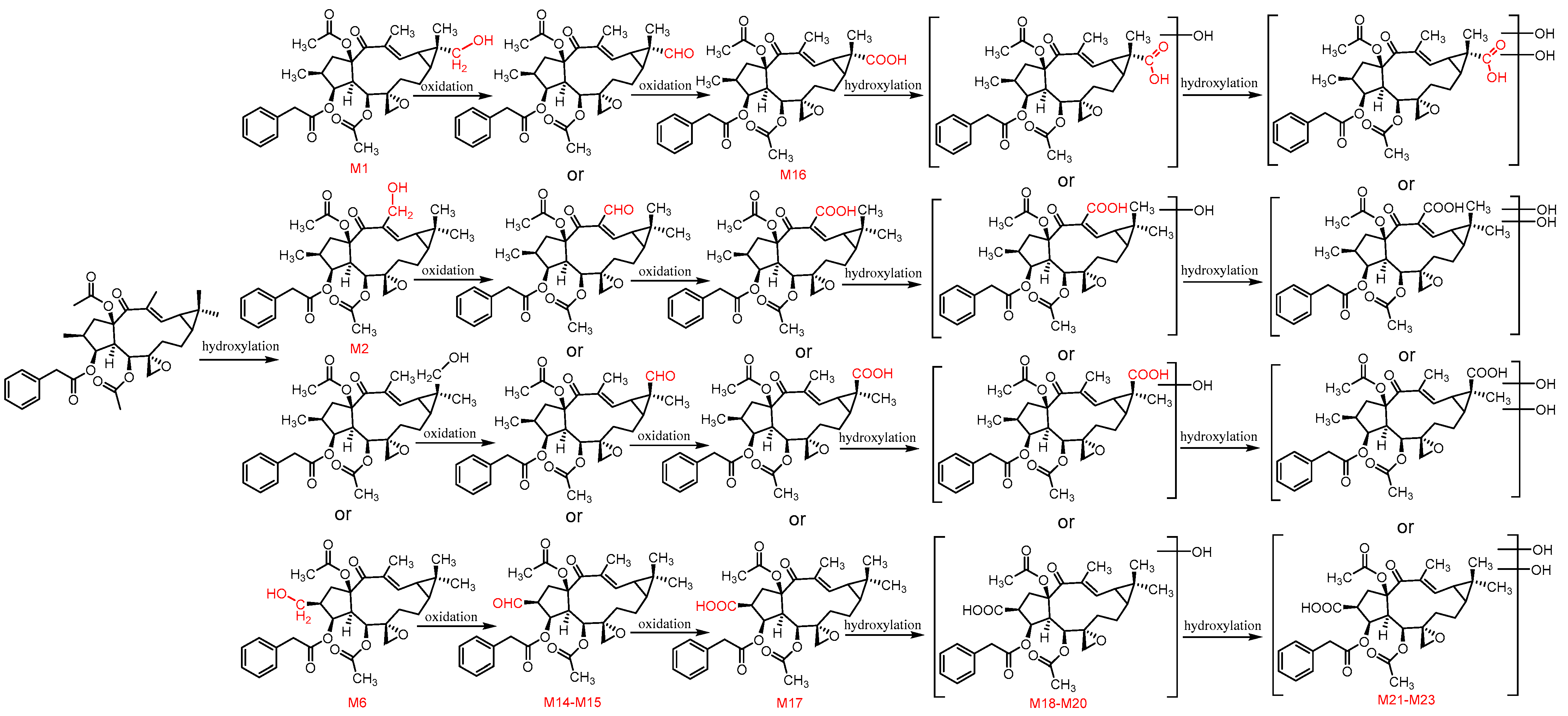
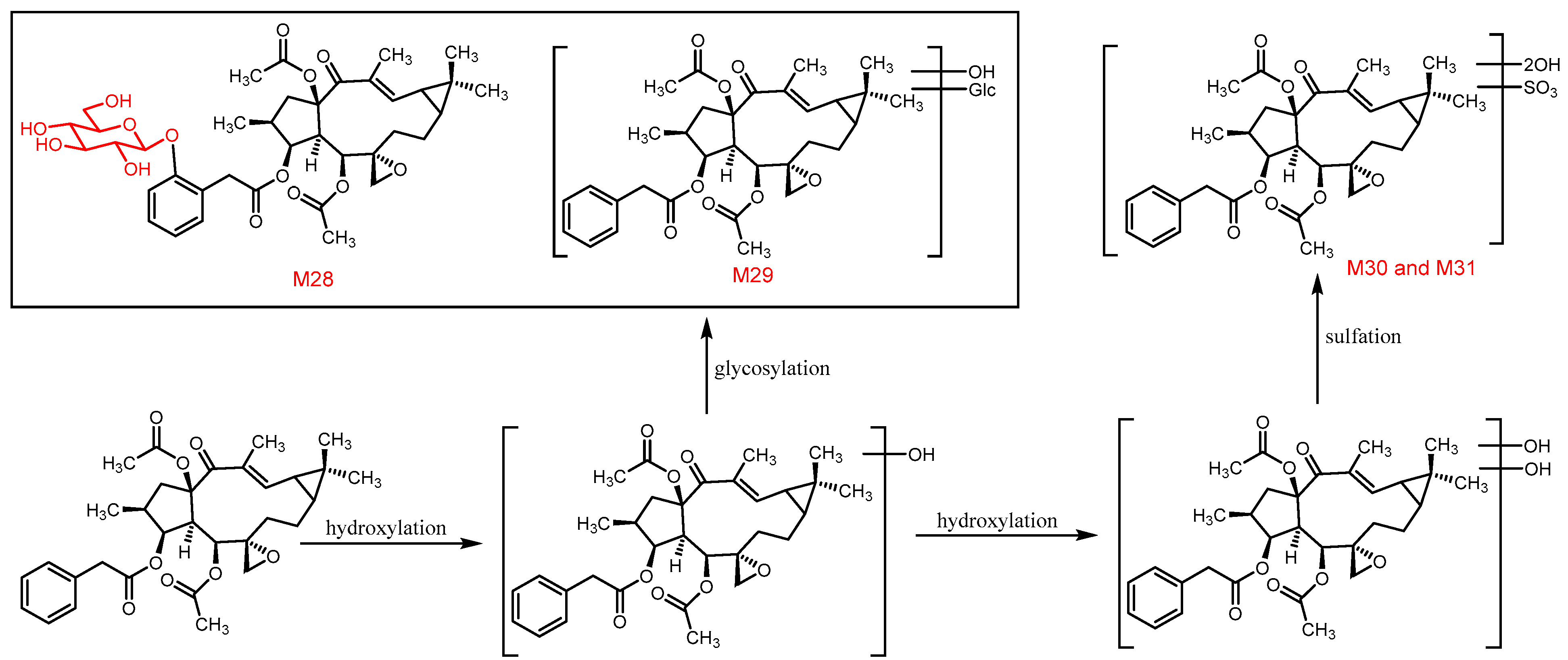
3.3. The Structure Elucidation of Transformation Products
3.4. Comparison of Metabolite Formation In Vitro and In Vivo
3.5. Proposed Metabolic Pathways of Euphorbiasteroid In Vivo
3.6. Cytotoxicity of Euphorbiasteroid and Its Metabolites
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- An, Z.; Tao, Z.; Qi, W. The phytochemistry, pharmacokinetics, pharmacology and toxicity of Euphorbia semen. J. Ethnopharmacol. 2018, 227, 41–55. [Google Scholar]
- Jaretzky, R.; Köhler, W. Euphorbia lathyris L. als Heilpflanze. Pharmakologische Prüfungen, insonderheit des fetten Samenöls. Arch. Pharm. 1943, 281, 265–269. [Google Scholar] [CrossRef]
- Choi, J.S.; Kang, N.S.; Yong, K.M.; Kim, S.H. Euphorbiasteroid reverses P-glycoprotein-mediated multi-drug resistance in human sarcoma cell line MES-SA/Dx5. Phytother. Res. 2010, 24, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Park, J.H.; Han, A.; Davaatseren, M.; Kwon, D.Y. Euphorbiasteroid, a component of Euphorbia lathyris L., inhibits adipogenesis of 3T3-L1 cells via activation of AMP-activated protein kinase. Cell Biochem. Funct. 2015, 33, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.W.; Guo, F.; Zhang, C.; Ren, X.; Jiang, G.S. Mechanism of euphorbiasteroid inducing apoptosis of HL-60 cells. China J. Cancer Prev. Treat. 2014, 21, 1865–1870. [Google Scholar]
- De Sousa, I.P.; Sousa Teixeira, M.V.; Jacometti Cardoso Furtado, N.A. An Overview of Biotransformation and Toxicity of Diterpenes. Molecules 2018, 23, 1387. [Google Scholar] [CrossRef]
- Blanz, J.; Délémonté, T.; Pearson, D.; Luneau, A.; Ritzau, M.; Gertsch, W.; Ramstein, P.; Dayer, J.; Desrayaud, S.; Braun, E. Micropreparative isolation and NMR structure elucidation of metabolites of the drug candidate 1-isopropyl-4-(4-isopropylphenyl)-6-(prop-2-yn-1-yloxy) quinazolin-2(1H)-one from rat bile and urine. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2015, 989, 1–10. [Google Scholar] [CrossRef]
- Beccaria, M.; Cabooter, D. Current developments in LC-MS for pharmaceutical analysis. Analyst 2020, 145, 1129–1157. [Google Scholar] [CrossRef]
- Xie, C.; Zhong, D.; Yu, K.; Chen, X. Recent advances in metabolite identification and quantitative bioanalysis by LC–Q-TOF MS. Bioanalysis 2012, 4, 937–959. [Google Scholar] [CrossRef]
- Xu, L.; Liu, Y.; Wu, H.; Zhou, A. Rapid identification of absorbed components and metabolites of Gandou decoction in rat plasma and liver by UPLC-Q-TOF-MSE. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2019, 1137, 121934. [Google Scholar] [CrossRef]
- Li, K.; Qin, F.; Jing, L.; Li, F.; Guo, X. In vivo and In vitro metabolism of a novel beta2-adrenoceptor agonist, trantinterol: Metabolites isolation and identification by LC-MS/MS and NMR. Anal. Bioanal. Chem. 2013, 405, 2619–2634. [Google Scholar] [CrossRef]
- Gao, Q.; Wang, D.; Shao, S.; Xue, Y.; Zhang, Y.; Chen, C.; Tang, F.; Sun, J.; Li, Y.; Guo, Q. Identification and quantitation of the actual active components in bamboo juice and its oral liquid by NMR and UPLC-Q-TOF-MS. Sci. Rep. 2020, 10, 19664. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhang, M.; Yang, Q.; Wang, Q.; Ma, B.; Li, Z.; Cheng, W.; Tang, H.; Feng, S.; Wang, Z. Metabolomic profiling of M. speciosa champ at different growth stages. Food Chem. 2021, 376, 131941. [Google Scholar] [CrossRef]
- Wang, Y.D.; Song, C.G.; Yang, J.; Zhou, T.; Zhao, Y.Y.; Qin, J.C.; Guo, L.P.; Ding, G. Accurate Identification of Degraded Products of Aflatoxin B1 Under UV Irradiation Based on UPLC-Q-TOF-MS/MS and NMR Analysis. Food Chem. 2021, 9, 789249. [Google Scholar] [CrossRef] [PubMed]
- Duan, M.; Li, Q.; Zhong, D.; Peng, Z. Identification of novel ondansetron metabolites using LC/MS n and NMR. J. Chromatogr. B 2018, 1095, 138–141. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Fan, J.; Chen, R.; Guo, D.; Zhao, M.; Zhang, Z.; Liang, C.; Chen, M.; Song, H.; Zhang, W. Stereoselective in vitro metabolism of cyproconazole in rat liver microsomes and identification of major metabolites. Chemosphere 2021, 264, 128495. [Google Scholar] [CrossRef]
- Gaunitz, F.; Dahm, P.; Mogler, L.; Thomas, A.; Thevis, M.; Mercer-Chalmers-Bender, K. In vitro metabolic profiling of synthetic cannabinoids by pooled human liver microsomes, cytochrome P450 isoenzymes, and Cunninghamella elegans and their detection in urine samples. Anal. Bioanal. Chem. 2019, 411, 3561–3579. [Google Scholar] [CrossRef]
- Nguyen, T.; Yeom, S.-J.; Yun, C.-H. Production of a Human Metabolite of Atorvastatin by Bacterial CYP102A1 Peroxygenase. Appl. Sci. 2021, 11, 603. [Google Scholar] [CrossRef]
- Lapham, K.; Callegari, E.; Cianfrogna, J.; Lin, J.; Niosi, M.; Orozco, C.C.; Sharma, R.; Goosen, T.C. In Vitro Characterization of Ertugliflozin Metabolism by UDP-Glucuronosyltransferase and Cytochrome P450 Enzymes. Drug Metab. Dispos. 2020, 48, 1350–1363. [Google Scholar] [CrossRef]
- Ma, Y.; Sun, P.; Zhao, Y.; Wang, K.; Chang, X.; Bai, Y.; Zhang, D.; Yang, L. A microbial transformation model for simulating mammal metabolism of Artemisinin. Molecules 2019, 24, 315. [Google Scholar] [CrossRef]
- Yildirim, K.; Kuru, A.; Küçükbaşol, E. Microbial transformation of androstenedione by Cladosporium sphaerospermum and Ulocladium chartarum. Biocatal. Biotransform. 2019, 38, 7–14. [Google Scholar] [CrossRef]
- Aziz, A.; Bano, S.; Atia Tul, W.; Choudhary, M.I. Microbial transformation of oral contraceptive ethisterone by Aspergillus niger and Cunninghamella blakesleeana. Steroids 2020, 154, 108467. [Google Scholar] [CrossRef] [PubMed]
- Sponchiado, R.; Sorrentino, J.M.; Olegario, N.; Oliveira, S.S.; Cordenonsi, L.M.; Silveira, G.P.; Fuentefria, A.M.; Mendez, A.S.L.; Steppe, M.; Garcia, C.V. Microbial transformation of ambrisentan to its glycosides by Cunninghamella elegans. Biomed. Chromatogr. 2019, 33, e4496. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Song, K.; Zhang, Y.; Chu, C.; Fan, B.; Song, Y.; Huang, H.; Chen, G. Biotransformation of betulinic acid by Circinella muscae and Cunninghamella echinulata to discover anti-inflammatory derivatives. Phytochemistry 2021, 182, 112608. [Google Scholar] [CrossRef]
- Palmer-Brown, W.; Miranda-CasoLuengo, R.; Wolfe, K.H.; Byrne, K.P.; Murphy, C.D. The CYPome of the model xenobiotic-biotransforming fungus Cunninghamella elegans. Sci. Rep. 2019, 9, 9240–9248. [Google Scholar] [CrossRef]
- Asha, S.; Vidyavathi, M. Cunninghamella—A microbial model for drug metabolism studies—A review. Biotechnol. Adv. 2009, 27, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.J.; Li, S.S.; Xie, B.; Chen, W.; Xu, X.K.; Zu, X.P.; Shen, Y.H. Systematic characterization of metabolic profiles of ingenol in rats by UPLC-Q/TOF-MS and NMR in combination with microbial biotransformation. RSC Adv. 2021, 11, 37752–37759. [Google Scholar] [CrossRef]
- Wintermeyer, A.; Möller, I.; Thevis, M.; Jübner, M.; Beike, J.; Rothschild, M.A.; Bender, K. In vitro phase I metabolism of the synthetic cannabimimetic JWH-018. Anal. Bioanal. Chem. 2010, 398, 2141–2153. [Google Scholar] [CrossRef]
- Baydoun, E.; Atia-tul-Wahab; Mehmood, H.; Ahmad, M.S.; Malik, R.; Smith, C.; Choudhary, M.I. Microbial transformation of danazol with Cunninghamella blakesleeana and anti-cancer activity of danazol and its transformed products. Steroids 2016, 105, 121–127. [Google Scholar] [CrossRef]
- Wang, Y.; Xiang, L.; Wang, Z.; Li, J.; Xu, J.; He, X. New anti-neuroinflammatory steroids against LPS induced NO production in BV2 microglia cells by microbial transformation of isorhodeasapogenin. Bioorg. Chem. 2020, 101, 103870. [Google Scholar] [CrossRef]
- Sun, J.H.; Yang, M.; Wang, X.M.; Xu, M.; Liu, A.H.; Guo, D.A. Identification of tanshinones and their metabolites in rat bile after oral administration of TTE-50, a standardized extract of Salvia miltiorrhiza by HPLC-ESI-DAD-MSn. J. Pharm. Biomed. Anal. 2007, 44, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jiang, M.; Yue, Z.; Wang, Z.; Wang, H.; Chen, M.; Wei, X.; Shi, S.; Wang, M.; Wang, Y. Characterization of diterpene metabolism in rats with ingestion of seed products from Euphorbia lathyris L. (Semen Euphorbiae and Semen Euphorbiae Pulveratum) using UHPLC-Q-Exactive MS. Biomed. Chromatogr. 2022, 36, e5394. [Google Scholar] [CrossRef] [PubMed]
- Šuláková, A.; Nykodemová, J.; Palivec, P.; Jurok, R.; Rimpelová, S.; Leonhardt, T.; Šíchová, K.; Páleníček, T.; Kuchař, M. 25CN-NBOMe Metabolites in Rat Urine, Human Liver Microsomes and C.elegans-Structure Determination and Synthesis of the Most Abundant Metabolites. Metabolites 2021, 11, 212. [Google Scholar] [CrossRef]
- Nykodemová, J.; Šuláková, A.; Palivec, P.; Češková, H.; Rimpelová, S.; Šíchová, K.; Leonhardt, T.; Jurásek, B.; Hájková, K.; Páleníček, T.; et al. 2C-B-Fly-NBOMe Metabolites in Rat Urine, Human Liver Microsomes and C. elegans: Confirmation with Synthesized Analytical Standards. Metabolites 2021, 11, 775. [Google Scholar] [CrossRef] [PubMed]
- Grafinger, K.E.; Stahl, K.; Wilke, A.; König, S.; Weinmann, W. In vitro phase I metabolism of three phenethylamines 25D-NBOMe, 25E-NBOMe and 25N-NBOMe using microsomal and microbial models. Drug Test. Anal. 2018, 10, 1607–1626. [Google Scholar] [CrossRef]
- Bicchi, C.; Appendino, G.; Cordero, C.; Rubiolo, P.; Ortelli, D.; Veuthey, J.L. HPLC-UV and HPLC-positive-ESI-MS analysis of the diterpenoid fraction from caper spurge (Euphorbia lathyris) seed oil. Phytochem. Anal. 2001, 12, 255–262. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | M0 a | 1 a | 2 a | 3 a | 4 a | 5 a | 6 b |
|---|---|---|---|---|---|---|---|
| 1a | 3.32 (dd, 8.15, 13.68) | 3.29 (dd, 8.29, 14.22) | 3.32 (dd, 8.38, 14.26) | 3.30 (dd, 8.67, 14,43) | 3.19 (dd, 8.25, 14.37) | 3.35 (dd, 8.25, 14.28) | 3.20 (dd, 8.16, 12.94) |
| 1b | 1.35 (dd, 12.01, 13.68) | 1.34 (dd, 12.42, 14.22) | 1.37 (dd, 12.22, 14.26) | 1.36 (dd, 12.80, 14.43) | 1.04 (dd, 12.60, 14.37) | 1.42 (dd, 12.26, 14.28) | 1.41 (t, 12.94) |
| 2 | 2.08 (m) | 2.06 (overlap) | 2.07 (m) | 2.07 (m) | 2.01 (m) | 2.10 (overlap) | 2.05 (overlap) |
| 3 | 5.48 (brt., 3.32) | 5.47 (brt., 3.15) | 5.47 (brt., 3.05) | 5.50 (s) | 5.49 (brt., 2.84) | 5.50 (brt, 3.03) | 5.37 (brt, 2.95) |
| 4 | 1.86 (dd, 3.32, 9.28) | 1.85 (dd, 3.20, 9.35) | 1.86 (dd, 3.05, 9.30) | 1.92 (d, 9.66) | 1.84 (dd, 2.84, 9.11) | 1.88 (dd, 3.03, 9.17) | 1.83 (dd, 2.95, 9.02) |
| 5 | 6.24 (d, 9.28) | 6.23 (d, 9.23) | 6.23 (d, 9.30) | 6.25 (d, 9.66) | 6.28 (d, 9.11) | 6.25 (d, 9.17) | 6.29 (d, 9.02) |
| 7a | 1.87 (m) | 2.14 (m) | 2.12 (m) | 2.14 (m) | 2.12 (overlap) | 2.10 (overlap) | 2.10 (m) |
| 7b | 0.92 (m) | 0.94 (t, 13.16) | 0.93 (m) | 0.98 (m) | 0.94 (m) | 0.93 (m) | 0.97 (t, 13.93) |
| 8a | 2.10 (m) | 2.06 (overlap) | 2.07 (m) | 2.12 (overlap) | 2.09 (m) | 2.07 (m) | 2.00 (m) |
| 8b | 1.72 (m) | 1.77 (m) | 1.72 (m) | 1.75 (m) | 1.72 (m) | 1.68 (m) | 1.80 (m) |
| 9 | 1.09 (m) | 1.21 (m) | 1.09 (ddd, 3.78, 8.08, 12.07) | 1.21 (overlap) | 1.09 (m) | 1.09 (m) | 1.33 (m) |
| 11 | 1.48 (dd, 8.11, 11.34) | 1.65 (dd, 8.39, 11.13) | 1.48 (dd, 8.08, 11.35) | 1.71 (m) | 1.48 (dd, 8.20, 11.42) | 1.47 (dd, 8.10, 11.39) | 1.78 (overalp) |
| 12 | 6.59 (d, 11.34) | 6.59 (d, 11.23) | 6.59 (dd, 1.05, 11.35) | 6.71 (d, 12.41) | 6.59 (dd, 1.05, 11.30) | 6.59 (d, 11.39) | 6.69 (d, 11.28) |
| 16 | 0.66 (d, 6.63) | 0.65 (d, 6.65) | 0.67 (d, 6.66) | 0.67 (d, 6.63) | 0.33 (d, 6.70) | 0.69 (d, 6.69) | 0.76 (d, 6.72) |
| 17a | 2.48 (d, 3.37) | 2.49 (d, 3.35) | 2.49 (d, 3.40) | 2.62 (s) | 2.50 (d, 3.24) | 2.50 (d, 3.24) | 2.56 (d, 3.54) |
| 17b | 2.30 (brt., 3.37) | 2.29 (brt., 3.35) | 2.31 (brt., 3.40) | 2.50 (s) | 2.28 (brt., 3.24) | 2.21 (brt., 3.24) | 2.20 (t, 3.54) |
| 18a | 1.20 (s) | 3.52 (d, 11.25) | 1.20 (s) | 1.21 (s) | 1.21 (s) | 1.20 (s) | 3.48 (d, 11.25) |
| 18b | 3.44 (d, 11.25) | 3.38 (d, 11.25) | |||||
| 19 | 1.21 (s) | 1.27 (s) | 1.21 (s) | 1.22 (s) | 1.22 (s) | 1.21 (s) | 1.27 (s) |
| 20a | 1.84 (s) | 1.84 (s) | 1.84 (brd, 0.81) | 4.42 (d, 12.38) | 1.83 (d, 1.04) | 1.84 (s) | 1.79 (d, 0.93) |
| 20b | 4.28 (d, 12.38) | ||||||
| 3-O-phenylacetyl | |||||||
| 2′ | 7.25 (overlap) | 7.25 (overlap) | 7.13 (d, 7.92) | 7.27 (overlap) | 7.37 (m) | ||
| 3′ | 7.30 (m) | 7.29 (td, 1.21, 7.19) | 6.76 (d, 7.92) | 7.31 (t, 7.26) | 7.32 (overlap) | 6.90 (overlap) | 6.86 (dd, 0.96, 7.35) |
| 4′ | 7.27 (t, 7.76) | 7.24 (m) | 7.26 (overlap) | 7.30 (overlap) | 7.18 (t, 7.45) | 7.08 (t, 7.35) | |
| 5′ | 7.30 (m) | 7.29 (td, 1.21, 7.19) | 6.76 (d, 7.92) | 7.31 (t, 7.26) | 7.32 (overlap) | 6.88 (overlap) | 6.78 (t, 7.35) |
| 6′ | 7.25 (overlap) | 7.25 (overlap) | 7.13 (d, 7.92) | 7.27 (overlap) | 7.37 (m) | 7.11 (dd, 1.45, 7.45) | 7.18 (d, 7.35) |
| 7′a | 3.58 (2H, d, 3.21) | 3.56 (2H, 2.71) | 3.50 (2H, d, 3.57) | 3.58 (2H, s) | 5.10 (2H, s) | 3.70 (d, 15.10) | 3.65 (d, 15.64) |
| 7′b | 3.55 (d, 15.10) | 3.55 (d, 15.64) | |||||
| 8′ | |||||||
| 5-OAc | |||||||
| C=O | |||||||
| CH3 | 2.02 (s) | 2.00 (s) | 2.01 (s) | 2.03 (s) | 2.12 (s) | 1.94 (s) | 2.06 (s) |
| 15-OAc | |||||||
| C=O | |||||||
| CH3 | 2.12 (s) | 2.12 (s) | 2.13 (s) | 2.12 (s) | 2.19 (s) | 2.17 (s) | 2.08 (s) |
| Position | M0 a | 7 a | 8 a | 9 a | 10 a | 11 a | 12 a |
|---|---|---|---|---|---|---|---|
| 1a | 3.32 (dd, 8.15, 13.68) | 3.40 (dd, 8.21, 14.17) | 3.39 (dd, 8.03, 13.95) | 3.30 (dd, 8.24,14.25) | 3.00 (dd, 9.58, 13.84) | 3.15 (dd, 9.37, 14.18) | 3.21 (dd, 7.65, 13.47) |
| 1b | 1.35 (dd, 12.01, 13.68) | 1.52 (dd, 12.25, 14.17) | 1.50 (dd, 12.69, 13.95) | 1.36 (dd, 12.35, 14.25) | 2.26 (dd, 6.08, 13.84) | 0.91 (dd, 11.72, 14.18) | 1.16 (overlap) |
| 2 | 2.08 (m) | 2.16 (m) | 2.16 (m) | 2.08 (m) | 2.04 (m) | 2.04 (m) | 1.96 (m) |
| 3 | 5.48 (brt., 3.32) | 5.51 (brt., 3.08) | 5.53 (brs.) | 5.49 (brt., 2.80) | 4.25 (brs) | 5.46 (s) | 5.37 (s) |
| 4 | 1.86 (dd, 3.32, 9.28) | 1.89 (dd, 3.08, 9.06) | 1.89 (m) | 1.87 (overlap) | 1.28 (brs) | 1.76 (dd, 2.84, 9.91) | 1.79 (overlap) |
| 5 | 6.24 (d, 9.28) | 6.22 (d, 9.06) | 6.24 (d, 8.82) | 6.22 (d, 9.10) | 4.39 (brs) | 5.92 (d, 9.91) | 6.21 (d, 8.90) |
| 7a | 1.87 (m) | 2.08 (m) | 2.08 (m) | 2.17 (m) | 2.04 (m) | 2.12 (dd, 6.42, 13.12) | 2.08 (overlap) |
| 7b | 0.92 (m) | 0.92 (t, 13.69) | 0.93 (t, 12.07) | 1.01 (t, 13.36) | 1.05 (m) | 0.87 (m) | 0.90 (m) |
| 8a | 2.10 (m) | 2.04 (m) | 2.05 (m) | 2.11 (m) | 1.77 (m) | 2.08 (overlap) | 2.08 (overlap) |
| 8b | 1.72 (m) | 1.73 (m) | 1.71 (m) | 1.77 (m) | 1.67 (dd, 10.35,13.80) | 1.69 (m) | 1.69 (m) |
| 9 | 1.09 (m) | 1.08 (m) | 1.08 (m) | 1.90 (overlap) | 1.16 (m) | 1.09 (m) | 1.07 (brt., 8.28) |
| 11 | 1.48 (dd, 8.11, 11.34) | 1.48 (dd,8.07, 11.30) | 1.48 (dd,8.37, 11.17) | 2.44 (dd, 9.04, 11.20) | 1.42 (dd, 8.48, 11.02) | 1.48 (dd, 8.21, 11.60) | 1.46 (dd, 8.28, 11.11) |
| 12 | 6.59 (d, 11.34) | 6.60 (d, 11.03) | 6.60 (d, 11.17) | 6.47 (d, 11.20) | 6.68 (d, 8.48) | 7.32 (overlap) | 6.56 (d, 11.11) |
| 16 | 0.66 (d, 6.63) | 0.87 (d, 6.72) | 0.85 (d, 6.39) | 0.67 (d, 6.58) | 1.11 (d, 6.84) | 0.81 (d, 6.69) | 0.42 (d, 6.46) |
| 17a | 2.48 (d, 3.37) | 2.48 (d, 3.46) | 2.49 (d, 3.33) | 2.53 (d, 3.11) | 2.64 (d, 3.79) | 2.41 (d, 2.24) | 2.46 (s) |
| 17b | 2.30 (brt., 3.37) | 2.32 (brt, 3.46) | 2.32 (brs) | 2.29 (brt., 3.11) | 2.59 (d, 3.79) | 2.27 (s) | 2.26 (s) |
| 18 | 1.20 (s) | 1.19 (s) | 1.19 (s) | 1.14 (s) | 1.21 (s) | 1.19 (s) | |
| 19 | 1.21 (s) | 1.19 (s) | 1.19 (s) | 1.44 (s) | 1.15 (s) | 1.25 (s) | 1.20 (s) |
| 20 | 1.84 (s) | 1.85 (s) | 1.85 (s) | 1.88 (s) | 1.89 (s) | 1.78 (s) | 1.81 (s) |
| 3-O-phenylacetyl | |||||||
| 2′ | 7.25 (overlap) | 5.54 (brs) | 5.66 (s) | 7.27 (overlap) | 7.32 (overlap) | ||
| 3′ | 7.30 (m) | 7.54 (d, 10.30) | 3.72 (brs) | 7.31 (td, 0.83, 7.12) | 7.36 (t, 7.35) | 7.01 (d, 7.36) | |
| 4′ | 7.27 (t, 7.76) | 6.07 (dt, 1.81, 10.30) | 4.25 (brd., 5.56) | 7.26 (m) | 7.28 (t, 7.21) | 7.20 (t, 7.36) | |
| 5′ | 7.30 (m) | 4.26 (d, 7.49) | 6.07 (m) | 7.31 (td, 0.83, 7.12) | 7.36 (t, 7.35) | 6.92 (t, 7.36) | |
| 6′ | 7.25 (overlap) | 3.76 (m) | 6.15 (d, 10.14) | 7.27 (overlap) | 7.32 (overlap) | 7.12 (d, 7.36) | |
| 7′a | 3.58 (2H, d, 3.21) | 2.67 (m) | 3.69 (m) | 3.58 (2H, s) | 3.67 (d, 16.65) | 3.81 (d, 13.39) | |
| 7′b | 2.54 (m) | 2.55 (m) | 7.27 (overlap) | 3.64 (d, 16.65) | 3.35 (d, 13.39) | ||
| 8′ | |||||||
| 5-OAc | |||||||
| C=O | |||||||
| CH3 | 2.02 (s) | 2.00 (s) | 2.00 (s) | 2.02 (s) | 2.08 (s) | 2.04 (s) | |
| 15-OAc | |||||||
| C=O | |||||||
| CH3 | 2.12 (s) | 2.13 (s) | 2.13 (s) | 2.13 (s) | 2.17 (s) | ||
| Glc | |||||||
| 1′′ | 4.81 (d, 6.77) | ||||||
| 2′′ | 3.69 (overlap) | ||||||
| 3′′ | 3.49 (m) | ||||||
| 4′′ | 3.69 (overlap) | ||||||
| 5′′ | 3.65 (m) | ||||||
| 6′′a | 3.89 (d, 13.27) | ||||||
| 6′′b | 3.85 (d, 13.27) | ||||||
| Position | M0 a | 1 a | 2 a | 3 a | 4 a | 5 a | 6 b | 7 a | 8 a | 9 a | 10 a | 11 a | 12 a |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 47.9(d) | 47.8 (t) | 47.9 (t) | 47.3 (t) | 47.5 (t) | 47.7(t) | 48.3 (t) | 47.9 (t) | 47.9 (t) | 47.9 (t) | 48.2 (t) | 49.9 (t) | 47.7 (t) |
| 2 | 37.7 (s) | 37.7 (d) | 37.8 (d) | 37.8 (d) | 37.8 (d) | 37.8 (d) | 38.5 (d) | 37.8 (d) | 37.9 (d) | 37.7 (d) | 37.6 (d) | 37.2 (d) | 37.6 (d) |
| 3 | 80.6(d) | 80.6 (d) | 80.6 (d) | 80.6 (d) | 82.6 (d) | 81.5 (d) | 80.9 (d) | 79.8 (d) | 79.8 (d) | 80.6 (d) | 78.9 (d) | 82.8 (d) | 81.0 (d) |
| 4 | 49.9 (s) | 49.9 (d) | 49.9 (d) | 49.9 (d) | 49.7 (d) | 49.7 (d) | 50.6 (d) | 49.9 (d) | 49.7 (d) | 50.0 (d) | 53.4 (d) | 51.6 (d) | 49.7 (d) |
| 5 | 65.2(d) | 65.1 (d) | 65.2 (d) | 65.1 (d) | 65.1 (d) | 65.3 (d) | 65.9 (d) | 65.2 (d) | 65.2 (d) | 65.0 (d) | 66.5 (d) | 66.0 (d) | 65.2 (d) |
| 6 | 58.9 (s) | 58.9 (s) | 59.0 (s) | 58.6 (s) | 58.9 (s) | 58.9 (s) | 59.4 (s) | 59.0 (s) | 59.0 (s) | 58.7 (s) | 60.8 (s) | 58.9 (s) | 58.9 (s) |
| 7 | 33.5(d) | 33.4 (t) | 33.5 (t) | 33.4 (t) | 33.4 (t) | 33.5 (t) | 34.1 (t) | 33.5 (t) | 33.4 (t) | 32.9 (t) | 32.1 (t) | 33.9 (t) | 33.4 (t) |
| 8 | 20.0(d) | 19.7 (t) | 20.1 (t) | 20.0 (t) | 20.1 (t) | 20.1 (t) | 20.6 (t) | 20.0 (t) | 20.0 (t) | 19.3 (t) | 19.7 (t) | 20.0 (t) | 20.0 (t) |
| 9 | 34.8 (s) | 30.2 (d) | 34.8 (d) | 35.8 (d) | 34.8 (d) | 34.8 (d) | 31.1 (d) | 34.8 (d) | 34.8 (d) | 32.9 (d) | 34.7 (d) | 35.7 (d) | 34.8 (d) |
| 10 | 25.6 (s) | 31.0 (s) | 25.7 (s) | 26.9 (s) | 25.7 (s) | 25.7 (s) | 32.1 (s) | 25.6 (s) | 25.6 (s) | 29.9 (s) | 25.1 (s) | 26.4 (s) | 25.6 (s) |
| 11 | 29.0 (d) | 25.2 (d) | 29.0 (d) | 29.0 (d) | 29.1 (d) | 29.0 (d) | 26.2 (d) | 29.1 (d) | 29.1 (d) | 30.1 (d) | 28.7 (d) | 29.3 (d) | 29.0 (d) |
| 12 | 143.7 (t) | 142.1(d) | 143.8(d) | 147.6(d) | 143.8(d) | 143.7(d) | 144.1(d) | 143.7(d) | 143.7(d) | 137.9(d) | 144.6(d) | 150.8(d) | 143.7(d) |
| 13 | 136.0(d) | 136.7 (s) | 136.0 (s) | 138.4 (s) | 136.0 (s) | 136.0 (s) | 136.7 (s) | 136.0 (s) | 135.9 (s) | 139.0 (s) | 136.2 (s) | 135.1 (s) | 135.9 (s) |
| 14 | 196.9(d) | 196.9 (s) | 196.9(s) | 198.4 (s) | 196.6(s) | 196.7(s) | 197.3 (s) | 196.9 (s) | 196.8 (s) | 197.2 (s) | 202.8 (s) | 199.3 (s) | 197.3 (s) |
| 15 | 91.7 (s) | 91.7 (s) | 91.8 (s) | 91.5 (s) | 91.5 (s) | 91.6 (s) | 92.2 (s) | 91.8 (s) | 91.7 (s) | 91.7 (s) | 88.4 (s) | 88.7 (s) | 91.6 (s) |
| 16 | 13.5(q) | 13.5 (q) | 13.6 (q) | 13.5 (q) | 12.8 (q) | 13.4 (q) | 13.8 (q) | 14.0 (q) | 14.0 (q) | 13.5 (q) | 13.9 (q) | 14.2 (q) | 13.1 (q) |
| 17 | 55.4 (t) | 55.3 (t) | 55.4 (t) | 56.2 (t) | 55.3 (t) | 55.3 (t) | 55.3 (t) | 55.3 (t) | 55.3 (t) | 55.2 (t) | 53.4 (t) | 55.4 (t) | 55.3 (t) |
| 18 | 28.9(q) | 71.2 (t)) | 28.9 (q) | 28.8 (q) | 28.9 (q) | 28.9 (q) | 70.9 (t) | 16.7 (q) | 16.7 (q) | 179.8 (s) | 28.7 (q) | 29.0 (q) | 28.9 (q) |
| 19 | 16.8(q) | 12.5 (q) | 16.8 (q) | 16.7 (q) | 16.8 (q) | 16.8 (q) | 12.9 (q) | 28.9 (q) | 28.9 (q) | 10.4 (q) | 15.8 (q) | 16.3 (q) | 16.7 (q) |
| 20 | 12.3 (t) | 12.4 (q) | 12.4 (q) | 58.1 (t) | 12.3 (q) | 12.4 (q) | 12.5 (q) | 12.4 (q) | 12.4 (q) | 12.5 (q) | 13.0 (q) | 12.2 (t) | 12.3 (q) |
| 3-O-phenylacetyl | |||||||||||||
| 1′ | 133.8 (s) | 133.7 (s) | 125.8 (s) | 133.7 (s) | 138.4 (s) | 120.3 (s) | 121.1 (s) | 150.2 (s) | 151.0 (s) | 133.6 (s) | 133.8 (s) | 123.1 (s) | |
| 2′ | 129.4(d) | 129.4(d) | 130.6(d) | 129.4(d) | 126.5(d) | 154.7 (s) | 156.3 (s) | 115.5(d) | 117.6(d) | 129.4 (s) | 129.6(d) | 155.3 (s) | |
| 3′ | 128.5(d) | 128.5(d) | 115.4(d) | 128.5(d) | 128.4(d) | 117.4(d) | 115.8(d) | 125.5(d) | 72.3 (d) | 128.5(d) | 128.9(d) | 114.5(d) | |
| 4′ | 127.2(d) | 127.2(d) | 154.9 (s) | 127.3(d) | 128.7(d) | 129.2(d) | 129.1(d) | 138.1(d) | 72.2 (d) | 127.3(d) | 127.6(d) | 129.2(d) | |
| 5′ | 128.5(d) | 128.5(d) | 115.4(d) | 128.5(d) | 128.4(d) | 120.9(d) | 120.1(d) | 73.5 (d) | 137.1(d) | 128.5(d) | 128.9(d) | 122.5(d) | |
| 6′ | 129.4(d) | 129.4(d) | 130.6(d) | 129.4(d) | 126.5(d) | 131.1(d) | 132.3(d) | 72.9 (d) | 130.7(d) | 129.4(d) | 129.4(d) | 131.2(d) | |
| 7′ | 41.53 (t) | 41.5 (t) | 40.6 (t) | 41.5 (t) | 72.8 (d) | 37.3 (t) | 36.4 (t) | 38.5 (t) | 32.4 (t) | 41.5 (t) | 41.5 (t) | 36.4 (t) | |
| 8′ | 170.9(s) | 170.9 (s) | 171.3 (s) | 170.9 (s) | 173.7 (s) | 172.1 (s) | 171.7 (s) | 165.8 (s) | 165.8 (s) | 170.9 (s) | 169.9 (s) | 172.0 (s) | |
| 5-OAc | |||||||||||||
| C=O | 170.8(s) | 170.7 (s) | 170.8 (s) | 170.7 (s) | 170.8 (s) | 171.2 (s) | 170.9 (s) | 170.6 (s) | 170.7 (s) | 170.8 (s) | 171.0 (s) | 171.1 (s) | |
| CH3 | 21.0(q) | 21.0 (q) | 21.0 (q) | 21.0 (q) | 20.9 (q) | 20.8 (q) | 21.1 (q) | 20.9 (q) | 20.8 (q) | 21.0 (q) | 21.1 (q) | 20.9 (q) | |
| 15-OAc | |||||||||||||
| C=O | 169.6(s) | 169.6 (s) | 169.7 (s) | 169.6 (s) | 169.4 (s) | 169.8 (s) | 170.2 (s) | 169.7 (s) | 169.8 (s) | 169.6 (s) | 169.6 (s) | ||
| CH3 | 21.9 (s) | 21.9 (q) | 21.9 (q) | 21.8 (q) | 21.8 (q) | 21.9 (q) | 22.0 (q) | 21.9 (q) | 21.9 (q) | 21.8 (q) | 21.9 (q) | ||
| Glc | |||||||||||||
| 1′′ | 101.2(d) | ||||||||||||
| 2′′ | 73.5 (d) | ||||||||||||
| 3′′ | 75.9 (d) | ||||||||||||
| 4′′ | 69.6 (d) | ||||||||||||
| 5′′ | 75.9 (d) | ||||||||||||
| 6′′ | 61.8 (t) | ||||||||||||
| No. | Component Name | RT (min) | Formula [M + H]+ | Observed m/z | Error (ppm) | MS n | Distribution | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Rat | RLMs | Fungi | |||||||||
| Plasma | Urine | Faeces | |||||||||
| M1 | M + O | 8.49 | C32H41O9 | 569.2746 | 0.18 | 569.2746, 509.2533, 449.2336, 433.2231, 373.2012, 355.1910, 313.1801, 295.1697, 277.1595, 267.1741, 249.1643 | √ | √ | √ | √ | √ |
| M2 | M + O | 9.79 | C32H41O9 | 569.2748 | 0.53 | 569.2748, 509.2543, 449.2332, 417.2275, 375.2172, 357.2069, 315.1955, 297.1854, 279.1748, 269.1904, 251.1803 | - | √ | √ | √ | √ |
| M3 | M + O | 9.99 | C32H41O9 | 569.2747 | 0.35 | 569.2747, 551.2642, 509.2538, 491.2438,449.2336, 431.2226, 373.2020, 355.1910, 313.1805, 295.1699, 277.1592, 267.1745, 249.1645 | √ | √ | √ | √ | √ |
| M4 | M + O | 10.34 | C32H41O9 | 569.2756 | 1.93 | 569.2756, 509.2549, 449.2331, 417.2286, 375.2184, 357.2071, 315.1959, 297.1858, 279.1751, 269.1907, 251.1805 | √ | √ | √ | √ | √ |
| M5 | M + O | 10.66 | C32H41O9 | 569.2754 | 1.58 | 569.2754, 509.2547, 449.2329, 417.2288, 375.2181, 357.2071, 315.1959, 297.1865, 279.1751, 269.1912 | √ | - | √ | √ | √ |
| M6 | M + O | 11.38 | C32H41O9 | 569.2747 | 0.35 | 569.2747, 509.2542, 449.2339, 431.2230, 373.2020, 355.1910, 313.1806, 295.1699, 277.1592, 267.1745 | √ | - | √ | √ | - |
| M7 | M + 2O | 6.06 | C32H41O10 | 585.2688 | −1.03 | 585.2688, 525.2480, 507.2377, 433.2221, 373.1995, 331.1906, 313.1796, 295.1701, 285.1841, 277.1594 | √ | √ | - | √ | - |
| M8 | M + 2O | 6.36 | C32H41O10 | 585.2704 | 1.71 | 585.2704, 525.2490, 507.2382, 465.2271, 433.2222, 391.2116, 373.2011, 355.1904, 313.1805, 295.1698, 285.1847, 267.1743, 255.1382 | √ | √ | √ | √ | √ |
| M9 | M + 2O | 6.59 | C32H41O10 | 585.2689 | −0.85 | 585.2689, 525.2485, 507.2382, 465.2272, 447.2169, 433.2221, 391.2114, 373.2005, 355.1900, 313.1806, 295.1689, 285.1847, 277.1591, 267.1749 | √ | √ | √ | √ | √ |
| M10 | M + 2O | 6.92 | C32H41O10 | 585.2692 | −0.34 | 585.2692, 525.2480, 507.2377, 465.2278, 391.2118, 373.2010, 355.1907, 313.1805, 295.1695, 267.1747 | √ | - | √ | √ | - |
| M11 | M + 2O | 7.12 | C32H41O10 | 585.2714 | 3.42 | 585.2714, 525.2496, 465.2284, 373.1997, 355.1906, 313.1802, 295.1706 | √ | - | - | √ | - |
| M12 | M + 2O + 2H | 7.35 | C32H43O10 | 587.2858 | 0.17 | 587.2858, 527.2645, 467.2433, 357.2069, 315.1961, 297.1860, 279.1752, 269.1905, 251.1796 | √ | - | √ | √ | √ |
| M13 | M + 2O + 2H | 7.49 | C32H43O10 | 587.2867 | 2.72 | 587.2867, 527.2661, 509.2549, 467.2442, 449.2337, 417.2285, 357.2074, 315.1963, 297.1858, 279.1757, 269.1906 | √ | - | √ | √ | √ |
| M14 | M + O-2H | 8.80 | C32H39O9 | 567.2598 | 1.59 | 567.2598, 507.2377, 447.2166, 371.1853, 329.1760, 311.1644, 293.1538, 283.1700, 265.1597, 255.1747 | √ | √ | √ | - | √ |
| M15 | M + O-2H | 8.95 | C32H39O9 | 567.2598 | 1.59 | 567.2598, 507.2377, 447.2166, 371.1852, 329.1761, 311.1654, 293.1547, 283.1700, 265.1597 | √ | √ | √ | - | - |
| M16 | M + 2O-2H | 8.71 | C32H39O10 | 583.2530 | −1.37 | 583.2530, 523.2330, 463.2126, 447.2032, 387.1815, 345.1707, 327.1595, 309.1498, 299.1645, 281.1541, 263.1438, 253.1591 | √ | √ | √ | √ | √ |
| M17 | M + 2O-2H | 9.49 | C32H39O10 | 583.2548 | 1.71 | 583.2548, 523.2332, 463.2132, 387.1814, 345.1710, 327.1600, 309.1490, 299.1647, 281.1540, 263.1449, 253.1596 | √ | - | √ | √ | √ |
| M18 | M + 3O-2H | 5.96 | C32H39O11 | 599.2497 | 1.67 | 599.2497, 539.2279, 387.1814, 345.1704, 327.1600, 309.1490, 299.1645, 281.1540, 263.1425, 253.1594 | √ | √ | √ | - | - |
| M19 | M + 3O-2H | 6.37 | C32H39O11 | 599.2505 | 3.00 | 599.2505, 539.2284, 479.2076, 447.2024, 405.1918, 387.1812, 345.1705, 327.1601, 309.1494, 299.1647, 281.1541, 263.1436, 253.1585 | √ | √ | √ | √ | - |
| M20 | M + 3O-2H | 6.75 | C32H39O11 | 599.2500 | 2.17 | 599.2500, 539.2286, 387.1811, 345.1711, 327.1602, 309.1496, 299.1648, 281.1547, 263.1438, 253.1591 | √ | √ | √ | - | - |
| M21 | M + 4O-2H | 4.61 | C32H39O12 | 615.2451 | 2.44 | 615.2451, 597.2335, 555.2226, 537.2123, 477.1913, 385.1654, 343.1542, 325.1440, 307.1334, 297.1497, 279.1386, 261.1280, 251.1426 | √ | √ | √ | - | - |
| M22 | M + 4O-2H | 5.04 | C32H39O12 | 615.2460 | 3.90 | 615.2460, 555.2236, 537.2133, 477.1919, 343.1549, 325.1448, 297.1494, 279.1389, 261.1285, 251.1437 | √ | √ | √ | - | - |
| M23 | M + 4O-2H | 5.77 | C32H39O12 | 615.2453 | 2.76 | 615.2453, 555.2231, 537.2125, 477.1916, 343.1549, 325.1440, 307.1335, 297.1493, 279.1390, 261.1286, 251.1439 | √ | √ | √ | - | - |
| M24 | M-2CH2CO-C8H6O | 5.84 | C20H31O5 | 351.2166 | 0.00 | 351.2166, 333.2059, 315.1951, 297.1849, 279.1746, 269.1898, 251.1802 | √ | √ | √ | √ | - |
| M25 | M-C2H2O | 12.49 | C30H39O7 | 511.2694 | 0.78 | 511.2694, 451.2482, 433.2373, 375.2171, 357.2068, 315.1959, 297.1858, 279.1750, 269.1902, 251.1431 | √ | √ | √ | √ | - |
| M26 | M-C2H2O + O | 7.66 | C30H39O8 | 527.2661 | 4.17 | 527.2661, 509.2540, 467.2440, 449.2337, 391.2120, 373.2017, 331.1910, 313.1803, 295.1696 | √ | √ | √ | - | - |
| M27 | M-C2H2O + O | 9.14 | C30H39O8 | 527.2647 | 1.52 | 527.2647, 509.2553, 467.2435, 449.2330, 391.2122, 373.2020, 331.1909, 313.1804, 295.1706 | √ | √ | √ | - | √ |
| M28 | M + O+ C6H10O5 | 5.23 | C38H51O14 | 731.3280 | 0.96 | 731.3280, 569.2768, 509.2549, 449.2335, 357.2074, 315.1971, 297.1868, 279.1754, 269.1911 | - | - | √ | - | - |
| M29 | M + O+ C6H10O5 | 6.97 | C38H51O14 | 731.3286 | 1.78 | 731.3286, 671.3065, 569.2745, 509.2544, 449.2331, 375.2173, 357.2073, 315.1963, 297.1855, 279.1752, 269.1902 | √ | - | √ | - | √ |
| M30 | M + 2O+ SO3 | 2.87 | C32H41O13S | 665.2271 | 1.35 | 665.2271, 647.2167, 605.2055, 587.1955, 545.1848, 393.1376, 313.1788, 295.1683, 277.1589, 267.1744 | - | - | √ | - | - |
| M31 | M + 2O+ SO3 | 3.19 | C32H41O13S | 665.2267 | 0.75 | 665.2267, 605.2061, 545.1843, 393.1371, 313.1805, 295.1691, 277.1580 | - | - | √ | - | - |
| Compound | IC50 (μM) | |||
|---|---|---|---|---|
| SH-SY5Y | LO2 | AC-16 | HK-2 | |
| M0 | 54.95 ± 1.20 | >100 | 97.72 ± 2.13 | >100 |
| M3 | 39.63 ± 0.43 | 37.41 ± 0.41 | 17.86 ± 0.19 | 22.65 ± 0.25 |
| M24 | 40.74 ± 0.44 | 38.9 ± 0.42 | 21.73 ± 0.24 | 26.49 ± 0.29 |
| M25 | 33.27 ± 0.36 | 30.13 ± 0.33 | 3.60 ± 0.04 | 16.11 ± 0.18 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, S.; Xu, X.; Wei, X.; Xin, J.; Li, S.; Lv, Y.; Chen, W.; Yuan, W.; Xie, B.; Zu, X.; et al. Comprehensive Metabolic Profiling of Euphorbiasteroid in Rats by Integrating UPLC-Q/TOF-MS and NMR as Well as Microbial Biotransformation. Metabolites 2022, 12, 830. https://doi.org/10.3390/metabo12090830
Xiao S, Xu X, Wei X, Xin J, Li S, Lv Y, Chen W, Yuan W, Xie B, Zu X, et al. Comprehensive Metabolic Profiling of Euphorbiasteroid in Rats by Integrating UPLC-Q/TOF-MS and NMR as Well as Microbial Biotransformation. Metabolites. 2022; 12(9):830. https://doi.org/10.3390/metabo12090830
Chicago/Turabian StyleXiao, Sijia, Xike Xu, Xintong Wei, Jiayun Xin, Shanshan Li, Yanhui Lv, Wei Chen, Wenlin Yuan, Bin Xie, Xianpeng Zu, and et al. 2022. "Comprehensive Metabolic Profiling of Euphorbiasteroid in Rats by Integrating UPLC-Q/TOF-MS and NMR as Well as Microbial Biotransformation" Metabolites 12, no. 9: 830. https://doi.org/10.3390/metabo12090830