Glycomics Approaches for the Bioassay and Structural Analysis of Heparin/Heparan Sulphates

Abstract
:1. Introduction—The Complexity and Challenges Associated with Glycomics
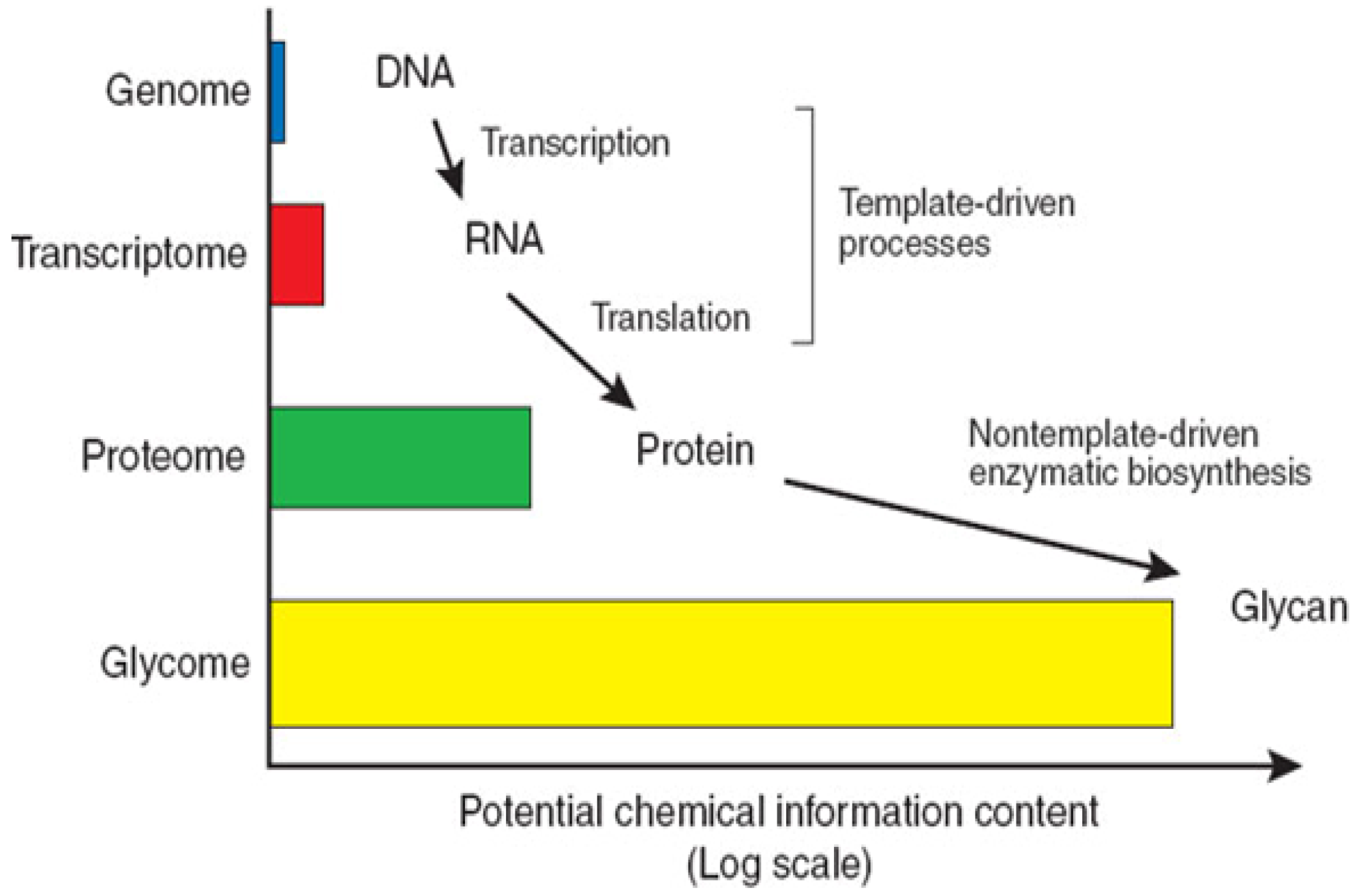
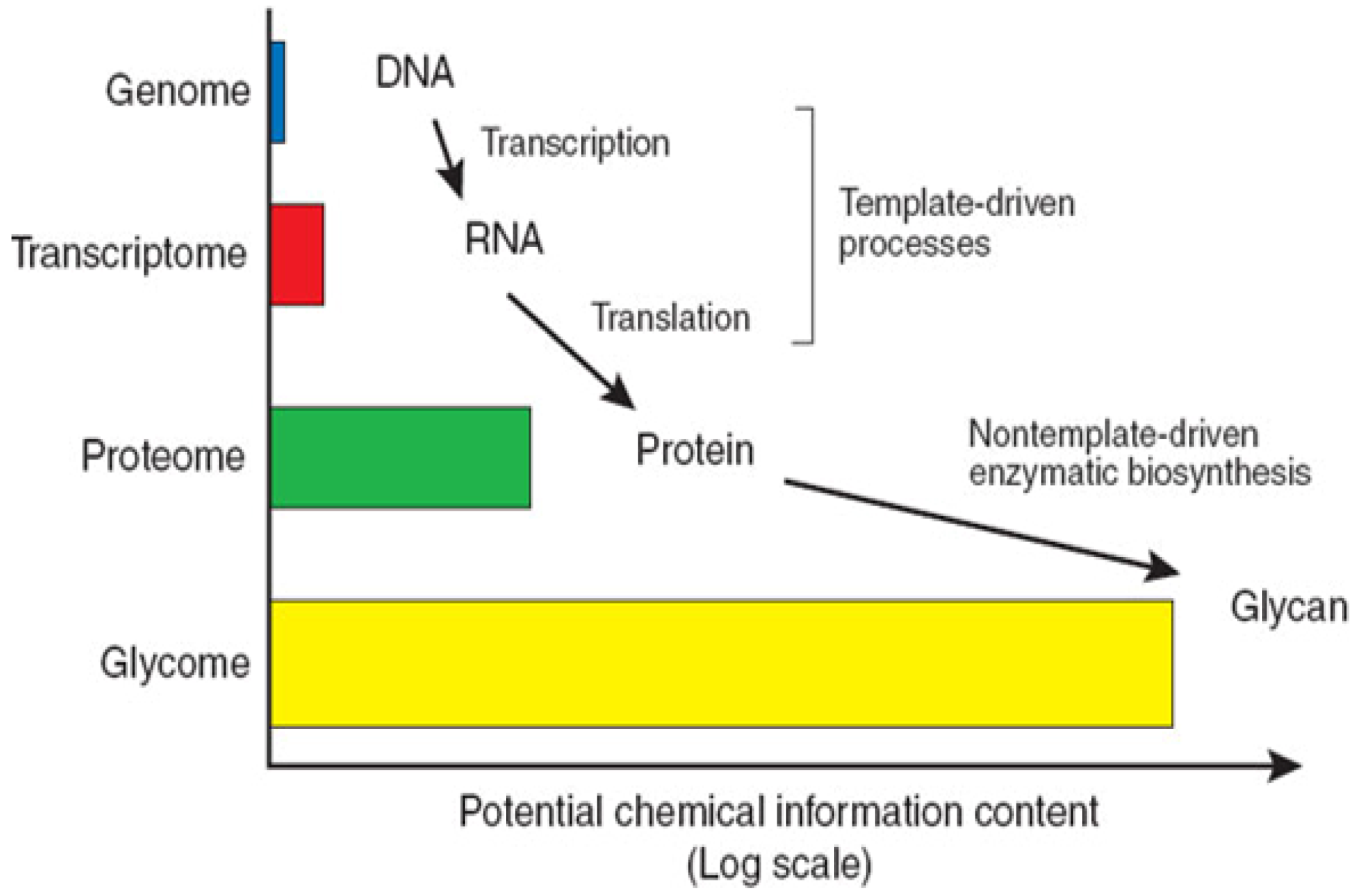
1.1. Structural Characteristics of Proteoglycans
1.2. Heparan Sulphate Structure and Biosynthesis
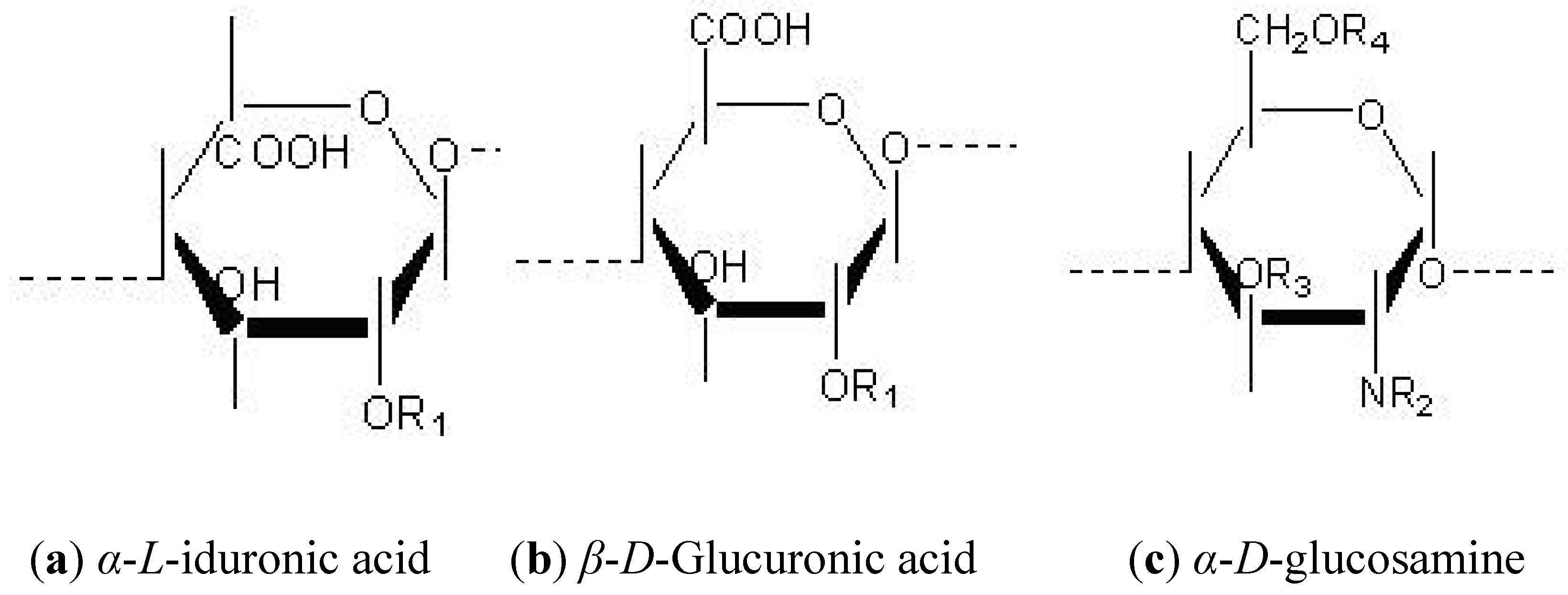
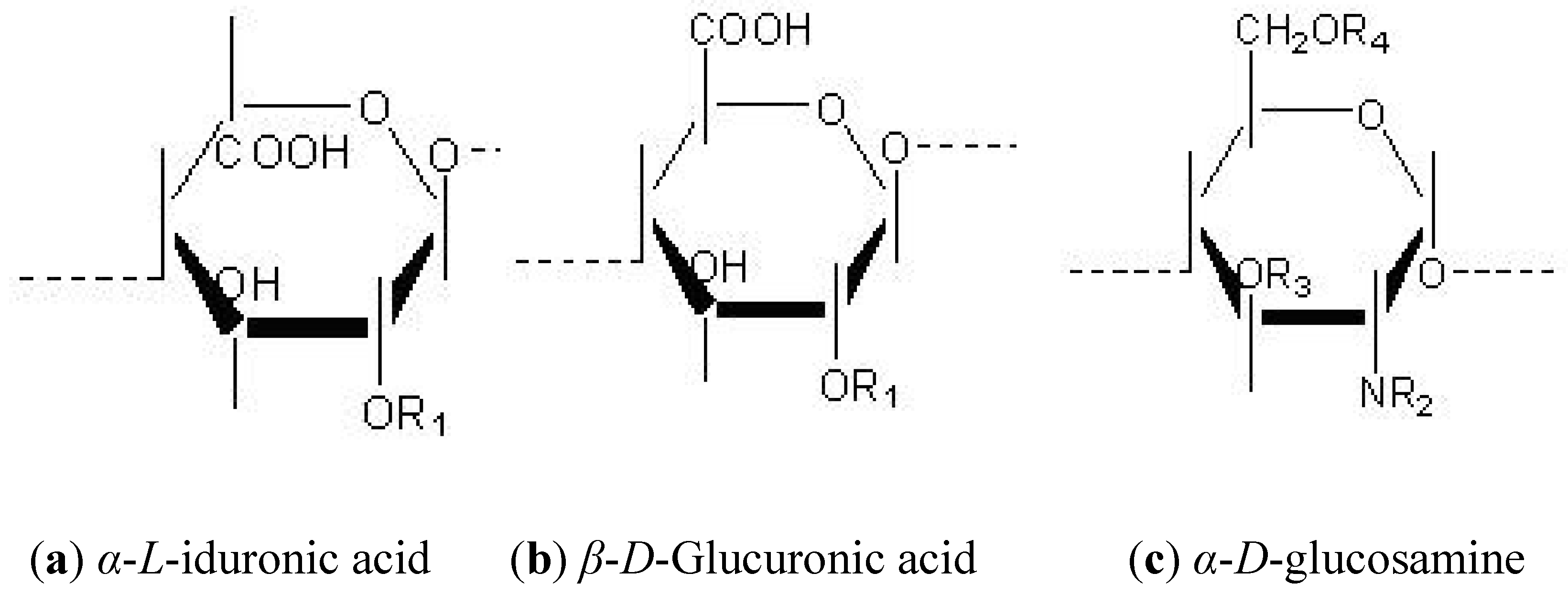
1.3. Heterogeneity Supplied by the Domain Structure of HS Polysaccharides
1.4. Flexibility of HS Chains

1.5. Diverse Functions of HS
1.6. Variation in Endogenous Tissue HS Structures
2. Structural Analysis of HS Glycan Chains
2.1. Indirect Methods To Study HS Structure
2.2. Studying the Domain Structure of HS
2.3. Study of HS from Tissues

{kind=link}
{kind=link}
{kind=link}
| Enzyme | Substrate Specificity |
|---|---|
| Bacterial Exoenzyme | |
| Δ4,5-glycuronate-2-sulphatase | ΔUA(2S) |
| Δ4,5-glycuronidase | ΔUA |
| Exoglycosidases | |
| Iduronidase | IdoA |
| Glucuronidase | GlcA |
| α- N-acetylglucaminidase | GlcNAc |
| Exosulphatases | |
| Iduronate-2-sulphatase | IdoA(2S) |
| Glucosamine-6-sulphatase | GlcNAc(6S). GlcNS (6S) |
| Sulphaminidase (glucosamine N –sulphatase) | GlcNS |
| Glucuronate -2-sulphatase | GlcA(2S) |
| Glucosamine -3-sulphatase | GlcNS(3S) |
2.4. Traditional Methods for Extraction and Purification of HS
2.5. Structural Analysis of Cellular and Tissue Derived HS
2.6. Fluorescent Tags Which Offer Higher Sensitivity
2.7. Use of Separations Techniques for Structural Analysis
2.7.1. Strong Anion Exchange Chromatography
2.7.2. Polyacrylamide Gel Separation
2.7.3. Capillary Electrophoresis
2.7.4. Integral Glycan Sequencing (IGS) Techniques
2.8. The Use of Mass Spectrometry for Sequence Information
3. Current Methods for Analysis of Large Data Sets Obtained from Structural Studies
3.1. The Need for Chemometric Techniques for HS Structural Analysis
- (1). The sensitivity of the methods means they are good at sample analysis, especially when comparing with standards (if possible). But, when standards are not available (which is common in HS oligosaccharide analysis), large differences in spectral results are caused from small changes in sample processing and handling. This makes comparison of results very difficult.
- (2). The high dimensionality of the data from experimental techniques results in ‘noisy’ spectra (white noise) which may require pre-processing. This is a challenge as one has to be careful not to remove significant components.
- (3). One of the most common problems for all techniques that study HS oligosaccharides is that simultaneous and routine analysis provides a vast quantity of data.
3.2. Existing Chemometric Methods
3.3. Databases Crucial in the Intercalation of Data
4. Functional Analysis of HS Structues
- -Direct extraction and purification of HS populations from tissues or culture cells arising from different organs.
4.1. Techniques for Studying Structural Aspects of HS and Protein Interactions
4.2. Investigating Effects on Cellular Signalling
4.3. Cellular Signalling Assays
4.3.1. Cell Proliferation
4.3.2. Cell Adhesion and Invasion Assays
4.3.3. Angiogenesis Assays
4.3.4. Cellular Signalling Assays
4.4. The Need for Higher Throughput Biological Assays for the Study of HS
4.5. Use of Cell Based Screening Techniques
4.6. Glycoarrays-Carbohydrate Microarrays
4.7. Fabrication of Carbohydrate Microarrays
- -Reducing-end aldehyde linked to amino and hydrazide surfaces groups on the surface.
- -Immobilization using Diels-Alder reaction.
- -SH-/malemide.
- -Site specific and non-covalent immobilization.
4.8. Glycobioarrays
5. Concluding Remarks
Acknowledgments
Conflict of Interest
References
- Turnbull, J.E.; Field, R.A. Emerging glycomics technologies. Nat. Chem. Biol. 2007, 3, 74–77. [Google Scholar] [CrossRef]
- Wang, D. Carbohydrate microarrays. Proteomics 2003, 3, 2167–2175. [Google Scholar] [CrossRef]
- Bourin, M.C.; Lindahl, U. Glycosaminoglycans and the regulation of blood coagulation. Biochem.J. 1993, 289, 313–330. [Google Scholar]
- van Horssen, J.; Wesseling, P.; van den Heuvel, L.P.; de Waal, R.M.; Verbeek, M.M. Heparan sulphate proteoglycans in alzheimer's disease and amyloid-related disorders. Lancet Neurol. 2003, 2, 482–492. [Google Scholar] [CrossRef]
- Whitelock, J.M.; Iozzo, R.V. Heparan sulfate: A complex polymer charged with biological activity. Chem. Rev. 2005, 105, 2745–2764. [Google Scholar] [CrossRef]
- Esko, J.D.; Lindahl, U. Molecular diversity of heparan sulfate. J. Clin. Invest. 2001, 108, 169–173. [Google Scholar]
- Westling, C.; Lindahl, U. Location of n-unsubstituted glucosamine residues in heparan sulfate. J. Biol. Chem. 2002, 277, 49247–49255. [Google Scholar] [CrossRef]
- Powell, A.K.; Yates, E.A.; Fernig, D.G.; Turnbull, J.E. Interactions of heparin/heparan sulfate with proteins: Appraisal of structural factors and experimental approaches. Glycobiology 2004, 14, 17R–30R. [Google Scholar] [CrossRef]
- Yates, E.A.; Guimond, S.E.; Turnbull, J.E. Highly diverse heparan sulfate analogue libraries: Providing access to expanded areas of sequence space for bioactivity screening. J. Med. Chem. 2004, 47, 277–280. [Google Scholar] [CrossRef]
- Turnbull, J.E.; Gallagher, J.T. Molecular organization of heparan sulphate from human skin fibroblasts. Biochem. J. 1990, 265, 715–724. [Google Scholar]
- Gallagher, J. Multiprotein signalling complexes: Regional assembly on heparan sulphate. Biochem. Soci. Trans. 2006, 34, 438–441. [Google Scholar]
- Conrad, H.E. Heparin-Binding Proteins; Academic Press: New York, NY, USA, 1998; p. 527. [Google Scholar]
- Lindahl, U.; Kusche-Gullberg, M.; Kjellén, L. Regulated diversity of heparan sulfate. J. Biol. Chem. 1998, 273, 24979–24982. [Google Scholar]
- Van Vactor, D.; Wall, D.P.; Johnson, K.G. Heparan sulfate proteoglycans and the emergence of neuronal connectivity. Curr. Opin. Neurobiol. 2006, 16, 40–51. [Google Scholar] [CrossRef]
- Iozzo, R.V.; Sanderson, R.D. Proteoglycans in cancer biology, tumour microenvironment and angiogenesis. J. Cell. Mol. Med. 2010, 15, 1013–1031. [Google Scholar]
- Bishop, J.R.; Schuksz, M.; Esko, J.D. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 2007, 446, 1030–1037. [Google Scholar] [CrossRef]
- Lindahl, U.; Thunberg, L.; Backstrom, G.; Riesenfeld, J.; Nordling, K.; Bjork, I. Extension and structural variability of the antithrombin-binding sequence in heparin. J. Biol. Chem. 1984, 259, 12368–12376. [Google Scholar]
- Park, Y.; Yu, G.; Gunay, N.S.; Linhardt, R.J. Purification and characterization of heparan sulphate proteoglycan from bovine brain. Biochem. J. 1999, 344, 723–730. [Google Scholar]
- Lindahl, B.; Lindahl, U. Amyloid-specific heparan sulfate from human liver and spleen. J. Biol. Chem. 1997, 272, 26091–26094. [Google Scholar] [CrossRef]
- Lindahl, B.; Eriksson, L.; Lindahl, U. Structure of heparan sulphate from human brain, with special regard to alzheimer's disease. Biochem. J. 1995, 306, 177–184. [Google Scholar]
- Ledin, J.; Staatz, W.; Li, J.P.; Gotte, M.; Selleck, S.; Kjellen, L.; Spillmann, D. Heparan sulfate structure in mice with genetically modified heparan sulfate production. J. Biol. Chem. 2004, 279, 42732–42741. [Google Scholar]
- Ruoslahti, E. Proteoglycans in cell regulation. J. Biol. Chem. 1989, 264, 13369–13372. [Google Scholar]
- van Kuppevelt, T.H.; Dennissen, M.A.; van Venrooij, W.J.; Hoet, R.M.; Veerkamp, J.H. Generation and application of type-specific anti-heparan sulfate antibodies using phage display technology. Further evidence for heparan sulfate heterogeneity in the kidney. J. Biol. Chem. 1998, 273, 12960–12966. [Google Scholar]
- Dennissen, M.A.B.A.; Jenniskens, G.J.; Pieffers, M.; Versteeg, E.M.M.; Petitou, M.; Veerkamp, J.H.; van Kuppevelt, T.H. Large, tissue-regulated domain diversity of heparan sulfates demonstrated by phage display antibodies. J. Biol. Chem. 2002, 277, 10982–10986. [Google Scholar]
- David, G.; Bai, X.M.; van der Schueren, B.; Cassiman, J.J.; van den Berghe, H. Developmental changes in heparan sulfate expression: In situ detection with mabs. J. Cell Biol. 1999, 119, 961–975. [Google Scholar]
- Thompson, S.M.; Connell, M.G.; Fernig, D.G.; Ten Dam, G.B.; van Kuppevelt, T.H.; Turnbull, J.E.; Jesudason, E.C.; Losty, P.D. Novel 'phage display antibodies identify distinct heparan sulfate domains in developing mammalian lung. Pediatr. Surg. Int. 2007, 23, 411–417. [Google Scholar] [CrossRef]
- Feyzi, E.; Saldeen, T.; Larsson, E.; Lindahl, U.; Salmivirta, M. Age-dependent modulation of heparan sulfate structure and function. J. Biol. Chem. 1998, 273, 13395–13398. [Google Scholar]
- Esko, J.D.; Lindahl, U. Molecular diversity of heparan sulfate. J. Clin. Invest. 2001, 108, 169–173. [Google Scholar]
- Turnbull, J.E.; Gallagher, J.T. Oligosaccharide mapping of heparan sulphate by polyacrylamide-gradient-gel electrophoresis and electrotransfer to nylon membrane. Biochem. J. 1988, 251, 597–608. [Google Scholar]
- Turnbull, J.E. Mapping and sequencing of heparan sulphate. PhD thesis, University of Manchester, Manchester, UK, 1990. [Google Scholar]
- Turnbull, J.E.; Gallagher, J.T. Sequence analysis of heparan sulphate indicates defined location of N-sulphated glucosamine and iduronate 2-sulphate residues proximal to the protein-linkage region. Biochem. J. 1991, 277, 297–303. [Google Scholar]
- Turnbull, J.E.; Hopwood, J.J.; Gallagher, J.T. A strategy for rapid sequencing of heparan sulfate and heparin saccharides. Proc. Nat. Acad. Sci. USA 1999, 96, 2698–2703. [Google Scholar] [CrossRef]
- Burdon, R.H.; Van Knippenberg, P.H. Glycoprotein and proteoglycan techniques. In Laboratory Techniques in Biochemistry and Molecular Biology; Elsevier: Amsterdam, The Netherlands, 1985; Volume 16, p. 462. [Google Scholar]
- Shively, J.E.; Conrad, H.E. Formation of anhydrosugars in the chemical depolymerisation of heparin. Biochemistry 1976, 15, 3932–3942. [Google Scholar] [CrossRef]
- Hovingh, P.; Linker, A. The enzymatic degradation of heparin and heparitin sulfate. 3. Purification of a heparitinase and a heparinase from flavobacteria. J. Biol. Chem. 1970, 245, 6170–6175. [Google Scholar]
- Linhardt, R.J.; Rice, K.G.; Kim, Y.S.; Lohse, D.L.; Wang, H.M.; Loganathan, D. Mapping and quantification of the major oligosaccharide components of heparin. Biochem. J. 1988, 254, 781–787. [Google Scholar]
- LeBrun, L.A.; Linhardt, R.J. Degradadation of heparan sulphate with heparin lyases. In Methods in Molecular Biology: Proteoglycan Protocols; Iozzo, R., Ed.; Humana Press Inc.: Totowa, NJ, USA, 2001; p. 171. [Google Scholar]
- Skidmore, M.A.; Guimond, S.E.; Dumax-Vorzet, A.F.; Atrih, A.; Yates, E.A.; Turnbull, J.E. High sensitivity separation and detection of heparan sulfate disaccharides. J. Chromatogr. A 2006, 1135, 52–56. [Google Scholar] [CrossRef]
- Drummond, K.J.; Yates, E.A.; Turnbull, J.E. Electrophoretic sequencing of heparin/heparan sulfate oligosaccharides using a highly sensitive fluorescent end label. Proteomics 2001, 1, 304–310. [Google Scholar] [CrossRef]
- Phelps, C.F. Polysaccharides; Oxford University Press: London, UK, 1972. [Google Scholar]
- Ha, Y.W.; Jeon, B.T.; Moon, S.H.; Toyoda, H.; Toida, T.; Linhardt, R.J.; Kim, Y.S. Characterization of heparan sulfate from the unossified antler of cervus elaphus. Carbohydr. Res. 2005, 340, 411–416. [Google Scholar]
- Tekotte, H.; Engel, M.; Margolis, R.U.; Margolis, R.K. Disaccharide composition of heparan sulfates: Brain, nervous tissue storage organelles, kidney, and lung. J. Neurochem. 1994, 62, 1126–1130. [Google Scholar]
- Maccarana, M.; Sakura, Y.; Tawada, A.; Yoshida, K.; Lindahl, U. Domain structure of heparan sulfates from bovine organs. J. Biol. Chem. 1996, 271, 17804–17810. [Google Scholar]
- Lamanna, W.C.; Baldwin, R.J.; Padva, M.; Kalus, I.; Ten Dam, G.; van Kuppevelt, T.H.; Gallagher, J.T.; von Figura, K.; Dierks, T.; Merry, C.L.R. Heparan sulfate 6-o-endosulfatases: Discrete in vivo activities and functional co-operativity. Biochem. J. 2006, 400, 63–73. [Google Scholar] [CrossRef]
- Guimond, S.; Turner, K.; Kita, M.; Ford-Perriss, M.; Turnbull, J. Dynamic biosynthesis of heparan sulphate sequences in developing mouse brain: A potential regulatory mechanism during development. Biochem. Soc. Trans. 2001, 29, 177–181. [Google Scholar]
- Winterbourne, D.J.; Mora, P.T. Cells selected for high tumorigenicity or transformed by simian virus 40 synthesize heparan sulfate with reduced degree of sulfation. J. Biol. Chem. 1981, 256, 4310–4320. [Google Scholar]
- Brickman, Y.G.; Nurcombe, V.; Ford, M.D.; Gallagher, J.T.; Bartlett, P.F.; Turnbull, J.E. Structural comparison of fibroblast growth factor-specific heparan sulfates derived from a growing or differentiating neuroepithelial cell line. Glycobiology 1998, 8, 463–471. [Google Scholar]
- Yanagishita, M. Isolation of proteoglycans from cell cultures and tissues. In Methods in Molecular Biology: Proteoglycan Protocols; Iozzo, R.V., Ed.; Humana Press: Totowa, NJ, USA, 2001; Volume 171, pp. 3–8. [Google Scholar]
- Lyon, M.; Gallagher, J.T. Purification and partial characterization of the major cell-associated heparan sulphate proteoglycan of rat liver. Biochem. J. 1991, 273, 415–422. [Google Scholar]
- Yanagishita, M.; Midura, R.J.; Hascall, V.C. Proteoglycans: Isolation and purification from tissue cultures. Methods Enzymol. 1987, 138, 279–289. [Google Scholar]
- Inoue, Y.; Nagasawa, K. Selective n-desulfation of heparin with dimethyl sulfoxide containing water or methanol. Carbohydr. Res. 1976, 46, 87–95. [Google Scholar] [CrossRef]
- Stenstad, T.; Magnus, J.H.; Husby, G.; Kolset, S.O. Purification of amyloid-associated heparan sulphate proteoglycans and galactosaminoglycan free chains from human tissues. Scand. J. Immunol. 1993, 37, 227–235. [Google Scholar] [CrossRef]
- Jalkanen, M.; Rapraeger, A.; Bernfield, M. Mouse mammary epithelial cells produce basement membrane and cell surface heparan sulfate proteoglycans containing distinct core proteins. J. Cell Biol. 1988, 106, 953–962. [Google Scholar] [CrossRef]
- Misra, K.B.; Kim, K.C.; Cho, S.; Low, M.G.; Bensadoun, A. Purification and characterization of adipocyte heparan sulfate proteoglycans with affinity for lipoprotein lipase. J. Biol. Chem. 1994, 269, 23838–23844. [Google Scholar]
- Yanagishita, M.; Hascall, V.C. Metabolism of proteoglycans in rat ovarian granulosa cell culture. Multiple intracellular degradative pathways and the effect of chloroquine. J. Biol. Chem. 1984, 259, 10270–10283. [Google Scholar]
- Sanderson, R.D.; Turnbull, J.E.; Gallagher, J.T.; Lander, A.D. Fine structure of heparan sulfate regulates syndecan-1 function and cell behavior. J. Biol. Chem. 1994, 269, 13100–13106. [Google Scholar]
- Ha, Y.W.; Jeon, B.T.; Moon, S.H.; Toyoda, H.; Toida, T.; Linhardt, R.J.; Kim, Y.S. Characterization of heparan sulfate from the unossified antler of cervus elaphus. Carbohydr. Res. 2005, 340, 411–416. [Google Scholar] [CrossRef]
- Toida, T.; Yoshida, H.; Toyoda, H.; Koshiishi, I.; Imanari, T.; Hileman, R.E.; Fromm, J.R.; Linhardt, R.J. Structural differences and the presence of unsubstituted amino groups in heparan sulphates from different tissues and species. Biochem. J. 1997, 322, 499–506. [Google Scholar]
- Kinoshita, A.; Sugahara, K. Microanalysis of glycosaminoglycan-derived oligosaccharides labeled with a fluorophore 2-aminobenzamide by high-performance liquid chromatography: Application to disaccharide composition analysis and exosequencing of oligosaccharides. Anal. Biochem. 1999, 269, 367–378. [Google Scholar] [CrossRef]
- Yamada, S.; Van Die, I.; Van den Eijnden, D.H.; Yokota, A.; Kitagawa, H.; Sugahara, K. Demonstration of glycosaminoglycans in caenorhabditis elegans. FEBS Lett. 1999, 459, 327–331. [Google Scholar] [CrossRef]
- Sato, K.; Okubo, A.; Yamazaki, S. Separation of 2-aminobenzoic acid-derivatized glycosaminoglycans and asparagine-linked glycans by capillary electrophoresis. Anal. Sci. 2005, 21, 21–24. [Google Scholar] [CrossRef]
- Lawrence, R.; Olson, S.K.; Steele, R.E.; Wang, L.; Warrior, R.; Cummings, R.D.; Esko, J.D. Evolutionary differences in glycosaminoglycan fine structure detected by quantitative glycan reductive isotope labeling. J. Biol. Chem. 2008, 283, 33674–33684. [Google Scholar]
- Holmborn, K.; Ledin, J.; Smeds, E.; Eriksson, I.; Kusche-Gullberg, M.; Kjellen, L. Heparan sulfate synthesized by mouse embryonic stem cells deficient in ndst1 and ndst2 is 6-o-sulfated but contains no n-sulfate groups. J. Biol. Chem. 2004, 279, 42355–42358. [Google Scholar]
- Toyoda, H.; Nagashima, T.; Hirata, R.; Toida, T.; Imanari, T. Sensitive high-performance liquid chromatographic method with fluorometric detection for the determination of heparin and heparan sulfate in biological samples: Application to human urinary heparan sulfate. J. Chromatogr. B Biomed. Sci. Appl. 1997, 704, 19–24. [Google Scholar]
- Kitagawa, H.; Kinoshita, A.; Sugahara, K. Microanalysis of glycosaminoglycan-derived disaccharides labeled with the fluorophore 2-aminoacridone by capillary electrophoresis and high-performance liquid chromatography. Anal. Biochem. 1995, 232, 114–121. [Google Scholar] [CrossRef]
- Militsopoulou, M.; Lamari, F.N.; Hjerpe, A.; Karamanos, N.K. Determination of twelve heparin- and heparan sulfate-derived disaccharides as 2-aminoacridone derivatives by capillary zone electrophoresis using ultraviolet and laser-induced fluorescence detection. Electrophoresis 2002, 23, 1104–1109. [Google Scholar] [CrossRef]
- Skidmore, M.A.; Guimond, S.E.; Dumax-Vorzet, A.F.; Atrih, A.; Yates, E.A.; Turnbull, J.E. High sensitivity separation and detection of heparan sulfate disaccharides. J. Chromatogr. A 2006, 1135, 52–56. [Google Scholar]
- Skidmore, M.A.; Guimond, S.E.; Dumax-Vorzet, A.F.; Yates, E.A.; Turnbull, J.E. Disaccharide compositional analysis of heparan sulfate and heparin polysaccharides using uv or high-sensitivity fluorescence (bodipy) detection. Nat. Protoc. 2010, 5, 1983–1992. [Google Scholar] [CrossRef]
- Guimond, S.E.; Puvirajesinghe, T.M.; Skidmore, M.A.; Kalus, I.; Dierks, T.; Yates, E.A.; Turnbull, J.E. Rapid purification and high sensitivity analysis of heparan sulfate from cells and tissues: Toward glycomics profiling. J. Biol. Chem. 2009, 284, 25714–25722. [Google Scholar]
- Dionex Propac pa1 column for hydrophilic anionic protein separation. Available online: http://www.dionex.com/en-us/columns-accessories/biocols/cons4783.html accessed on 27 November 2012.
- Vives, R.R.; Goodger, S.; Pye, D.A. Combined strong anion-exchange hplc and page approach for the purification of heparan sulphate oligosaccharides. Biochem. J. 2001, 354, 141–147. [Google Scholar] [CrossRef]
- Yang, B.; Solakyildirim, K.; Chang, Y.; Linhardt, R.J. Hyphenated techniques for the analysis of heparin and heparan sulfate. Anal. Bioanal. Chem. 2010, 399, 541–557. [Google Scholar]
- Hitchcock, A.M.; Yates, K.E.; Costello, C.E.; Zaia, J. Comparative glycomics of connective tissue glycosaminoglycans. Proteomics 2008, 8, 1384–1397. [Google Scholar] [CrossRef]
- Staples, G.O.; Bowman, M.J.; Costello, C.E.; Hitchcock, A.M.; Lau, J.M.; Leymarie, N.; Miller, C.; Naimy, H.; Shi, X.; Zaia, J. A chip-based amide-hilic lc/ms platform for glycosaminoglycan glycomics profiling. Proteomics 2009, 9, 686–695. [Google Scholar]
- McDevitt, C.A.; Muir, H. Gel electrophoresis of proteoglycans and glycosaminoglycans on large-pore composite polyacrylamide-agarose gels. Anal. Biochem. 1971, 44, 612–622. [Google Scholar]
- Cowman, M.K.; Slahetka, M.F.; Hittner, D.M.; Kim, J.; Forino, M.; Gadelrab, G. Polyacrylamide-gel electrophoresis and alcian blue staining of sulphated glycosaminoglycan oligosaccharides. Biochem. J. 1984, 221, 707–716. [Google Scholar]
- Dodge, G.R.; Heimer, R. Proteoglycans analyzed by composite gel electrophoresis and imunoblotting. Methods Mol. Biol. 2001, 171, 149–158. [Google Scholar]
- Karamanos, N.K.; Vanky, P.; Tzanakakis, G.N.; Hjerpe, A. High performance capillary electrophoresis method to characterize heparin and heparan sulfate disaccharides. Electrophoresis 1996, 17, 391–395. [Google Scholar]
- Ampofo, S.A.; Wang, H.M.; Linhardt, R.J. Disaccharide compositional analysis of heparin and heparan sulfate using capillary zone electrophoresis. Anal. Biochem. 1991, 199, 249–255. [Google Scholar]
- Merry, C.L.; Lyon, M.; Deakin, J.A.; Hopwood, J.J.; Gallagher, J.T. Highly sensitive sequencing of the sulfated domains of heparan sulfate. J. Biol. Chem. 1999, 274, 18455–18462. [Google Scholar]
- Vives, R.R.; Pye, D.A.; Salmivirta, M.; Hopwood, J.J.; Lindahl, U.; Gallagher, J.T. Sequence analysis of heparan sulphate and heparin oligosaccharides. Biochem. J. 1999, 339, 767–773. [Google Scholar] [CrossRef]
- Turnbull, J.E. Integral glycan sequencing of heparan sulphate and heparin saccharides. In Methods in Molecular Biology: Proteoglycan Protocols; Iozzo, R.V., Ed.; Humana Press: Totowa, NJ, USA, 2001; Volume 171. [Google Scholar]
- Venkataraman, G.; Shriver, Z.; Raman, R.; Sasisekharan, R. Sequencing complex polysaccharides. Science 1999, 286, 537–542. [Google Scholar] [CrossRef]
- Shriver, Z.; Raman, R.; Venkataraman, G.; Drummond, K.; Turnbull, J.; Toida, T.; Linhardt, R.; Biemann, K.; Sasisekharan, R. Sequencing of 3-o sulfate containing heparin decasaccharides with a partial antithrombin iii binding site. Proc. Nat. Acad. Sci. USA 2000, 97, 10359–10364. [Google Scholar]
- Shriver, Z.; Sundaram, M.; Venkataraman, G.; Fareed, J.; Linhardt, R.; Biemann, K.; Sasisekharan, R. Cleavage of the antithrombin iii binding site in heparin by heparinases and its implication in the generation of low molecular weight heparin. Proc. Natl. Acad. Sci.USA 2000, 97, 10365–10370. [Google Scholar]
- Lindhardt, R.J.; Hales, C.A. Chemistry and Biology of Heparin and Heparan Sulfate. Elsevier: Oxford, UK, 2005. [Google Scholar]
- Thanawiroon, C.; Rice, K.G.; Toida, T.; Linhardt, R.J. Liquid chromatography/mass spectrometry sequencing approach for highly sulfated heparin-derived oligosaccharides. J. Biol. Chem. 2004, 279, 2608–2615. [Google Scholar]
- Zaia, J. Mass spectrometry and glycomics. OMICS 2010, 14, 401–418. [Google Scholar] [CrossRef]
- Saad, O.M.; Leary, J.A. Delineating mechanisms of dissociation for isomeric heparin disaccharides using isotope labeling and ion trap tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2004, 15, 1274–1286. [Google Scholar] [CrossRef]
- Skidmore, M.; Atrih, A.; Yates, E.; Turnbull, J.E. Labelling heparan sulphate saccharides with chromophore, fluorescence and mass tags for hplc and ms separations. Methods Mol. Biol. 2009, 534, 157–169. [Google Scholar] [CrossRef]
- Hitchcock, A.M; Costello, C.E., Zaia. Glycoform quantification of chondroitin/dermatan sulfate using an LC/MS/MS platform. Biochemistry 2006, 45, 2350–2361. [Google Scholar]
- Henriksen, J.; Roepstorff, P.; Ringborg, L.H. Ion-pairing reversed-phased chromatography/mass spectrometry of heparin. Carbohydr. Res. 2006, 341, 382–387. [Google Scholar] [CrossRef]
- Linhardt, R.J.; Gu, K.N.; Loganathan, D.; Carter, S.R. Analysis of glycosaminoglycan-derived oligosaccharides using reversed-phase ion-pairing and ion-exchange chromatography with suppressed conductivity detection. Anal. Biochem. 1989, 181, 288–296. [Google Scholar]
- Turnbull, J.E.; Miller, R.L.; Ahmed, Y.; Puvirajesinghe, T.M.; Guimond, S.E. Glycomics profiling of heparan sulfate structure and activity. Methods Enzymol. 2010, 480, 65–85. [Google Scholar]
- Pope, R.M.; Raska, C.S.; Thorp, S.C.; Liu, J. Analysis of heparan sulfate oligosaccharides by nano-electrospray ionization mass spectrometry. Glycobiology 2001, 11, 505–513. [Google Scholar] [CrossRef]
- Zaia, J. Principles of mass spectrometry of glycosaminoglycans. J. Biomacromol Mass Spectrom. 2005, 1, 3–36. [Google Scholar]
- Kealey, D.; Haines, P.J. Analytical Chemistry; BIOS Scientific Publishers: Leeds, UK, 2002; p. 342. [Google Scholar]
- Yates, E.A.; Guimond, S.E.; Turnbull, J.E. Highly diverse heparan sulfate analogue libraries: Providing access to expanded areas of sequence space for bioactivity screening. J. Med. Chem. 2004, 47, 277–280. [Google Scholar] [CrossRef]
- Guimond, S.E.; Turnbull, J.E. Fibroblast growth factor receptor signalling is dictated by specific heparan sulphate saccharides. Curr. Biol. 1999, 9, 1343–1346. [Google Scholar]
- Rudd, T.R.; Guimond, S.E.; Skidmore, M.A.; Duchesne, L.; Guerrini, M.; Torri, G.; Cosentino, C.; Brown, A.; Clarke, D.T.; Turnbull, J.E.; et al. Influence of substitution pattern and cation binding on conformation and activity in heparin derivatives. Glycobiology 2007, 17, 983–993. [Google Scholar] [CrossRef]
- Tissot, B.; Ceroni, A.; Powell, A.K.; Morris, H.R.; Yates, E.A.; Turnbull, J.E.; Gallagher, J.T.; Dell, A.; Haslam, S.M. Software tool for the structural determination of glycosaminoglycans by mass spectrometry. Anal. Chem. 2008, 80, 9204–9212. [Google Scholar]
- Maxwell, E.; Tan, Y.; Hu, H.; Benson, G.; Aizikov, K.; Conley, S.; Staples, G.O.; Slysz, G.W.; Smith, R.D.; Zaia, J. Glycresoft: A software package for automated recognition of glycans from lc/ms data. PLoS One 2012, 7. [Google Scholar] [CrossRef]
- Raman, R.; Raguram, S.; Venkataraman, G.; Paulson, J.C.; Sasisekharan, R. Glycomics: An integrated systems approach to structure-function relationships of glycans. Nat. Methods 2005, 2, 817–824. [Google Scholar] [CrossRef]
- Tissot, B.; Ceroni, A.; Powell, A.K.; Morris, H.R.; Yates, E.A.; Turnbull, J.E.; Gallagher, J.T.; Dell, A.; Haslam, S.M. Software tool for the structural determination of glycosaminoglycans by mass spectrometry. Anal. Chem. 2008, 80, 9204–9212. [Google Scholar]
- Sasisekharan, R.; Raman, R.; Prabhakar, V. Glycomics approach to structure-function relationships of glycosaminoglycans. Annu. Rev. Biomed. Eng. 2006, 8, 181–231. [Google Scholar] [CrossRef]
- Puvirajesinghe, T.M.; Guimond, S.E.; Turnbull, J.E.; Guenneau, S. Chemometric analysis for comparison of heparan sulphate oligosaccharides. J. R. Soc. Interface 2009, 6, 997–1004. [Google Scholar]
- Grootenhuis, P.D.J.; Westerduin, P.; Meuleman, D.; Petitou, M.; van Boeckel, C.A.A. Rational design of synthetic heparin analogues with tailor-made coagulation factor inhibitory activity. Nat. Struct Mol. Biol. 1995, 2, 736–739. [Google Scholar]
- Seeberger, P.H.; Haase, W.C. Solid-phase oligosaccharide synthesis and combinatorial carbohydrate libraries. Chem. Rev. 2000, 100, 4349–4394. [Google Scholar] [CrossRef]
- Seeberger, P.H.; Werz, D.B. Automated synthesis of oligosaccharides as a basis for drug discovery. Nat. Rev. Drug Discov. 2005, 4, 751–763. [Google Scholar] [CrossRef]
- Vohra, Y.; Vasan, M.; Venot, A.; Boons, G.J. One-pot synthesis of oligosaccharides by combining reductive openings of benzylidene acetals and glycosylations. Org. Lett. 2008, 10, 3247–3250. [Google Scholar]
- Yates, E.A.; Terry, C.J.; Rees, C.; Rudd, T.R.; Duchesne, L.; Skidmore, M.A.; Levy, R.; Thanh, N.T.; Nichols, R.J.; Clarke, D.T.; et al. Protein-gag interactions: New surface-based techniques, spectroscopies and nanotechnology probes. Biochem. Soc. Trans. 2006, 34, 427–430. [Google Scholar] [CrossRef]
- Keller, C.A.; Kasemo, B. Surface specific kinetics of lipid vesicle adsorption measured with a quartz crystal microbalance. Biophys. J. 1998, 75, 1397–1402. [Google Scholar] [CrossRef]
- Rudd, T.R.; Skidmore, M.A.; Guimond, S.E.; Holman, J.; Turnbull, J.E.; Lauder, R.M.; Fernig, D.G.; Yates, E.A. The potential for circular dichroism as an additional facile and sensitive method of monitoring low-molecular-weight heparins and heparinoids. Thromb. Haemost. 2009, 102, 874–878. [Google Scholar]
- Powell, A.K.; Zhi, Z.L.; Turnbull, J.E. Saccharide microarrays for high-throughput interrogation of glycan-protein binding interactions. Methods Mol. Biol. 2009, 534, 313–329. [Google Scholar]
- Zhi, Z.; Powell, A.K.; Turnbull, J.E. Fabrication of carbohydrate microarrays on gold surface: Direct attachment of nonderivatized oligosaccharides to hydrazide-modified self- assembled monolayers. Anal. Chem. 2006, 78, 4786–4793. [Google Scholar]
- Powell, A.K.; Yates, E.A.; Fernig, D.G.; Turnbull, J.E. Interactions of heparin/heparan sulfate with proteins: appraisal of structural factors and experimental approaches. Glycobiology 2004, 14, R17–R30. [Google Scholar] [CrossRef]
- Krufka, A.; Guimond, S.; Rapraeger, A.C. Two hierarchies of fgf-2 signaling in heparin: Mitogenic stimulation and high-affinity binding/receptor transphosphorylation. Biochemistry 1996, 35, 11131–11141. [Google Scholar]
- Rapraeger, A.C.; Krufka, A.; Olwin, B.B. Requirement of heparan sulfate for bfgf-mediated fibroblast growth and myoblast differentiation. Science 1991, 252, 1705–1708. [Google Scholar]
- Ornitz, D.M.; Yayon, A.; Flanagan, J.G.; Svahn, C.M.; Levi, E.; Leder, P. Heparin is required for cell-free binding of basic fibroblast growth factor to a soluble receptor and for mitogenesis in whole cells. Mol. Cell. Biol. 1992, 12, 240–247. [Google Scholar]
- Guimond, S.; Maccarana, M.; Olwin, B.B.; Lindahl, U.; Rapraeger, A.C. Activating and inhibitory heparin sequences for fgf-2 (basic fgf). Distinct requirements for fgf-1, fgf-2, and fgf-4. J. Biol. Chem. 1993, 268, 23906–23914. [Google Scholar]
- Pye, D.A.; Vives, R.R.; Turnbull, J.E.; Hyde, P.; Gallagher, J.T. Heparan sulfate oligosaccharides require 6-o-sulfation for promotion of basic fibroblast growth factor mitogenic activity. J. Biol. Chem. 1998, 273, 22936–22942. [Google Scholar]
- Spivak-Kroizman, T.; Lemmon, M.A.; Dikic, I.; Ladbury, J.E.; Pinchasi, D.; Huang, J.; Jaye, M.; Crumley, G.; Schlessinger, J.; Lax, I. Heparin-induced oligomerization of fgf molecules is responsible for fgf receptor dimerization, activation, and cell proliferation. Cell 1994, 79, 1015–1024. [Google Scholar] [CrossRef]
- Rapraeger, A.C.; Krufka, A.; Olwin, B.B. Requirement of heparan sulfate for bfgf-mediated fibroblast growth and myoblast differentiation. Science 1991, 252, 1705–1708. [Google Scholar]
- Ornitz, D.M.; Xu, J.; Colvin, J.S.; McEwen, D.G.; MacArthur, C.A.; Coulier, F.; Gao, G.; Goldfarb, M. Receptor specificity of the fibroblast growth factor family. J. Biol. Chem. 1996, 271, 15292–15297. [Google Scholar]
- Lindhardt, R.J.; Hales, C.A. Chemistry and Biology of Heparin and Heparan Sulfate; Elsevier: San Diego, CA, USA, 2005. [Google Scholar]
- Smorenburg, S.M.; Van Noorden, C.J. The complex effects of heparins on cancer progression and metastasis in experimental studies. Pharmacol. Rev. 2001, 53, 93–105. [Google Scholar]
- Iozzo, R.V.; San Antonio, J.D. Heparan sulfate proteoglycans: Heavy hitters in the angiogenesis arena. J. Clin. Invest. 2001, 108, 349–355. [Google Scholar]
- Lundin, L.; Larsson, H.; Kreuger, J.; Kanda, S.; Lindahl, U.; Salmivirta, M.; Claesson-Welsh, L. Selectively desulfated heparin inhibits fibroblast growth factor-induced mitogenicity and angiogenesis. J. Biol. Chem. 2000, 275, 24653–24660. [Google Scholar]
- Guimond, S.E.; Turnbull, J.E.; Yates, E.A. Engineered bio-active polysaccharides from heparin. Macromol. Biosci. 2006, 6, 681–686. [Google Scholar]
- Lin, X. Functions of heparan sulfate proteoglycans in cell signaling during development. Development 2004, 131, 6009–6021. [Google Scholar] [CrossRef]
- Kapur, R. Whole cell microarrays. In Microarray Technology and Its Applications; Springer-Verlag: Berlin/Heidelberg, Germany, 2005. [Google Scholar]
- Schena, M.; Heller, R.A.; Theriault, T.P.; Konrad, K.; Lachenmeier, E.; Davis, R.W. Microarrays: biotechnology's discovery platform for functional genomics. Trends Biotechnol. 1998. [Google Scholar] [CrossRef]
- Ziauddin, J.; Sabatini, D.M. Microarrays of cells expressing defined cdnas. Nature 2001, 411, 107–110. [Google Scholar] [CrossRef]
- Erfle, H.; Neumann, B.; Liebel, U.; Rogers, P.; Held, M.; Walter, T.; Ellenberg, J.; Pepperkok, R. Reverse transfection on cell arrays for high content screening microscopy. Nat. Protoc. 2007, 2, 392–399. [Google Scholar]
- Wheeler, D.B.; Bailey, S.N.; Guertin, D.A.; Carpenter, A.E.; Higgins, C.O.; Sabatini, D.M. Rnai living-cell microarrays for loss-of-function screens in drosophila melanogaster cells. Nat. Methods 2004, 1, 127–132. [Google Scholar]
- Bailey, S.N.; Sabatini, D.M.; Stockwell, B.R. Microarrays of small molecules embedded in biodegradable polymers for use in mammalian cell-based screens. Proc. Natl. Acad. Sci. USA 2004, 101, 16144–16149. [Google Scholar]
- Palmer, E.L.; Miller, A.D.; Freeman, T.C. Identification and characterisation of human apoptosis inducing proteins using cell-based transfection microarrays and expression analysis. BMC Genomics 2006, 7, 145–162. [Google Scholar] [CrossRef]
- Kononen, J.; Bubendorf, L.; Kallioniemi, A.; Barlund, M.; Schraml, P.; Leighton, S.; Torhorst, J.; Mihatsch, M.J.; Sauter, G.; Kallioniemi, O.P. Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat. Med. 1998, 4, 844–847. [Google Scholar] [CrossRef]
- Ratner, D.M. Advancing the burgeoning field of glycomics. BTi 2005, 17, 8–11. [Google Scholar]
- Powell, A.K.; Zhi, Z.; Turnbull, J.E. Heparan sulphate saccharide microarrays for high-throughput interrogation of protein binding interactions. Methods Mol. Biol. 2009, 534, 313–329. [Google Scholar]
- Yates, E.A.; Jones, M.O.; Clarke, C.E.; Powell, A.K.; Johnson, S.R.; Porch, A.; Edwards, P.E; Turnbull, J.E. Microwave enhanced reaction of carbohydrates with aminoderivatised labels and glass surfaces. J. Mater. Chem. 2003, 13, 2061–2063. [Google Scholar]
- Houseman, B.T.; Mrksich, M. Carbohydrate arrays for the evaluation of protein binding and enzymatic modification. Chem. Biol. 2002, 9, 443–454. [Google Scholar] [CrossRef]
- Shin, I.; Park, S.; Lee, M.R. Carbohydrate microarrays: An advanced technology for functional studies of glycans. Chemistry 2005, 11, 2894–2901. [Google Scholar] [CrossRef]
- Feyzi, E.; Saldeen, T.; Larsson, E.; Lindahl, U.; Salmivirta, M. Age-dependent modulation of heparan sulfate structure and function. J. Biol. Chem. 1998, 273, 13395–13398. [Google Scholar]
- Fukui, S.; Feizi, T.; Galustian, C.; Lawson, A.M.; Chai, W. Oligosaccharide microarrays for high-throughput detection and specificity assignments of carbohydrate-protein interactions. Nat. Biotechnol. 2002, 20, 1011–1017. [Google Scholar] [CrossRef]
- Willats, W.G.; Rasmussen, S.E.; Kristensen, T.; Mikkelsen, J.D.; Knox, J.P. Sugar-coated microarrays: A novel slide surface for the high-throughput analysis of glycans. Proteomics 2002, 2, 1666–1671. [Google Scholar] [CrossRef]
- Erfle, H.; Neumann, B.; Liebel, U.; Rogers, P.; Held, M.; Walter, T.; Ellenberg, J.; Pepperkok, R. Reverse transfection on cell arrays for high content screening microscopy. Nat. Protoc. 2007, 2, 392–399. [Google Scholar]
- Wheeler, D.B.; Carpenter, A.E.; Sabatini, D.M. Cell microarrays and rna interference chip away at gene function. Nat. Genet. 2005, 37, S25–S30. [Google Scholar]
- Sardzik, R.; Sharma, R.; Kaloo, S.; Voglmeir, J.; Crocker, P.R.; Flitsch, S.L. Chemoenzymatic synthesis of sialooligosaccharides on arrays for studies of cell surface adhesion. Chem. Commun. 2011, 47, 5425–5427. [Google Scholar]
- Dickinson, L.E.; Gerecht, S. Micropatterned surfaces to study hyaluronic acid interactions with cancer cells. J. Vis. Exp. 2011, 46, 2413. [Google Scholar]
- Dickinson, L.E.; Ho, C.C.; Wang, G.M.; Stebe, K.J.; Gerecht, S. Functional surfaces for high-resolution analysis of cancer cell interactions on exogenous hyaluronic acid. Biomaterials 2010, 31, 5472–5478. [Google Scholar]
- Nimrichter, L.; Gargir, A.; Gortler, M.; Altstock, R.T.; Shtevi, A.; Weisshaus, O.; Fire, E.; Dotan, N.; Schnaar, R.L. Intact cell adhesion to glycan microarrays. Glycobiology 2004, 14, 197–203. [Google Scholar]
- Arndt, N.X.; Tiralongo, J.; Madge, P.D.; von Itzstein, M.; Day, C.J. Differential carbohydrate binding and cell surface glycosylation of human cancer cell lines. J. Cell Biochem. 2011, 112, 2230–2240. [Google Scholar]
- Puvirajesinghe, T.M.; Ahmed, Y.A.; Powell, A.K.; Fernig, D.G.; Guimond, S.E.; Turnbull, J.E. Array-based functional screening of heparin glycans. Chem. Biol. 2012, 19, 553–558. [Google Scholar] [CrossRef]
- Powell, A.K.; Ahmed, Y.A.; Yates, E.A.; Turnbull, J.E. Generating heparan sulfate saccharide libraries for glycomics applications. Nat. Protoc. 2010, 5, 821–833. [Google Scholar]
- Noti, C.; de Paz, J.L.; Polito, L.; Seeberger, P.H. Preparation and use of microarrays containing synthetic heparin oligosaccharides for the rapid analysis of heparin-protein interactions. Chemistry 2006, 12, 8664–8686. [Google Scholar] [CrossRef]
- de Paz, J.L.; Moseman, E.A.; Noti, C.; Polito, L.; von Andrian, U.H.; Seeberger, P.H. Profiling heparin-chemokine interactions using synthetic tools. ACS Chem. Biol. 2007, 2, 735–744. [Google Scholar] [CrossRef]
- Arungundram, S.; Al-Mafraji, K.; Asong, J.; Leach, F.E., 3rd; Amster, I.J.; Venot, A.; Turnbull, J.E.; Boons, G.J. Modular synthesis of heparan sulfate oligosaccharides for structure-activity relationship studies. J. Am. Chem. Soc. 2009, 131, 17394–17405. [Google Scholar]
- Zhi, Z.L.; Powell, A.K.; Turnbull, J.E. Fabrication of carbohydrate microarrays on gold surfaces: Direct attachment of nonderivatized oligosaccharides to hydrazide-modified self-assembled monolayers. Anal. Chem. 2006, 78, 4786–4793. [Google Scholar]
- Zhi, Z.L.; Laurent, N.; Powell, A.K.; Karamanska, R.; Fais, M.; Voglmeir, J.; Wright, A.; Blackburn, J.M.; Crocker, P.R.; Russell, D.A.; et al. A versatile gold surface approach for fabrication and interrogation of glycoarrays. Chembiochem 2008, 9, 1568–1575. [Google Scholar]
- Turnbull, J.E. Heparan sulfate glycomics: Towards systems biology strategies. Biochem. Soc. Trans. 2010, 38, 1356–1360. [Google Scholar] [CrossRef]
- Patey, S.J.; Edwards, E.A.; Yates, E.A.; Turnbull, J.E. Heparin derivatives as inhibitors of bace-1, the alzheimer's beta-secretase, with reduced activity against factor xa and other protease. J. Med. Chem. 2006, 49, 6129–6132. [Google Scholar]
- Higginson, J.; Thompson, S.M.; Santos-Silva, A.; Guimond, S.E.; Turnbull, J.E.; Barnett, S.C. Differential sulfation remodelling of heparan sulfate by extracellular 6-o-sulfatases regulates fibroblast growth factor-induced boundary formation by glial cells: Implications for glial cell transplantation. J. Neurosci. 2012, 32, 15902–15912. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Puvirajesinghe, T.M.; Turnbull, J.E. Glycomics Approaches for the Bioassay and Structural Analysis of Heparin/Heparan Sulphates. Metabolites 2012, 2, 1060-1089. https://doi.org/10.3390/metabo2041060
Puvirajesinghe TM, Turnbull JE. Glycomics Approaches for the Bioassay and Structural Analysis of Heparin/Heparan Sulphates. Metabolites. 2012; 2(4):1060-1089. https://doi.org/10.3390/metabo2041060
Chicago/Turabian StylePuvirajesinghe, Tania M., and Jeremy E. Turnbull. 2012. "Glycomics Approaches for the Bioassay and Structural Analysis of Heparin/Heparan Sulphates" Metabolites 2, no. 4: 1060-1089. https://doi.org/10.3390/metabo2041060
APA StylePuvirajesinghe, T. M., & Turnbull, J. E. (2012). Glycomics Approaches for the Bioassay and Structural Analysis of Heparin/Heparan Sulphates. Metabolites, 2(4), 1060-1089. https://doi.org/10.3390/metabo2041060