
Analysis of Mammalian Cell Proliferation and Macromolecule Synthesis Using Deuterated Water and Gas Chromatography-Mass Spectrometry

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods
2.1. Cell Culture
2.2. 2H2O Labelling
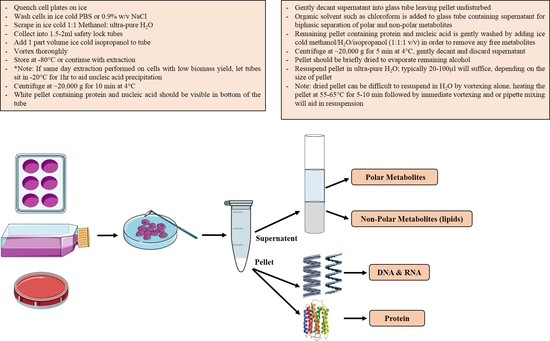
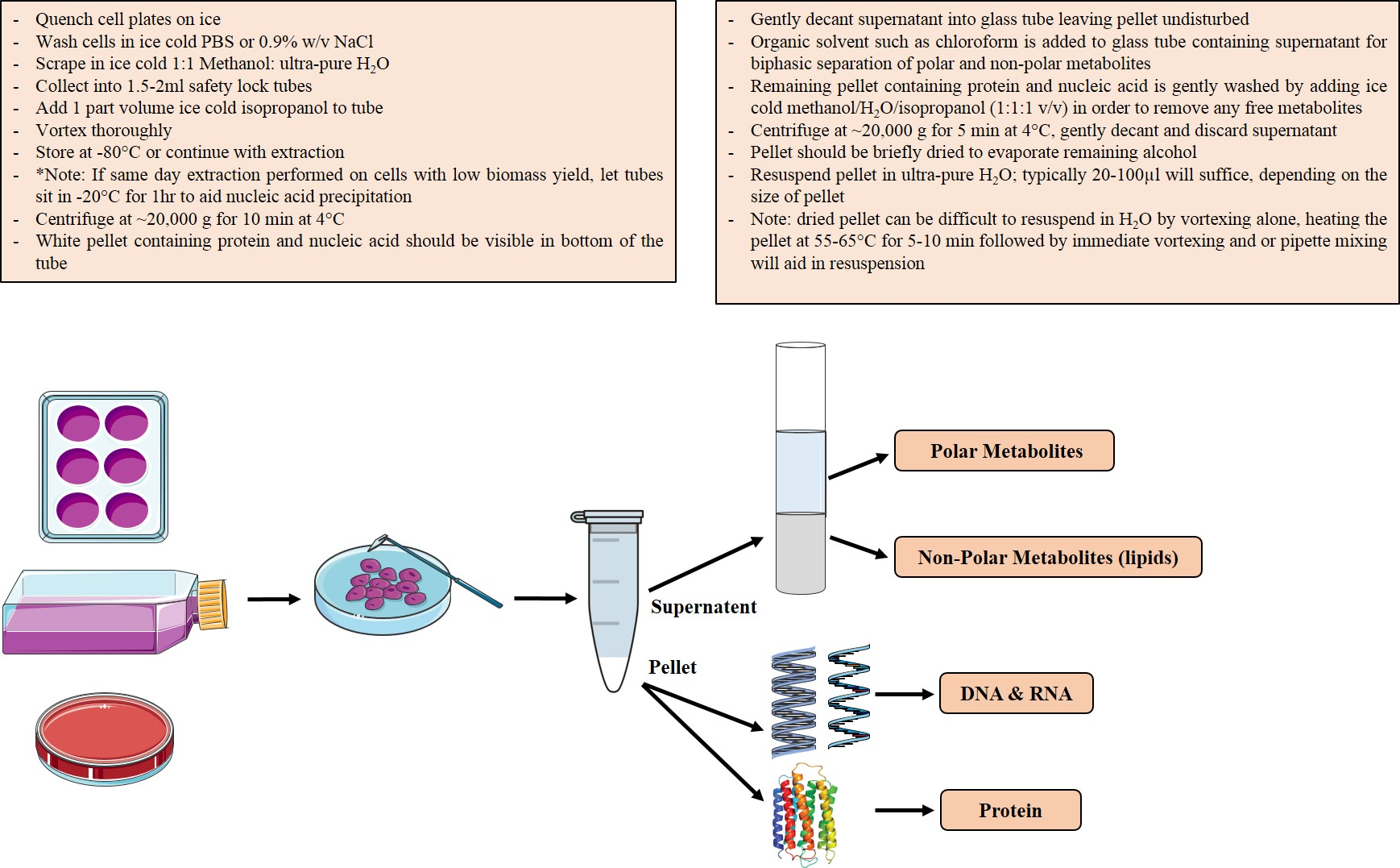
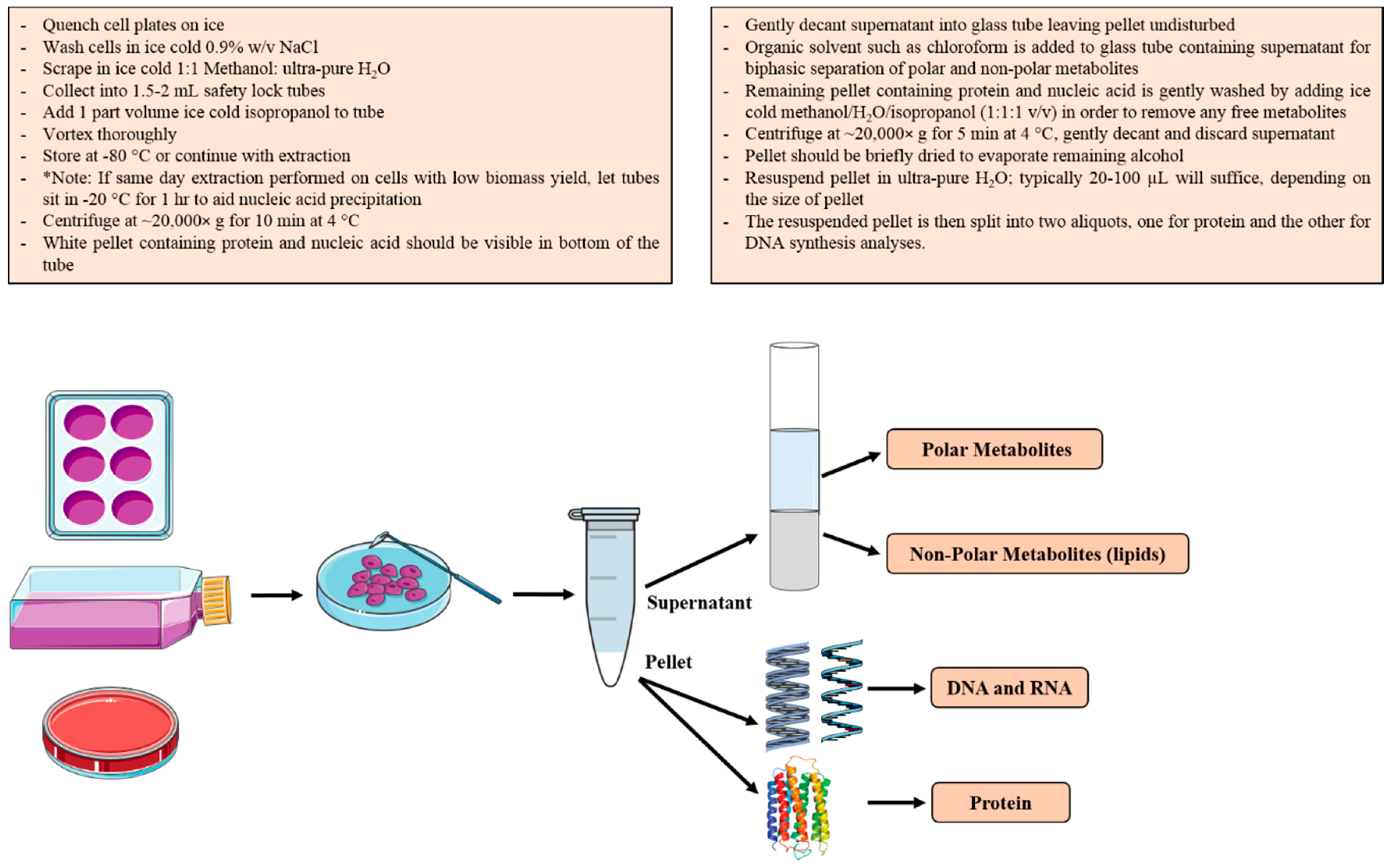
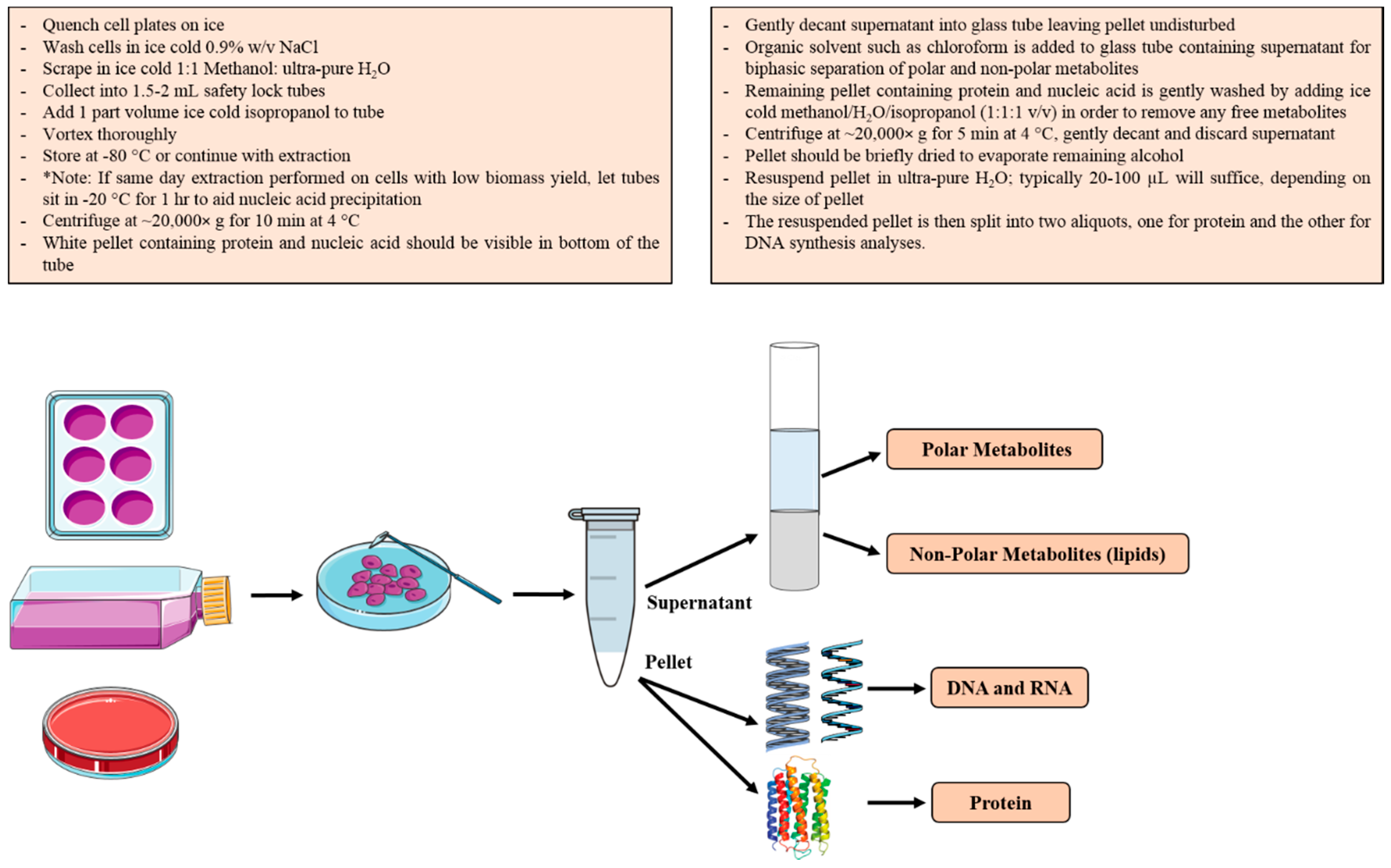
2.3. Biomass Fractionation
2.4. Analysis of Deuterium Incorporation into Protein
2.5. Analysis of Deuterium Incorporation into DNA
2.6. Analysis of Deuterium Incorporation into Lipids
2.7. GC-MS
2.8. Calculations
3. Results and Discussion
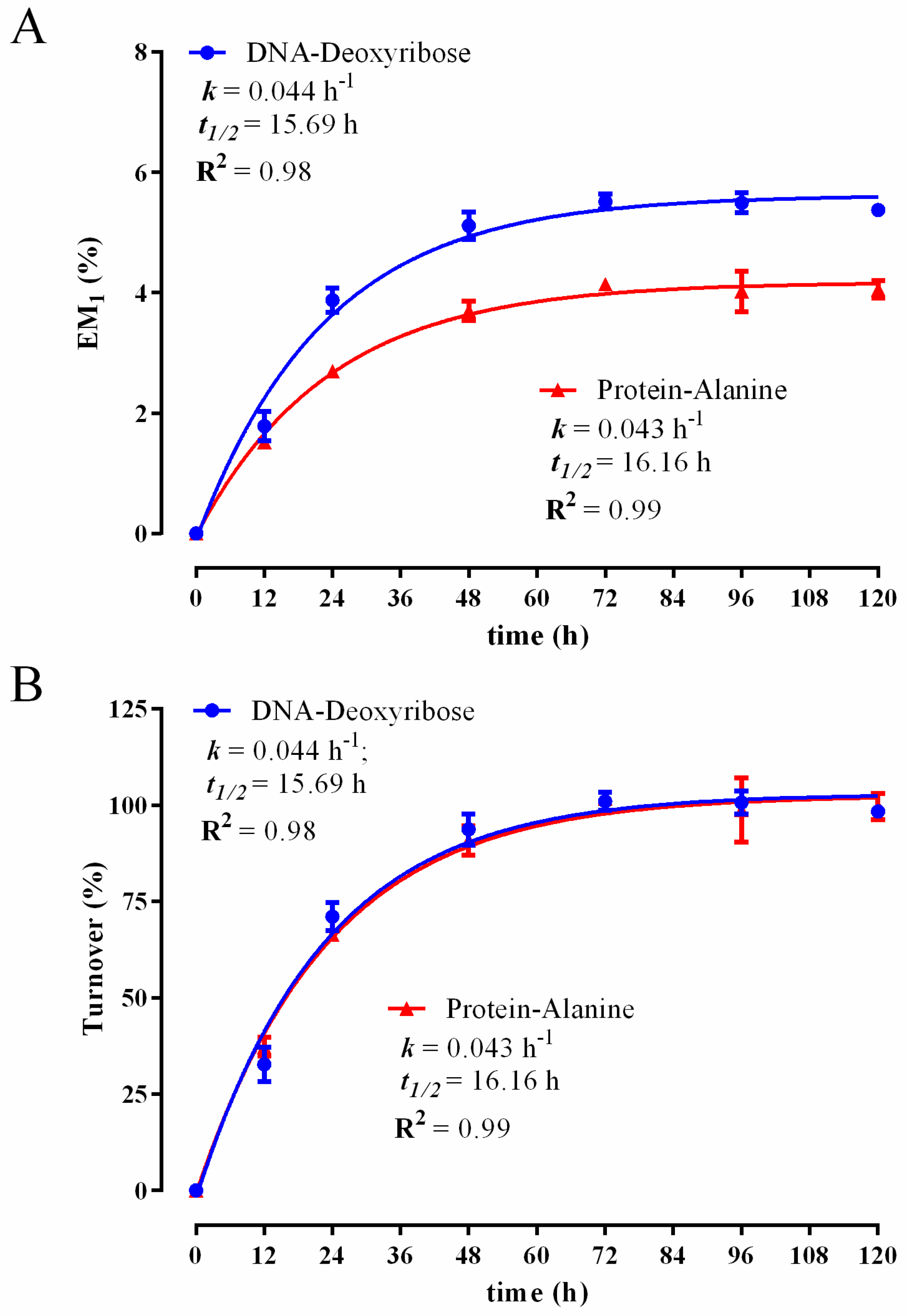
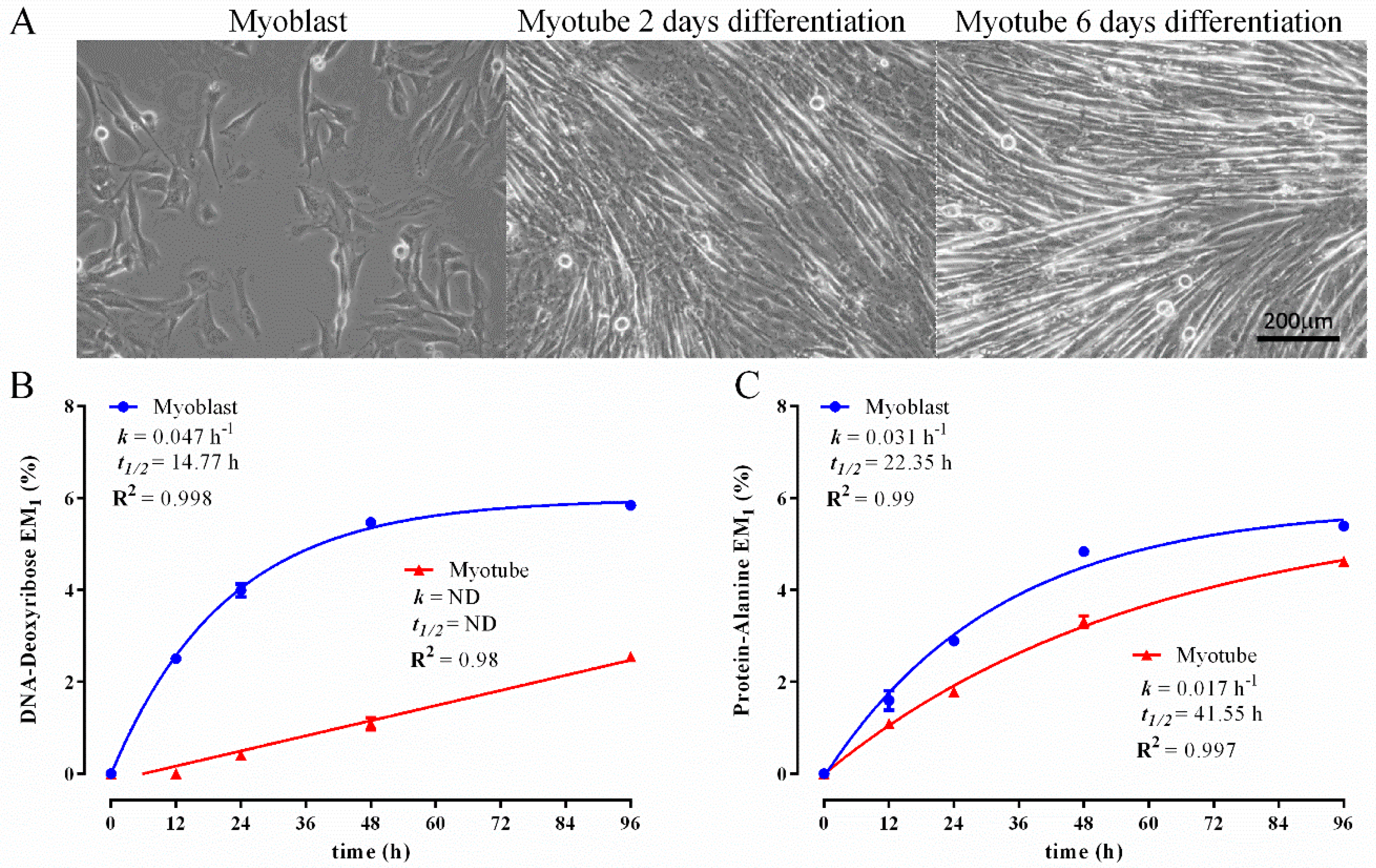
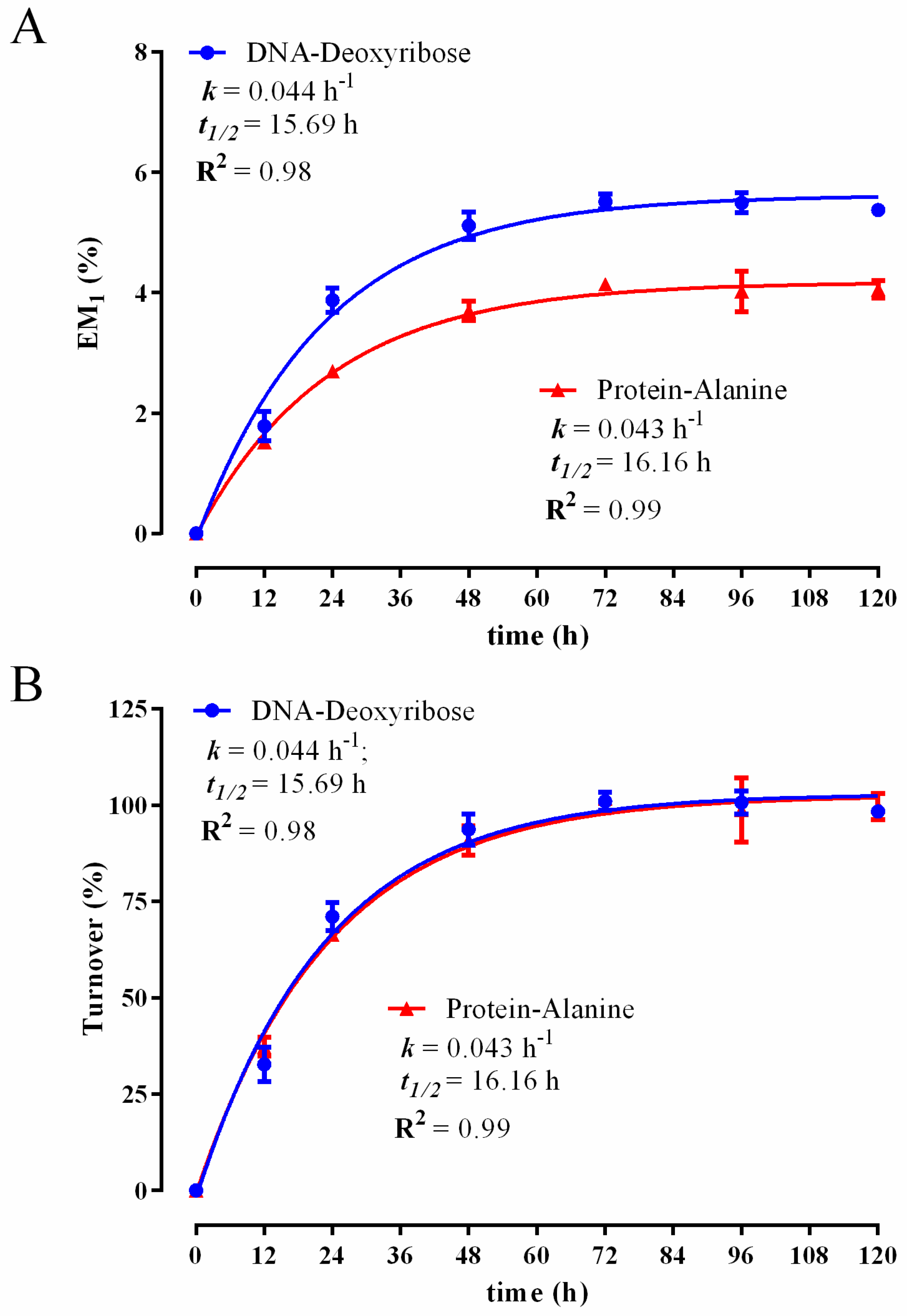
3.1. Establishing a DNA and Protein 2H Labelling Time Course in C2C12 Myoblasts
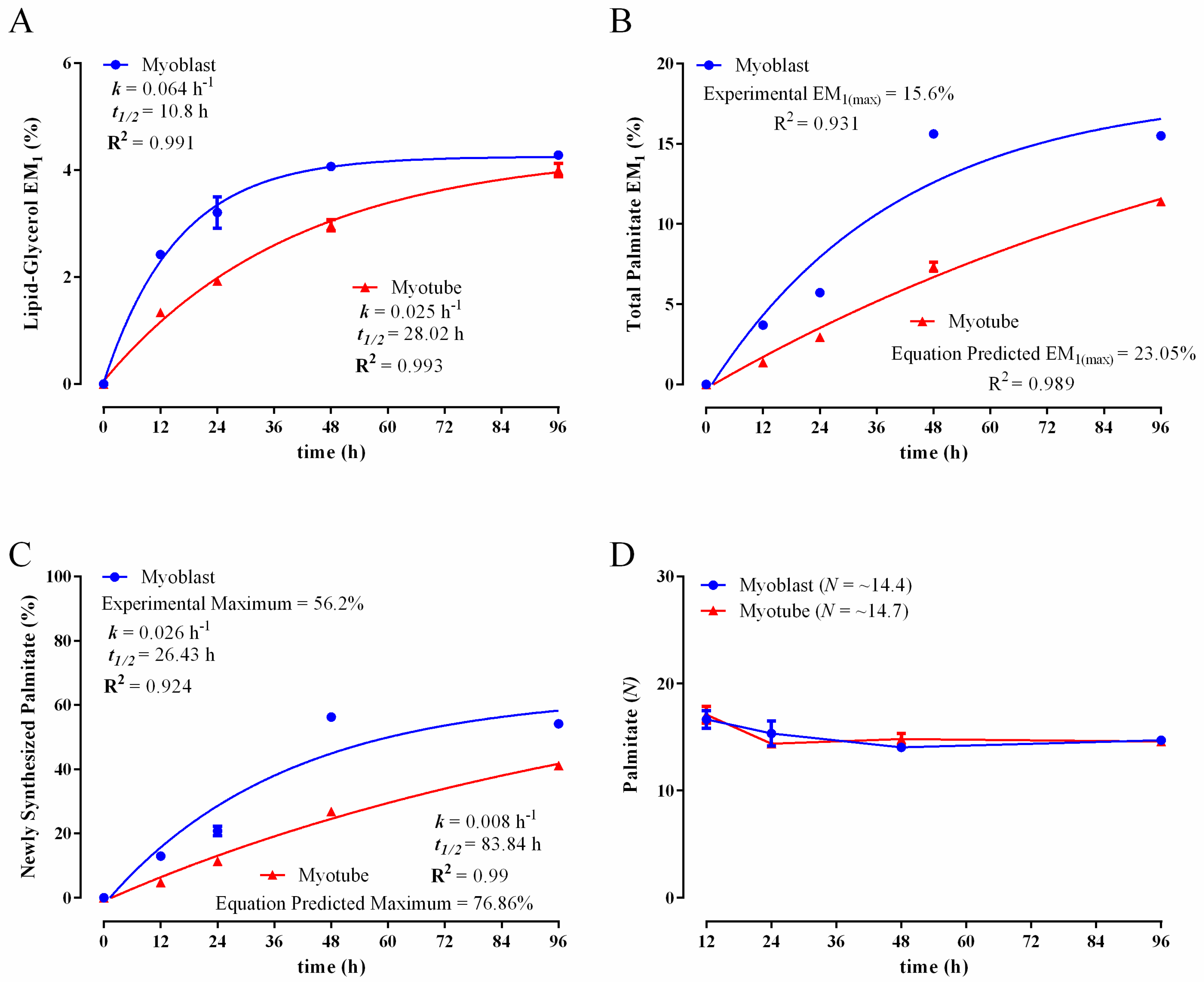
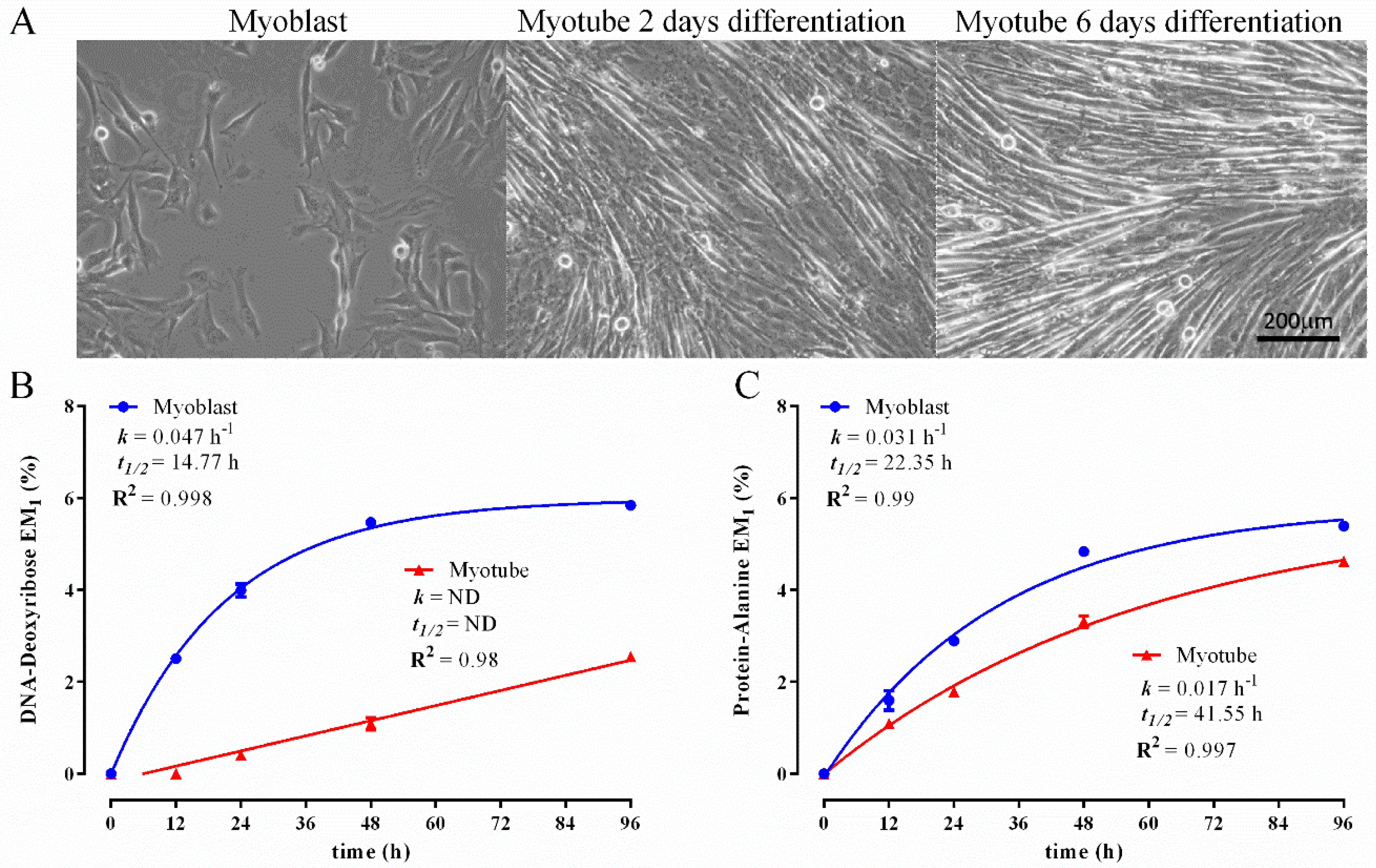
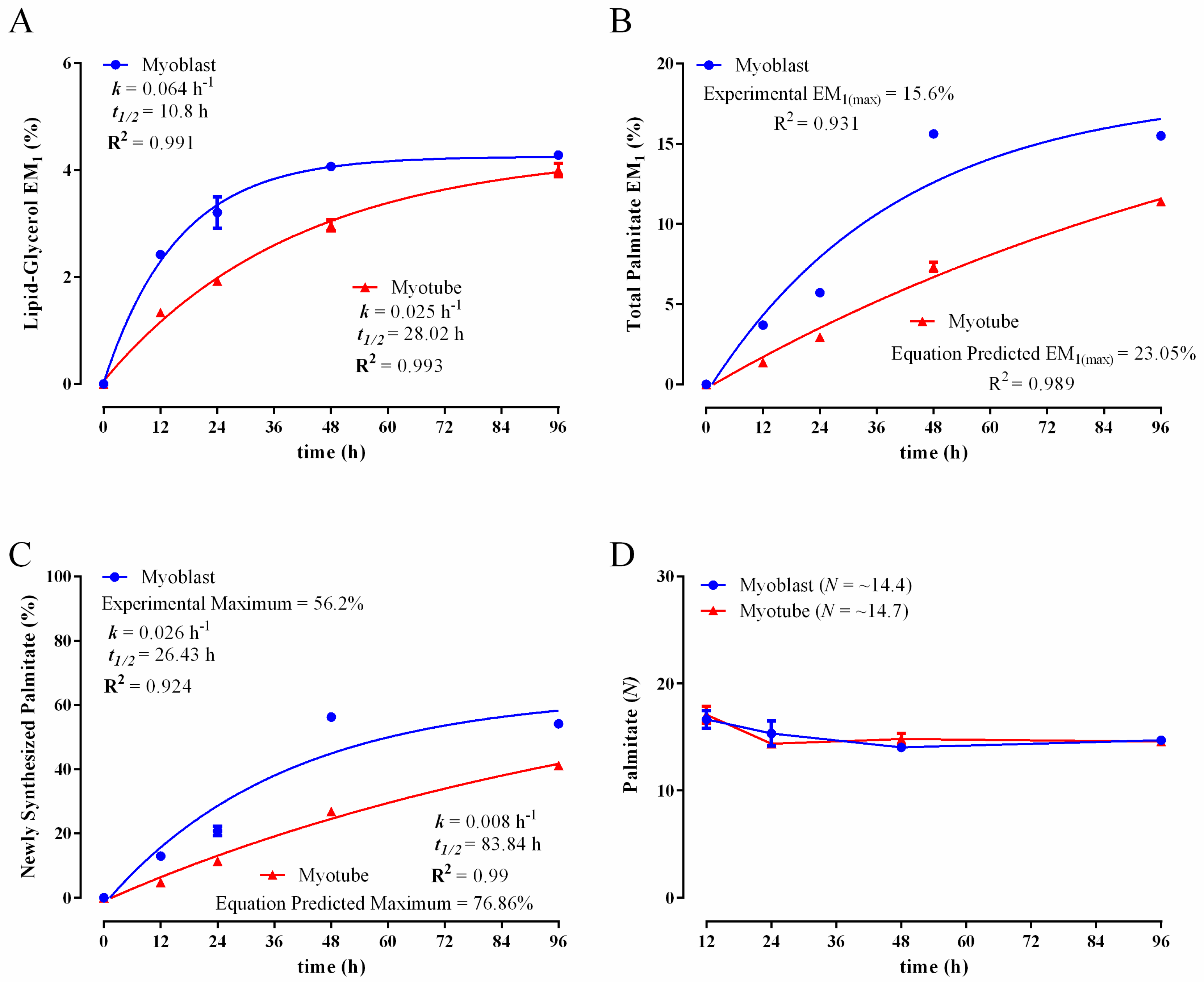
3.2. Comparison of Biomass Synthesis Rates under Mitotic and Post-Mitotic Conditions in C2C12 Cells
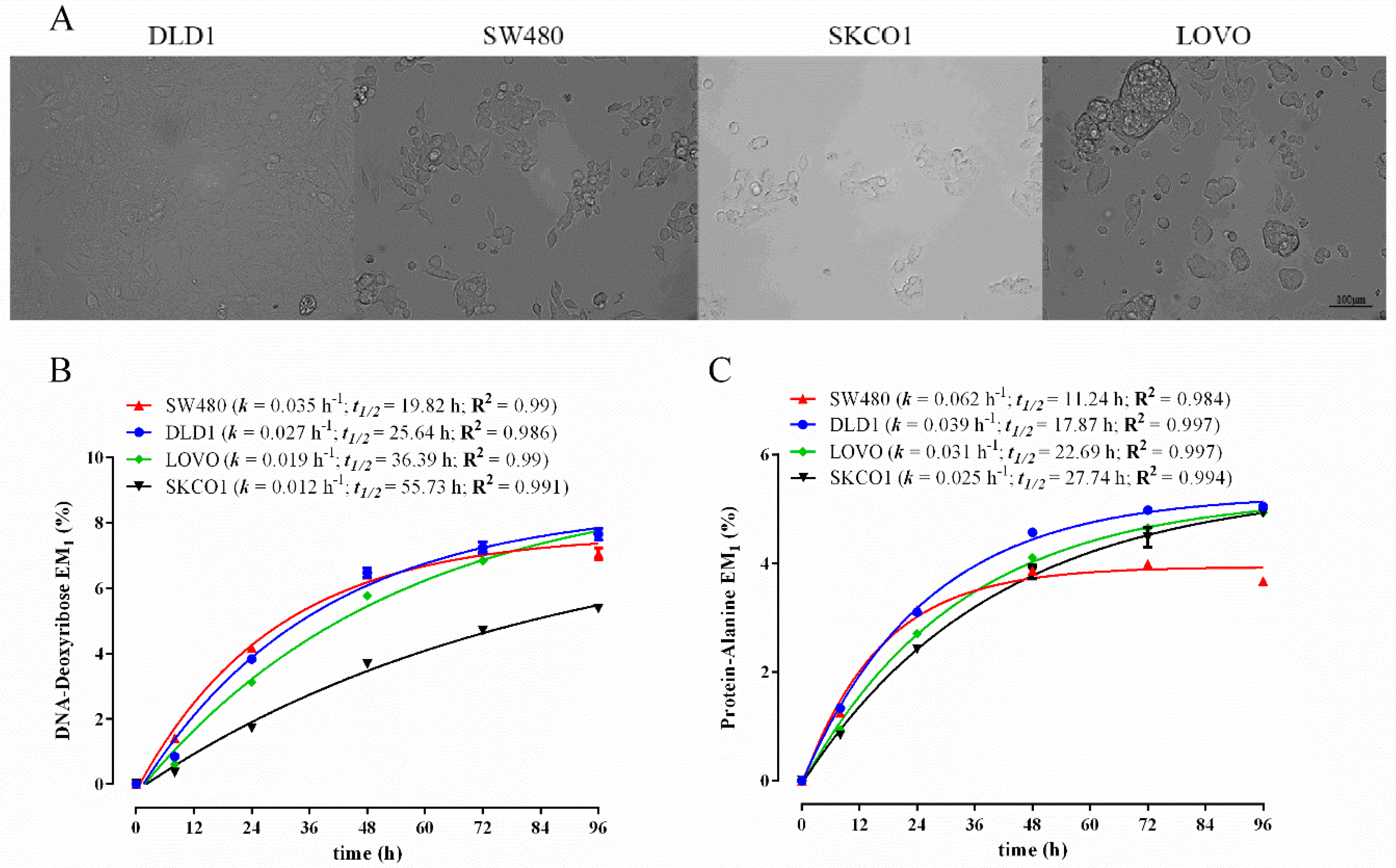
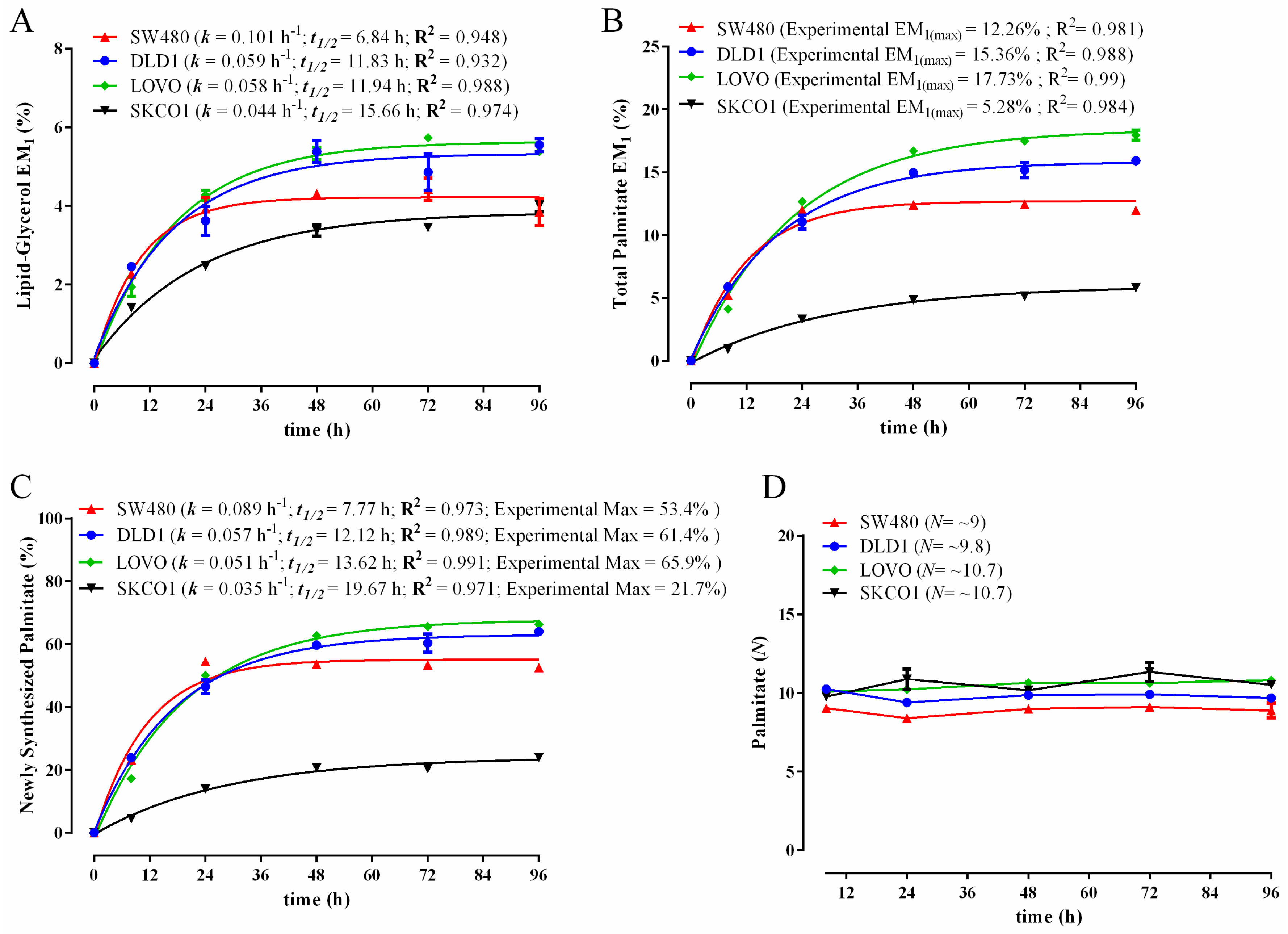
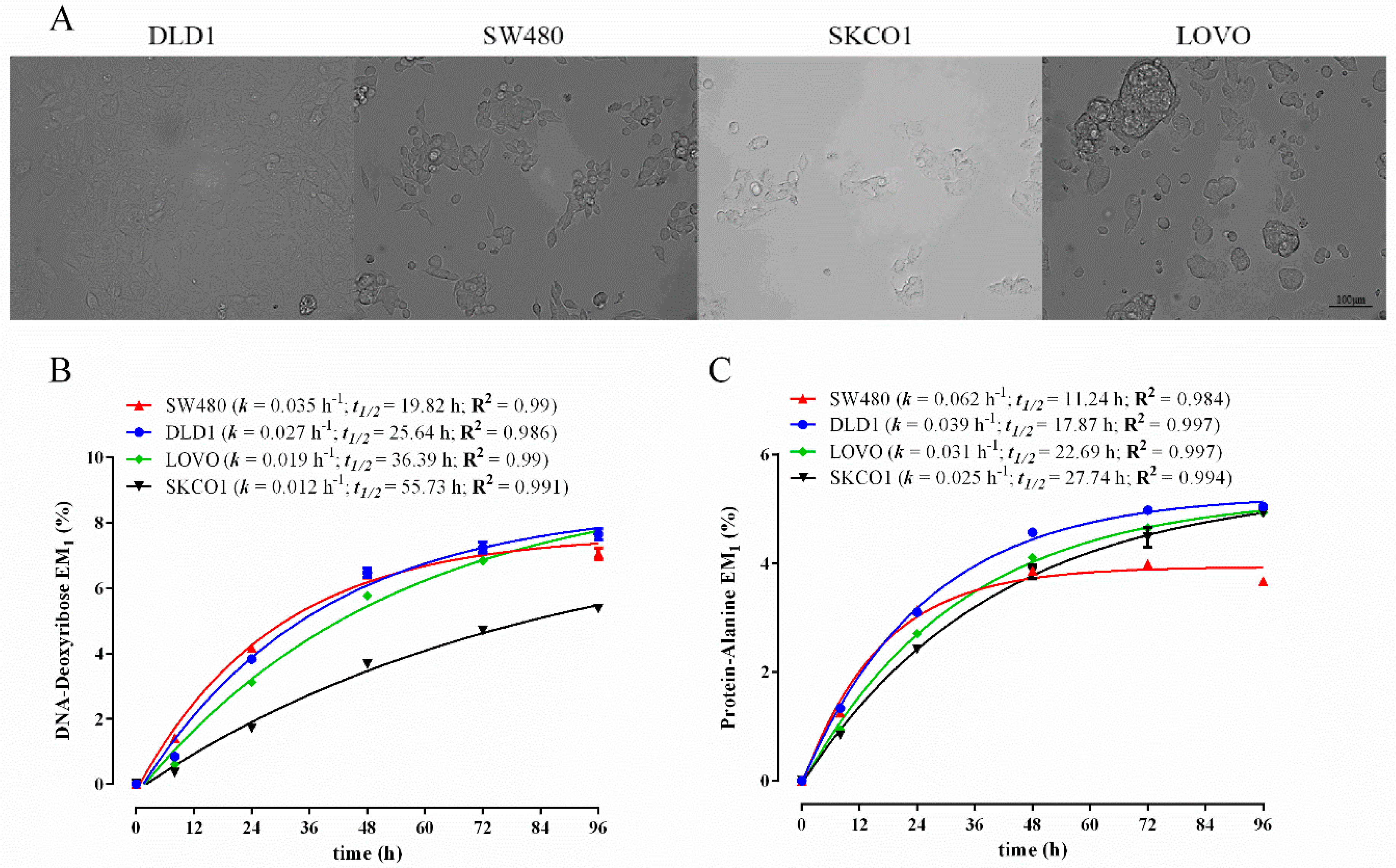
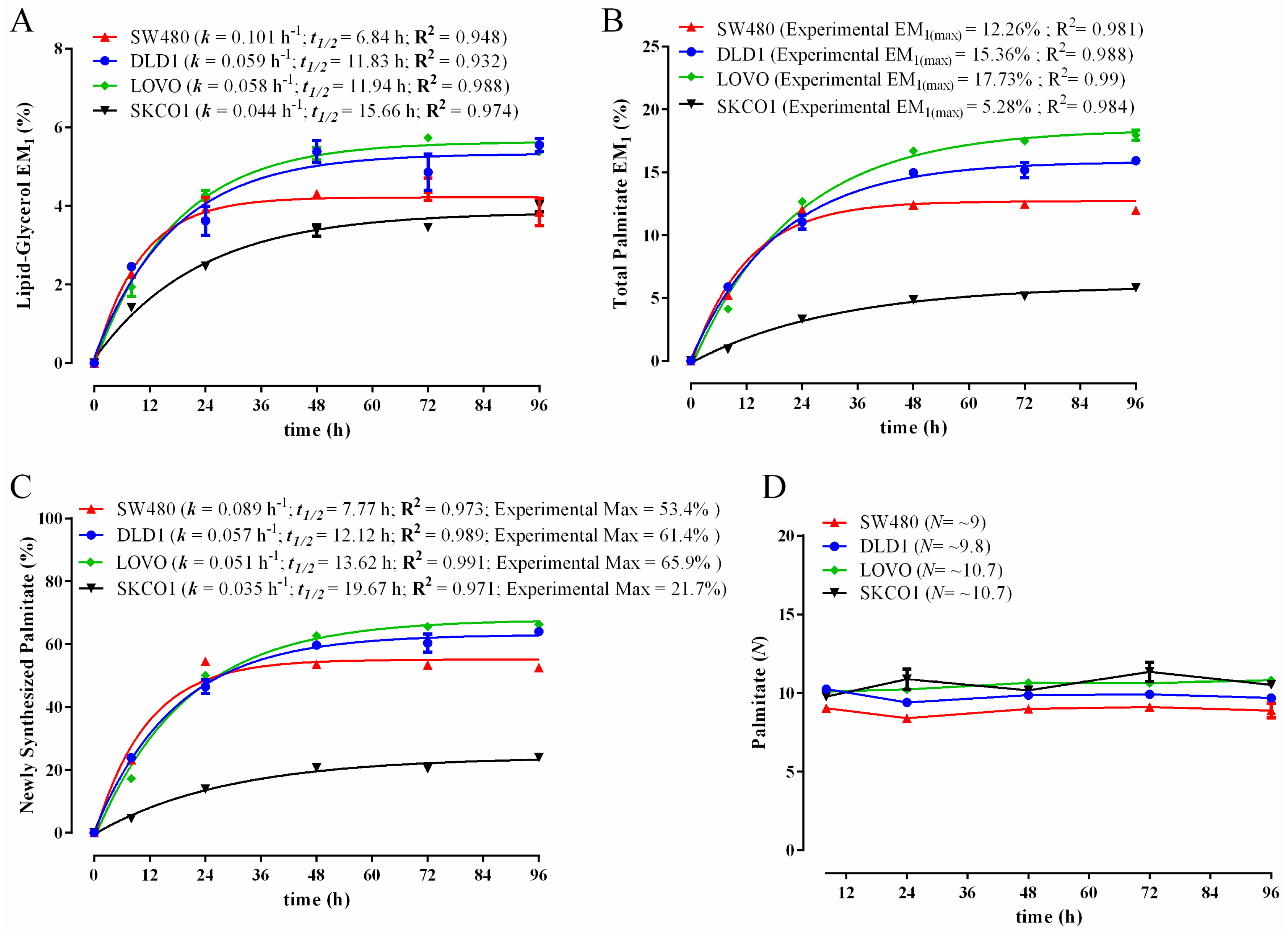
3.3. Comparison of Biomass Synthesis Rates in Colon Cancer Cell Lines
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wolfe, R.R.; Chinkes, D.L. Isotope Tracers in Metabolic Research: Principles and Practice of Kinetic Analysis, 2nd ed.; Wiley-Liss: Hoboken, NJ, USA, 2005. [Google Scholar]
- Hellerstein, M.K. In vivo measurement of fluxes through metabolic pathways: The missing link in functional genomics and pharmaceutical research. Annu. Rev. Nutr. 2003, 23, 379–402. [Google Scholar] [CrossRef] [PubMed]
- McCabe, B.J.; Previs, S.F. Using isotope tracers to study metabolism: Application in mouse models. Metab. Eng. 2004, 6, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Sauer, U. Metabolic networks in motion: 13c-based flux analysis. Mol. Syst. Biol. 2006, 2, 62. [Google Scholar] [CrossRef] [PubMed]
- Schoenheimer, R.; Rittenberg, D. Deuterium as an indicator in the study of intermediary metabolism. Science 1935, 82, 156–157. [Google Scholar] [CrossRef] [PubMed]
- Rittenberg, D.; Keston, A.S.; Schoenheimer, R.; Foster, G.L. Deuterium as an indicator in the study of intermediary metabolism xiii. The stability of hydrogen in amino acids. J. Biol. Chem. 1938, 125, 0001–0012. [Google Scholar]
- Foster, G.L.; Keston, A.S.; Rittenberg, D.; Schoenheimer, R. Deuterium as an indicator in the study of intermediary metabolism xii. The action of proteolytic enzymes on peptides in heavy water. J. Biol. Chem. 1938, 124, 159–161. [Google Scholar]
- Schoenheimer, R.; Rittenberg, D. Deuterium as an indicator in the study of intermediary metabolism ix. The conversion of stearic acid into palmitic acid in the organism. J. Biol. Chem. 1937, 120, 155–165. [Google Scholar]
- Rittenberg, D.; Schoenheimer, R. Deuterium as an indicator in the study of intermediary metabolism viii. Hydrogenation of fatty acids in the animal organism. J. Biol. Chem. 1937, 117, 485–490. [Google Scholar]
- Schoenheimer, R.; Rittenberg, D. Deuterium as an indicator in the study of intermediary metabolism vi. Synthesis and destruction of fatty acids in the organism. J. Biol. Chem. 1936, 114, 381–396. [Google Scholar]
- Schoenheimer, R.; Rittenberg, D. Deuterium as an indicator in the study of intermediary metabolism iii. The role of the fat tissues. J. Biol. Chem. 1935, 111, 175–181. [Google Scholar]
- Ussing, H.H. The glycogen-formation in the liver, studied by means of heavy hydrogen. Skand. Arch. Physiol. 1937, 77, 85–86. [Google Scholar]
- Ussing, H.H. The rate of protein renewal in mice and rats studied by means of heavy hydrogen. Acta Physiol. Scand. 1941, 2, 209–221. [Google Scholar] [CrossRef]
- Foster, G.L.; Rittenberg, D.; Schoenheimer, R. Deuterium as an indicator in the study of intermediary metabolism: Xiv. Biological formation of deuteroamino acids. J. Biol. Chem. 1938, 125, 13–22. [Google Scholar]
- Kloehn, J.; Saunders, E.C.; O’Callaghan, S.; Dagley, M.J.; McConville, M.J. Characterization of metabolically quiescent leishmania parasites in murine lesions using heavy water labeling. PLoS Pathog. 2015, 11, e1004683. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.M.; Murphy, E.J.; Neese, R.A.; Antelo, F.; Thomas, T.; Agarwal, A.; Go, C.; Hellerstein, M.K. Measurement of tg synthesis and turnover in vivo by 2H2O incorporation into the glycerol moiety and application of mida. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E790–E803. [Google Scholar] [CrossRef] [PubMed]
- Bederman, I.R.; Foy, S.; Chandramouli, V.; Alexander, J.C.; Previs, S.F. Triglyceride synthesis in epididymal adipose tissue: Contribution of glucose and non-glucose carbon sources. J. Biol. Chem. 2009, 284, 6101–6108. [Google Scholar] [CrossRef] [PubMed]
- Previs, S.F.; Mahsut, A.; Kulick, A.; Dunn, K.; Andrews-Kelly, G.; Johnson, C.; Bhat, G.; Herath, K.; Miller, P.L.; Wang, S.P.; et al. Quantifying cholesterol synthesis in vivo using (2)h(2)o: Enabling back-to-back studies in the same subject. J. Lipid Res. 2011, 52, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Lowenstein, J.M.; Brunengraber, H.; Wadke, M. Measurement of rates of lipogenesis with deuterated and tritiated water. Methods Enzymol. 1975, 35, 279–287. [Google Scholar] [PubMed]
- Diraison, F.; Pachiaudi, C.; Beylot, M. Measuring lipogenesis and cholesterol synthesis in humans with deuterated water: Use of simple gas chromatographic/mass spectrometric techniques. J. Mass Spectrom. JMS 1997, 32, 81–86. [Google Scholar] [CrossRef]
- Diraison, F.; Pachiaudi, C.; Beylot, M. In vivo measurement of plasma cholesterol and fatty acid synthesis with deuterated water: Determination of the average number of deuterium atoms incorporated. Metab. Clin. Exp. 1996, 45, 817–821. [Google Scholar] [CrossRef]
- Lee, W.N.; Bassilian, S.; Ajie, H.O.; Schoeller, D.A.; Edmond, J.; Bergner, E.A.; Byerley, L.O. In vivo measurement of fatty acids and cholesterol synthesis using d2o and mass isotopomer analysis. Am. J. Physiol. 1994, 266, E699–E708. [Google Scholar] [PubMed]
- Lee, W.N.; Bassilian, S.; Guo, Z.; Schoeller, D.; Edmond, J.; Bergner, E.A.; Byerley, L.O. Measurement of fractional lipid synthesis using deuterated water (2H2O) and mass isotopomer analysis. Am. J. Physiol. 1994, 266, E372–E383. [Google Scholar] [PubMed]
- Previs, S.F.; McLaren, D.G.; Wang, S.P.; Stout, S.J.; Zhou, H.; Herath, K.; Shah, V.; Miller, P.L.; Wilsie, L.; Castro-Perez, J.; et al. New methodologies for studying lipid synthesis and turnover: Looking backwards to enable moving forwards. Biochim. Biophys. Acta 2014, 1842, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, G.M.; Hamley, S.; Selathurai, A.; Kloehn, J.; De Souza, D.P.; O’Callaghan, S.; Nijagal, B.; Tull, D.L.; McConville, M.J.; Bruce, C.R. Reversing diet-induced metabolic dysregulation by diet switching leads to altered hepatic de novo lipogenesis and glycerolipid synthesis. Sci. Rep. 2016, 6, 27541. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, G.M.; Kloehn, J.; Burch, M.L.; Selathurai, A.; Hamley, S.; Bayol, S.A.; Lamon, S.; Watt, M.J.; Lee-Young, R.S.; McConville, M.J.; et al. Overexpression of sphingosine kinase 1 in liver reduces triglyceride content in mice fed a low but not high-fat diet. Biochim. Biophys. Acta 2015, 1851, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.A.; Carvalho, F.; Pearson, M.; Horton, J.D.; Browning, J.D.; Jones, J.G.; Burgess, S.C. A hfd suppresses de novo lipogenesis and desaturation, but not elongation and triglyceride synthesis in mice. J. Lipid Res. 2014, 55, 2541–2543. [Google Scholar] [CrossRef] [PubMed]
- Dufner, D.A.; Bederman, I.R.; Brunengraber, D.Z.; Rachdaoui, N.; Ismail-Beigi, F.; Siegfried, B.A.; Kimball, S.R.; Previs, S.F. Using 2H2O to study the influence of feeding on protein synthesis: Effect of isotope equilibration in vivo vs. In cell culture. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E1277–E1283. [Google Scholar] [CrossRef] [PubMed]
- Previs, S.F.; Fatica, R.; Chandramouli, V.; Alexander, J.C.; Brunengraber, H.; Landau, B.R. Quantifying rates of protein synthesis in humans by use of 2H2O: Application to patients with end-stage renal disease. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E665–E672. [Google Scholar] [CrossRef] [PubMed]
- Belloto, E.; Diraison, F.; Basset, A.; Allain, G.; Abdallah, P.; Beylot, M. Determination of protein replacement rates by deuterated water: Validation of underlying assumptions. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1340–E1347. [Google Scholar] [CrossRef] [PubMed]
- Busch, R.; Kim, Y.K.; Neese, R.A.; Schade-Serin, V.; Collins, M.; Awada, M.; Gardner, J.L.; Beysen, C.; Marino, M.E.; Misell, L.M.; et al. Measurement of protein turnover rates by heavy water labeling of nonessential amino acids. Biochim. Biophys. Acta 2006, 1760, 730–744. [Google Scholar] [CrossRef] [PubMed]
- Gasier, H.G.; Fluckey, J.D.; Previs, S.F. The application of 2H2O to measure skeletal muscle protein synthesis. Nutr. Metab. 2010, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Holmes, W.E.; Angel, T.E.; Li, K.W.; Hellerstein, M.K. Dynamic proteomics: In vivo proteome-wide measurement of protein kinetics using metabolic labeling. Methods Enzymol. 2015, 561, 219–276. [Google Scholar] [PubMed]
- Chan, X.C.; Black, C.M.; Lin, A.J.; Ping, P.; Lau, E. Mitochondrial protein turnover: Methods to measure turnover rates on a large scale. J. Mol. Cell. Cardiol. 2015, 78, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Busch, R.; Neese, R.A.; Awada, M.; Hayes, G.M.; Hellerstein, M.K. Measurement of cell proliferation by heavy water labeling. Nat. Protoc. 2007, 2, 3045–3057. [Google Scholar] [CrossRef] [PubMed]
- Hayes, G.M.; Simko, J.; Holochwost, D.; Kuchinsky, K.; Busch, R.; Misell, L.; Murphy, E.J.; Carroll, P.; Chan, J.; Shinohara, K.; et al. Regional cell proliferation in microdissected human prostate specimens after heavy water labeling in vivo: Correlation with prostate epithelial cells isolated from seminal fluid. Clin. Cancer Res. 2012, 18, 3250–3260. [Google Scholar] [CrossRef] [PubMed]
- Landau, B.R.; Wahren, J.; Chandramouli, V.; Schumann, W.C.; Ekberg, K.; Kalhan, S.C. Use of 2H2O for estimating rates of gluconeogenesis. Application to the fasted state. J. Clin. Investig. 1995, 95, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Landau, B.R.; Wahren, J.; Chandramouli, V.; Schumann, W.C.; Ekberg, K.; Kalhan, S.C. Contributions of gluconeogenesis to glucose production in the fasted state. J. Clin. Investig. 1996, 98, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Nuttall, F.Q.; Ngo, A.; Gannon, M.C. Regulation of hepatic glucose production and the role of gluconeogenesis in humans: Is the rate of gluconeogenesis constant? Diabetes Metab. Res. Rev. 2008, 24, 438–458. [Google Scholar] [CrossRef] [PubMed]
- Previs, S.F.; Brunengraber, H. Methods for measuring gluconeogenesis in vivo. Curr. Opin. Clin. Nutr. Metab. Care 1998, 1, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Delgado, T.C.; Martins, F.O.; Carvalho, F.; Goncalves, A.; Scott, D.K.; O’Doherty, R.; Macedo, M.P.; Jones, J.G. (2)h enrichment distribution of hepatic glycogen from 2H2O reveals the contribution of dietary fructose to glycogen synthesis. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E384–E391. [Google Scholar] [CrossRef] [PubMed]
- Soares, A.F.; Carvalho, R.A.; Veiga, F.J.; Alves, M.G.; Martins, F.O.; Viegas, I.; Gonzalez, J.D.; Meton, I.; Baanante, I.V.; Jones, J.G. Restoration of direct pathway glycogen synthesis flux in the stz-diabetes rat model by insulin administration. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E875–E885. [Google Scholar] [CrossRef] [PubMed]
- Viegas, I.; Rito, J.; Jarak, I.; Leston, S.; Carvalho, R.A.; Meton, I.; Pardal, M.A.; Baanante, I.V.; Jones, J.G. Hepatic glycogen synthesis in farmed European seabass (dicentrarchus labrax l.) is dominated by indirect pathway fluxes. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2012, 163, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Sapcariu, S.C.; Kanashova, T.; Weindl, D.; Ghelfi, J.; Dittmar, G.; Hiller, K. Simultaneous extraction of proteins and metabolites from cells in culture. MethodsX 2014, 1, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, G.M.; De Souza, D.P.; Burch, M.L.; Hamley, S.; Kloehn, J.; Selathurai, A.; Tull, D.; O’Callaghan, S.; McConville, M.J.; Bruce, C.R. Application of dynamic metabolomics to examine in vivo skeletal muscle glucose metabolism in the chronically high-fat fed mouse. Biochem. Biophys. Res. Commun. 2015, 462, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, G.M.; De Souza, D.P.; Risis, S.; Burch, M.L.; Hamley, S.; Kloehn, J.; Selathurai, A.; Lee-Young, R.S.; Tull, D.; O’Callaghan, S.; et al. In vivo cardiac glucose metabolism in the high-fat fed mouse: Comparison of euglycemic-hyperinsulinemic clamp derived measures of glucose uptake with a dynamic metabolomic flux profiling approach. Biochem. Biophys. Res. Commun. 2015, 463, 818–824. [Google Scholar] [CrossRef] [PubMed]
- Hellerstein, M.K.; Neese, R.A. Mass isotopomer distribution analysis at eight years: Theoretical, analytic, and experimental considerations. Am. J. Physiol. 1999, 276, E1146–E1170. [Google Scholar] [PubMed]
- Burattini, S.; Ferri, P.; Battistelli, M.; Curci, R.; Luchetti, F.; Falcieri, E. C2c12 murine myoblasts as a model of skeletal muscle development: Morpho-functional characterization. Eur. J. Histochem. EJH 2004, 48, 223–233. [Google Scholar] [PubMed]
- Elkalaf, M.; Andel, M.; Trnka, J. Low glucose but not galactose enhances oxidative mitochondrial metabolism in c2c12 myoblasts and myotubes. PLoS ONE 2013, 8, e70772. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, N.; Yoshida, S.; Koishi, K.; Masuda, K.; Nabeshima, Y. Cell heterogeneity upon myogenic differentiation: Down-regulation of myod and myf-5 generates ‘reserve cells’. J. Cell Sci. 1998, 111 Pt 6, 769–779. [Google Scholar] [PubMed]
- Stuelsatz, P.; Pouzoulet, F.; Lamarre, Y.; Dargelos, E.; Poussard, S.; Leibovitch, S.; Cottin, P.; Veschambre, P. Down-regulation of myod by calpain 3 promotes generation of reserve cells in c2c12 myoblasts. J. Biol. Chem. 2010, 285, 12670–12683. [Google Scholar] [CrossRef] [PubMed]
- Friday, B.B.; Pavlath, G.K. A calcineurin- and nfat-dependent pathway regulates myf5 gene expression in skeletal muscle reserve cells. J. Cell Sci. 2001, 114, 303–310. [Google Scholar] [PubMed]
- Mouradov, D.; Sloggett, C.; Jorissen, R.N.; Love, C.G.; Li, S.; Burgess, A.W.; Arango, D.; Strausberg, R.L.; Buchanan, D.; Wormald, S.; et al. Colorectal cancer cell lines are representative models of the main molecular subtypes of primary cancer. Cancer Res. 2014, 74, 3238–3247. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, S. Crosstalk between autophagy and proteasome protein degradation systems: Possible implications for cancer therapy. Folia Histochem. Cytobiol. 2013, 51, 249–264. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, J.V.; Brusselmans, K.; Verhoeven, G. Increased lipogenesis in cancer cells: New players, novel targets. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.H.; Fowle-Grider, R.; Mahieu, N.G.; Liu, G.Y.; Chen, Y.J.; Wang, R.; Singh, M.; Potter, G.S.; Gross, R.W.; Schaefer, J.; et al. Exogenous fatty acids are the preferred source of membrane lipids in proliferating fibroblasts. Cell Chem. Biol. 2016, 23, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Turner, S.; Killion, S.; Hellerstein, M.K. In vivo measurement of DNA synthesis rates of colon epithelial cells in carcinogenesis. Biochem. Biophys. Res. Commun. 2005, 331, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Louie, K.B.; Bowen, B.P.; McAlhany, S.; Huang, Y.; Price, J.C.; Mao, J.H.; Hellerstein, M.; Northen, T.R. Mass spectrometry imaging for in situ kinetic histochemistry. Sci. Rep. 2013, 3, 1656. [Google Scholar] [CrossRef] [PubMed]
- Messmer, B.T.; Messmer, D.; Allen, S.L.; Kolitz, J.E.; Kudalkar, P.; Cesar, D.; Murphy, E.J.; Koduru, P.; Ferrarini, M.; Zupo, S.; et al. In vivo measurements document the dynamic cellular kinetics of chronic lymphocytic leukemia b cells. J. Clin. Investig. 2005, 115, 755–764. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Foletta, V.C.; Palmieri, M.; Kloehn, J.; Mason, S.; Previs, S.F.; McConville, M.J.; Sieber, O.M.; Bruce, C.R.; Kowalski, G.M. Analysis of Mammalian Cell Proliferation and Macromolecule Synthesis Using Deuterated Water and Gas Chromatography-Mass Spectrometry. Metabolites 2016, 6, 34. https://doi.org/10.3390/metabo6040034
Foletta VC, Palmieri M, Kloehn J, Mason S, Previs SF, McConville MJ, Sieber OM, Bruce CR, Kowalski GM. Analysis of Mammalian Cell Proliferation and Macromolecule Synthesis Using Deuterated Water and Gas Chromatography-Mass Spectrometry. Metabolites. 2016; 6(4):34. https://doi.org/10.3390/metabo6040034
Chicago/Turabian StyleFoletta, Victoria C., Michelle Palmieri, Joachim Kloehn, Shaun Mason, Stephen F. Previs, Malcolm J. McConville, Oliver M. Sieber, Clinton R. Bruce, and Greg M. Kowalski. 2016. "Analysis of Mammalian Cell Proliferation and Macromolecule Synthesis Using Deuterated Water and Gas Chromatography-Mass Spectrometry" Metabolites 6, no. 4: 34. https://doi.org/10.3390/metabo6040034