
Regulation of Metabolic Activity by p53


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. p53 and Tumour Suppression
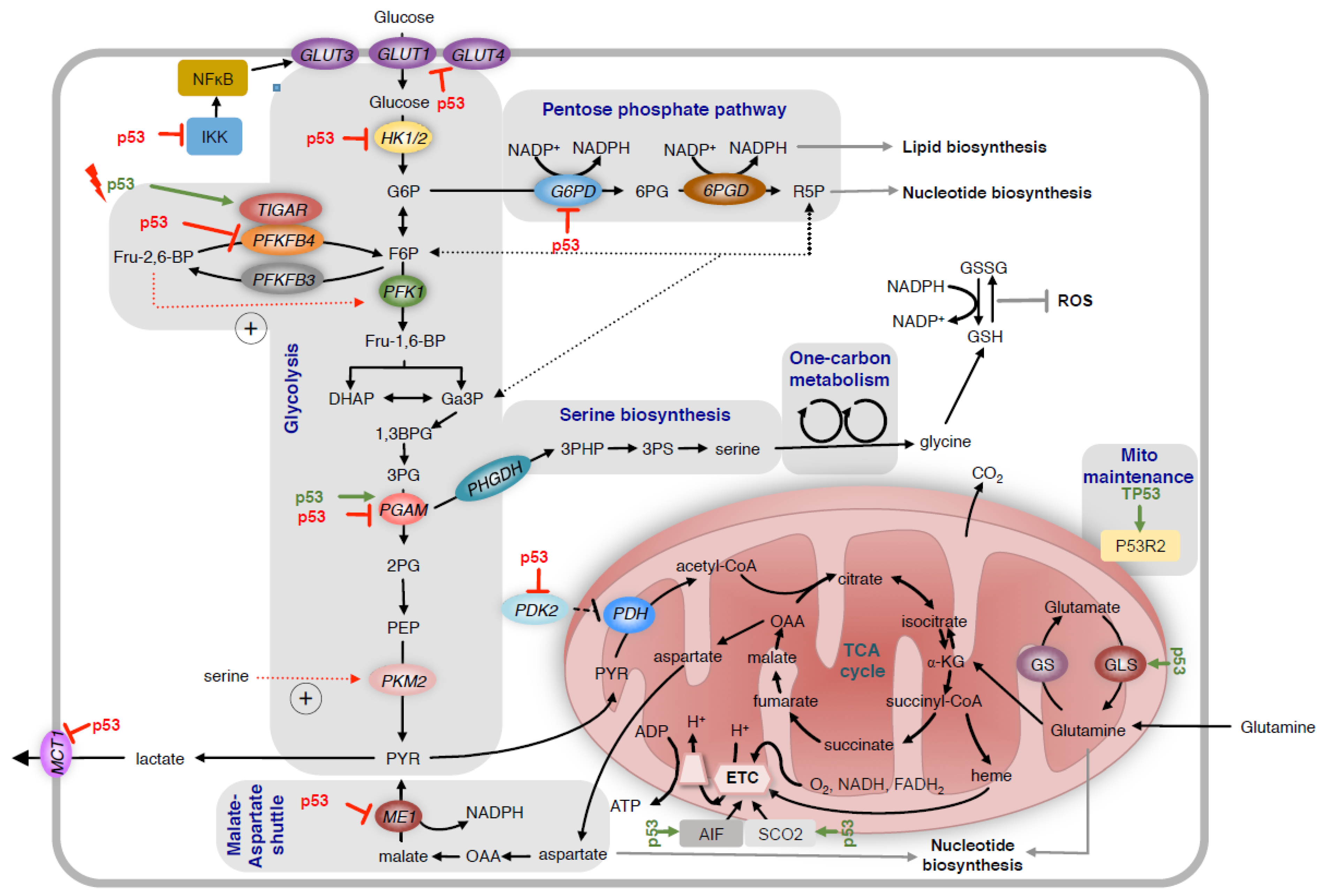
2. Regulation of Glycolysis and the Pentose Phosphate Pathway
3. Regulation of Serine Metabolism
4. Regulation of Mitochondrial Metabolism
5. Regulation of Oxidative Stress
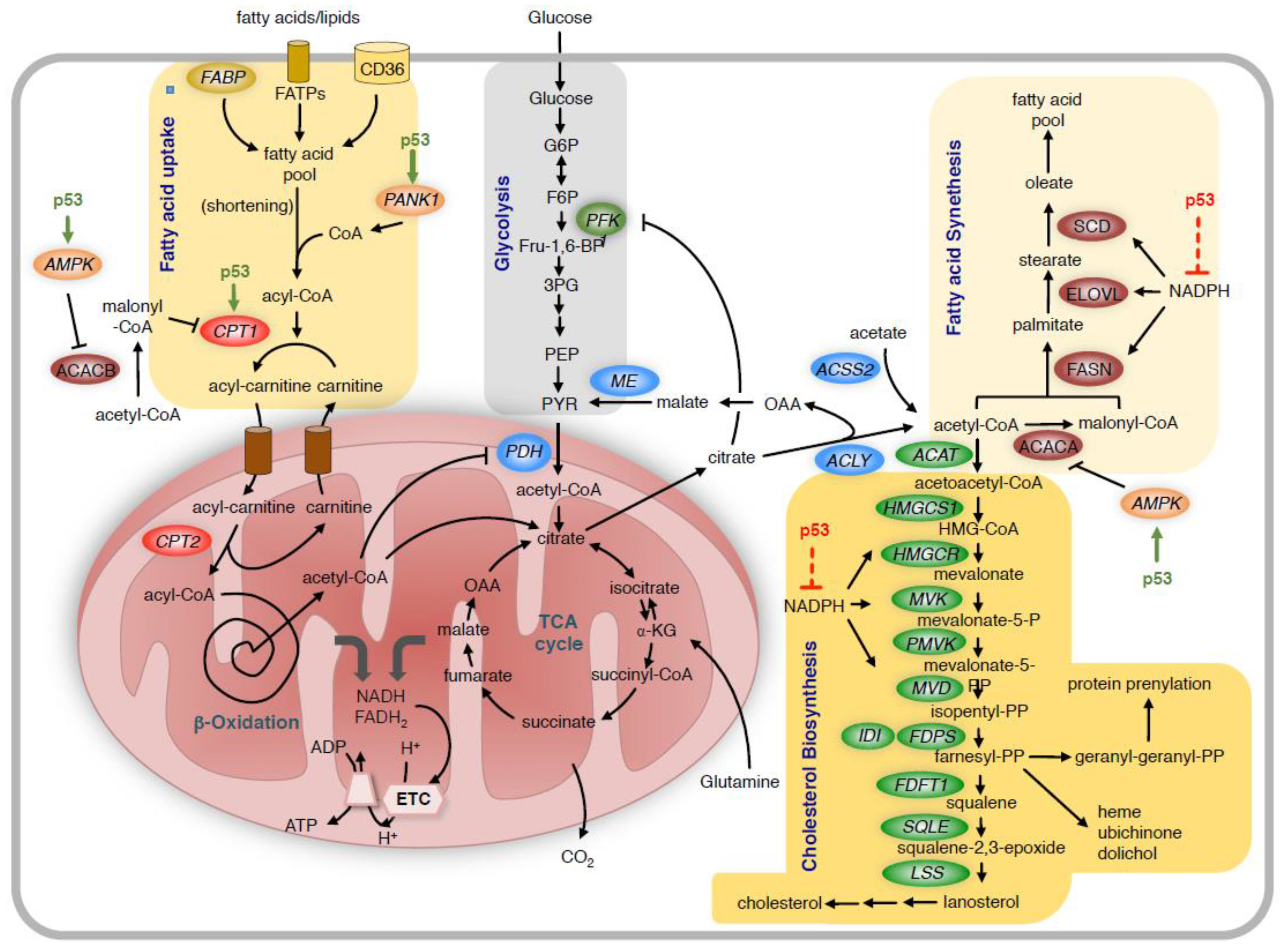
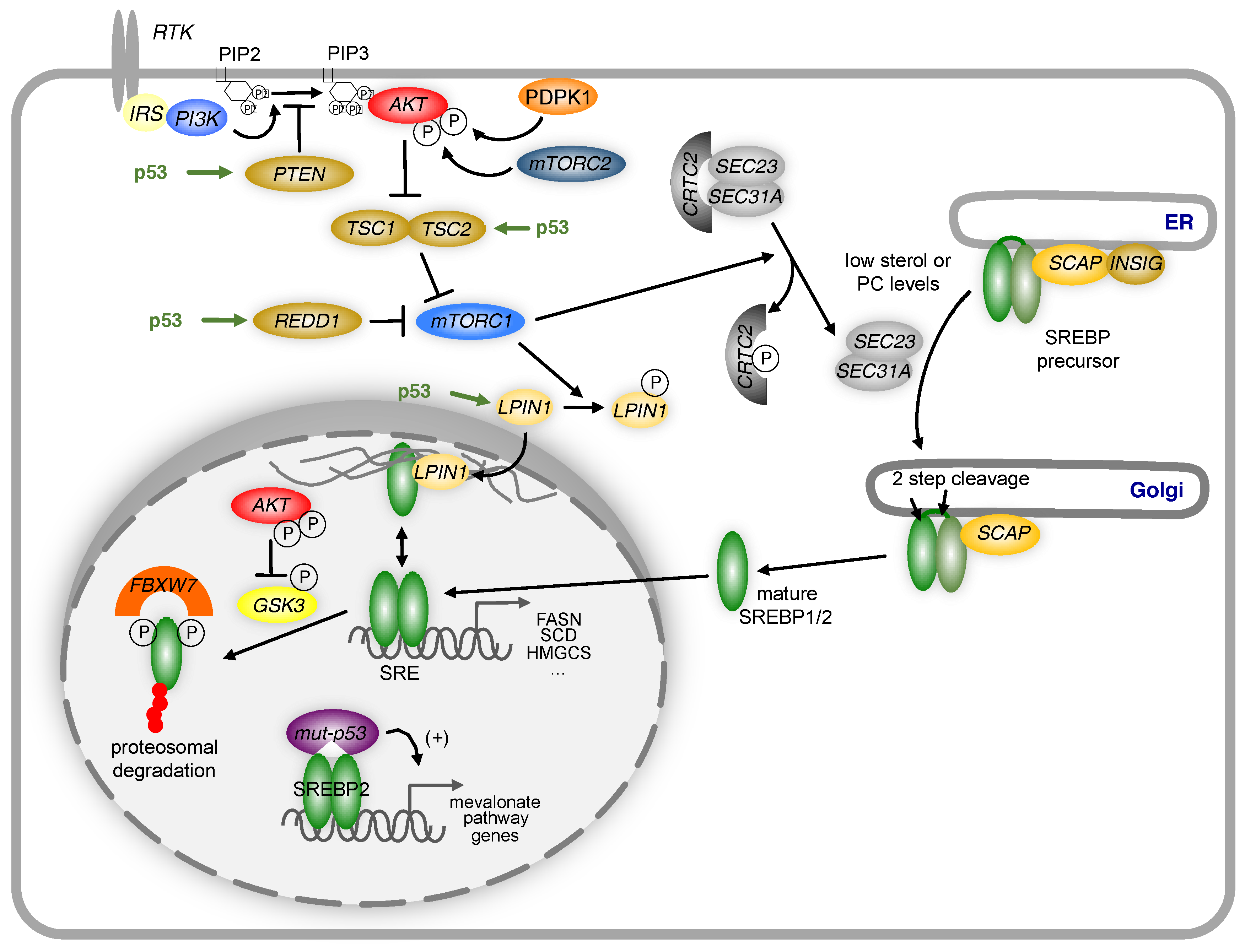
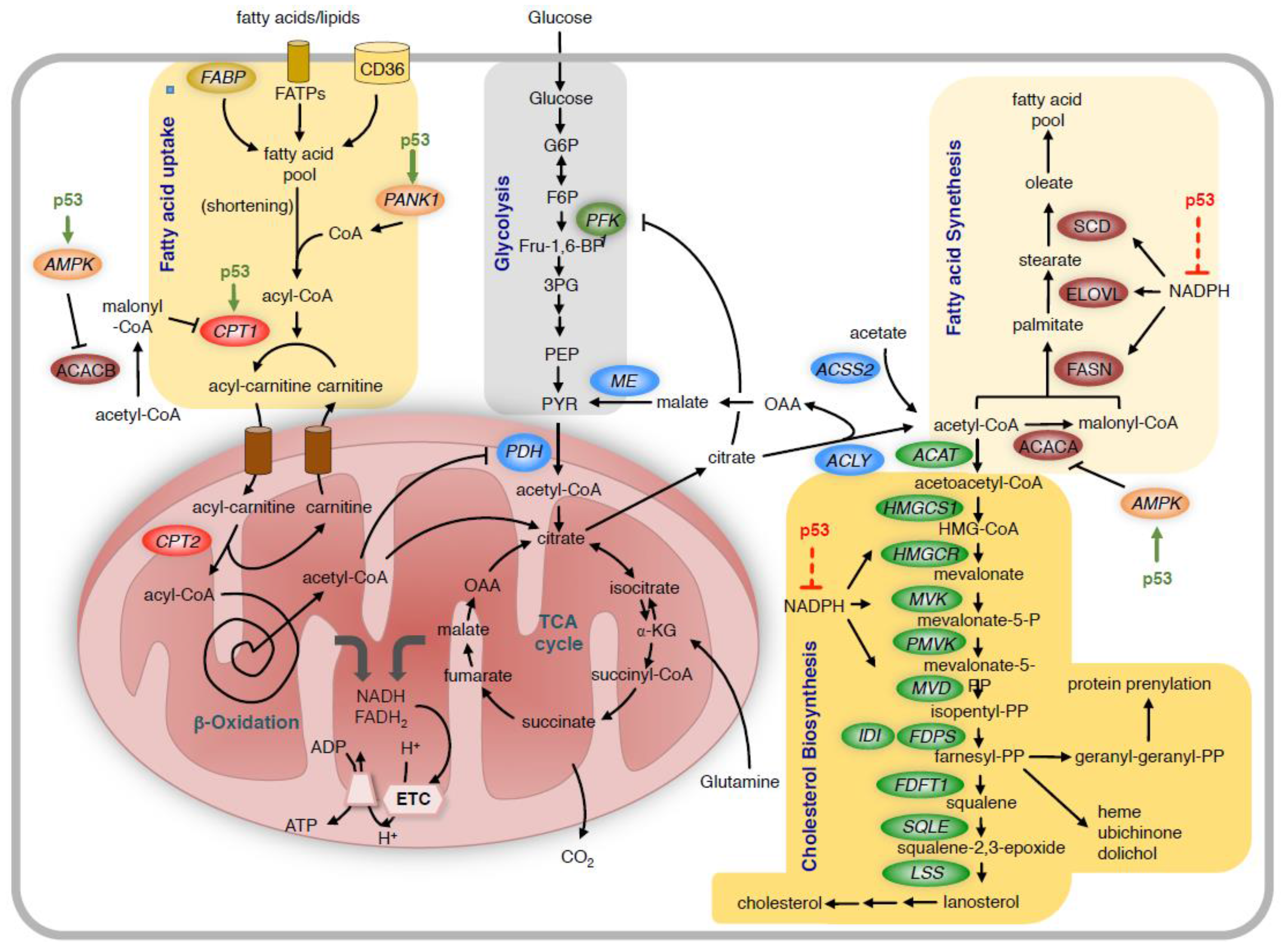
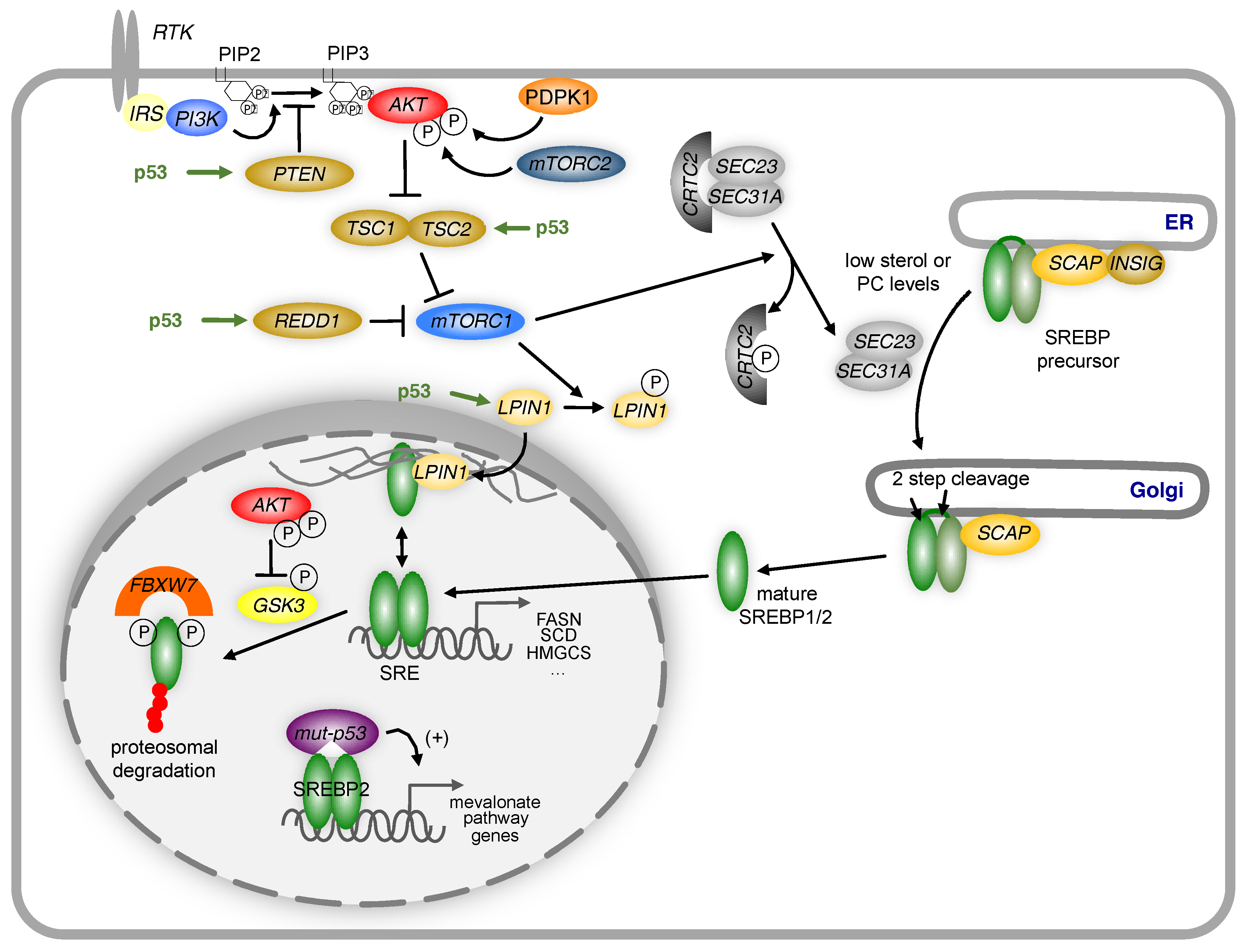
6. Regulation of Lipid Metabolism
7. Roles of p53 in the Tumour Microenvironment
8. Immune-Regulatory Functions of p53
9. Targeting Metabolism for the Treatment of p53 Deficient Cancer Cells
Acknowledgments
Conflicts of Interest
References
- Lane, D.P.; Crawford, L.V. T antigen is bound to a host protein in sy40-transformed cells. Nature 1979, 278, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.; Levine, A. P53 research: The past thirty years and the next thirty years. Cold Spring Harb. Perspect. Biol. 2010, 2, a000893. [Google Scholar] [CrossRef] [PubMed]
- Finlay, C.A.; Hinds, P.W.; Levine, A.J. The p53 proto-oncogene can act as a suppressor of transformation. Cell 1989, 57, 1083–1093. [Google Scholar] [CrossRef]
- Eliyahu, D.; Michalovitz, D.; Eliyahu, S.; Pinhasi-Kimhi, O.; Oren, M. Wild-type p53 can inhibit oncogene-mediated focus formation. Proc. Natl. Acad. Sci. USA 1989, 86, 8763–8767. [Google Scholar] [CrossRef] [PubMed]
- Soussi, T. P53 alterations in human cancer: More questions than answers. Oncogene 2007, 26, 2145–2156. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lane, D.P. P53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Prives, C. Blinded by the light: The growing complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Horn, H.F.; Vousden, K.H. Coping with stress: Multiple ways to activate p53. Oncogene 2007, 26, 1306–1316. [Google Scholar] [CrossRef] [PubMed]
- Marine, J.C.; Francoz, S.; Maetens, M.; Wahl, G.; Toledo, F.; Lozano, G. Keeping p53 in check: Essential and synergistic functions of mdm2 and mdm4. Cell Death. Differ. 2006, 13, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Wade, M.; Wang, Y.V.; Wahl, G.M. The p53 orchestra: Mdm2 and mdmx set the tone. Trends Cell Biol. 2010, 20, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Kruse, J.P.; Gu, W. Modes of p53 regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Giese, A.; Deppert, W. Wild-type p53 in cancer cells: When a guardian turns into a blackguard. Biochem. Pharmacol. 2009, 77, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Cui, Y. New perspective on targeting the tumor suppressor p53 pathway in the tumor microenvironment to enhance the efficacy of immunotherapy. J. Immunother. Cancer 2015, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Villunger, A.; Michalak, E.M.; Coultas, L.; Mullauer, F.; Bock, G.; Ausserlechner, M.J.; Adams, J.M.; Strasser, A. P53- and drug-induced apoptotic responses mediated by bh3-only proteins puma and noxa. Science 2003, 302, 1036–1038. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct activation of bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Soussi, T.; Dehouche, K.; Beroud, C. P53 website and analysis of p53 gene mutations in human cancer: Forging a link between epidemiology and carcinogenesis. Hum. Mutat. 2000, 15, 105–113. [Google Scholar] [CrossRef]
- Hollstein, M.; Rice, K.; Greenblatt, M.S.; Soussi, T.; Fuchs, R.; Sorlie, T.; Hovig, E.; Smith-Sorensen, B.; Montesano, R.; Harris, C.C. Database of p53 gene somatic mutations in human tumors and cell lines. Nucleic Acids Res. 1994, 22, 3551–3555. [Google Scholar] [PubMed]
- Hussain, S.P.; Amstad, P.; Raja, K.; Ambs, S.; Nagashima, M.; Bennett, W.P.; Shields, P.G.; Ham, A.J.; Swenberg, J.A.; Marrogi, A.J.; et al. Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: A cancer-prone chronic inflammatory disease. Cancer Res. 2000, 60, 3333–3337. [Google Scholar] [PubMed]
- Schetter, A.J.; Heegaard, N.H.; Harris, C.C. Inflammation and cancer: Interweaving microrna, free radical, cytokine and p53 pathways. Carcinogenesis 2010, 31, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Cooks, T.; Harris, C.C.; Oren, M. Caught in the cross fire: P53 in inflammation. Carcinogenesis 2014, 35, 1680–1690. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. P53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef]
- Wong, K.B.; DeDecker, B.S.; Freund, S.M.; Proctor, M.R.; Bycroft, M.; Fersht, A.R. Hot-spot mutants of p53 core domain evince characteristic local structural changes. Proc. Natl. Acad. Sci. USA 1999, 96, 8438–8442. [Google Scholar] [CrossRef] [PubMed]
- Friedman, P.N.; Chen, X.; Bargonetti, J.; Prives, C. The p53 protein is an unusually shaped tetramer that binds directly to DNA. Proc. Natl. Acad. Sci. USA 1993, 90, 3319–3323. [Google Scholar] [CrossRef] [PubMed]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H. P53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.W.; Cepero, E.; Evan, G. Intrinsic tumour suppression. Nature 2004, 432, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Parant, J.M.; Lang, G.; Chau, P.; Chavez-Reyes, A.; El-Naggar, A.K.; Multani, A.; Chang, S.; Lozano, G. Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in trp53 mutant mice. Nat. Genet. 2004, 36, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P. What has senescence got to do with cancer? Cancer Cell 2005, 7, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Kon, N.; Jiang, L.; Tan, M.; Ludwig, T.; Zhao, Y.; Baer, R.; Gu, W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012, 149, 1269–1283. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. Über den stoffwechsel der carcinomzelle. Die Naturwissenschaften 1924, 12, 1131–1137. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Schwartzenberg-Bar-Yoseph, F. The tumor suppressor p53 down-regulates glucose transporters glut1 and glut4 gene expression. Cancer Res. 2004, 64, 2627–2633. [Google Scholar] [CrossRef] [PubMed]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. P53 regulates glucose metabolism through an ikk-nf-kappab pathway and inhibits cell transformation. Nat. Cell Biol. 2008, 10, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Mathupala, S.P.; Heese, C.; Pedersen, P.L. Glucose catabolism in cancer cells. The type ii hexokinase promoter contains functionally active response elements for the tumor suppressor p53. J. Biol. Chem. 1997, 272, 22776–22780. [Google Scholar] [CrossRef] [PubMed]
- Rider, M.H.; Bertrand, L.; Vertommen, D.; Michels, P.A.; Rousseau, G.G.; Hue, L. 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase: Head-to-head with a bifunctional enzyme that controls glycolysis. Biochem. J. 2004, 381, 561–579. [Google Scholar] [CrossRef] [PubMed]
- Ros, S.; Schulze, A. Balancing glycolytic flux: The role of 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatases in cancer metabolism. Cancer Metab. 2013, 1, 8. [Google Scholar] [CrossRef] [PubMed]
- Atsumi, T.; Chesney, J.; Metz, C.; Leng, L.; Donnelly, S.; Makita, Z.; Mitchell, R.; Bucala, R. High expression of inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (ipfk-2; pfkfb3) in human cancers. Cancer Res. 2002, 62, 5881–5887. [Google Scholar] [PubMed]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. Tigar, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Jogl, G. Structural and biochemical studies of tigar (tp53-induced glycolysis and apoptosis regulator). J. Biol. Chem. 2009, 284, 1748–1754. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Cheung, E.C.; Vousden, K.H. Modulation of intracellular ros levels by tigar controls autophagy. EMBO J. 2009, 28, 3015–3026. [Google Scholar] [CrossRef] [PubMed]
- Muniyappa, H.; Song, S.; Mathews, C.K.; Das, K.C. Reactive oxygen species-independent oxidation of thioredoxin in hypoxia: Inactivation of ribonucleotide reductase and redox-mediated checkpoint control. J. Biol. Chem. 2009, 284, 17069–17081. [Google Scholar] [CrossRef] [PubMed]
- Sablina, A.A.; Budanov, A.V.; Ilyinskaya, G.V.; Agapova, L.S.; Kravchenko, J.E.; Chumakov, P.M. The antioxidant function of the p53 tumor suppressor. Nat. Med. 2005, 11, 1306–1313. [Google Scholar] [CrossRef] [PubMed]
- Gerin, I.; Noel, G.; Bolsee, J.; Haumont, O.; Van Schaftingen, E.; Bommer, G.T. Identification of tp53-induced glycolysis and apoptosis regulator (tigar) as the phosphoglycolate-independent 2,3-bisphosphoglycerate phosphatase. Biochem. J. 2014, 458, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.C.; Vousden, K.H. The role of p53 in glucose metabolism. Curr. Opin. Cell Biol. 2010, 22, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Hock, A.K.; Vousden, K.H.; Cheung, E.C. P53- and p73-independent activation of tigar expression in vivo. Cell Death Dis. 2015, 6, e1842. [Google Scholar] [CrossRef] [PubMed]
- Ros, S.; Floter, J.; Kaymak, I.; Da Costa, C.; Houddane, A.; Dubuis, S.; Griffiths, B.; Mitter, R.; Walz, S.; Blake, S.; et al. 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 is essential for p53-null cancer cells. Oncogene 2017. [Google Scholar] [CrossRef] [PubMed]
- Ros, S.; Santos, C.R.; Moco, S.; Baenke, F.; Kelly, G.; Howell, M.; Zamboni, N.; Schulze, A. Functional metabolic screen identifies 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 as an important regulator of prostate cancer cell survival. Cancer Discov. 2012, 2, 328–343. [Google Scholar] [CrossRef] [PubMed]
- Goidts, V.; Bageritz, J.; Puccio, L.; Nakata, S.; Zapatka, M.; Barbus, S.; Toedt, G.; Campos, B.; Korshunov, A.; Momma, S.; et al. Rnai screening in glioma stem-like cells identifies pfkfb4 as a key molecule important for cancer cell survival. Oncogene 2012, 31, 3235–3243. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. P53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, H.; Lleonart, M.E.; Gil, J.; Wang, J.; Degan, P.; Peters, G.; Martinez, D.; Carnero, A.; Beach, D. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005, 65, 177–185. [Google Scholar] [PubMed]
- Hitosugi, T.; Zhou, L.; Elf, S.; Fan, J.; Kang, H.B.; Seo, J.H.; Shan, C.; Dai, Q.; Zhang, L.; Xie, J.; et al. Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth. Cancer Cell 2012, 22, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Lozano, P.; Hixon, M.L.; Wagner, M.W.; Flores, A.I.; Ikawa, S.; Baldwin, A.S., Jr.; Chien, K.R.; Gualberto, A. P53 is a transcriptional activator of the muscle-specific phosphoglycerate mutase gene and contributes in vivo to the control of its cardiac expression. Cell Growth Differ. 1999, 10, 295–306. [Google Scholar] [PubMed]
- Chaneton, B.; Hillmann, P.; Zheng, L.; Martin, A.C.; Maddocks, O.D.; Chokkathukalam, A.; Coyle, J.E.; Jankevics, A.; Holding, F.P.; Vousden, K.H.; et al. Serine is a natural ligand and allosteric activator of pyruvate kinase m2. Nature 2012, 491, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Mancuso, A.; Tong, X.; Ward, P.S.; Fan, J.; Rabinowitz, J.D.; Thompson, C.B. Pyruvate kinase m2 promotes de novo serine synthesis to sustain mtorc1 activity and cell proliferation. Proc. Natl. Acad. Sci. USA 2012, 109, 6904–6909. [Google Scholar] [CrossRef] [PubMed]
- Anastasiou, D.; Poulogiannis, G.; Asara, J.M.; Boxer, M.B.; Jiang, J.K.; Shen, M.; Bellinger, G.; Sasaki, A.T.; Locasale, J.W.; Auld, D.S.; et al. Inhibition of pyruvate kinase m2 by reactive oxygen species contributes to cellular antioxidant responses. Science 2011, 334, 1278–1283. [Google Scholar] [CrossRef] [PubMed]
- Labuschagne, C.F.; van den Broek, N.J.; Mackay, G.M.; Vousden, K.H.; Maddocks, O.D. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 2014, 7, 1248–1258. [Google Scholar] [CrossRef] [PubMed]
- Maddocks, O.D.; Berkers, C.R.; Mason, S.M.; Zheng, L.; Blyth, K.; Gottlieb, E.; Vousden, K.H. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 2013, 493, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Maddocks, O.D.; Labuschagne, C.F.; Adams, P.D.; Vousden, K.H. Serine metabolism supports the methionine cycle and DNA/rna methylation through de novo atp synthesis in cancer cells. Mol. Cell 2016, 61, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Maddocks, O.D.K.; Athineos, D.; Cheung, E.C.; Lee, P.; Zhang, T.; van den Broek, N.J.F.; Mackay, G.M.; Labuschagne, C.F.; Gay, D.; Kruiswijk, F.; et al. Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 2017, 544, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, M.A.; Eaton, J.S.; Shadel, G.S. Loss of p53 causes mitochondrial DNA depletion and altered mitochondrial reactive oxygen species homeostasis. Biochim. Biophys. Acta 2009, 1787, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Kulawiec, M.; Ayyasamy, V.; Singh, K.K. P53 regulates mtdna copy number and mitocheckpoint pathway. J. Carcinog. 2009, 8, 8. [Google Scholar] [PubMed]
- Bourdon, A.; Minai, L.; Serre, V.; Jais, J.P.; Sarzi, E.; Aubert, S.; Chretien, D.; de Lonlay, P.; Paquis-Flucklinger, V.; Arakawa, H.; et al. Mutation of rrm2b, encoding p53-controlled ribonucleotide reductase (p53r2), causes severe mitochondrial DNA depletion. Nat. Genet. 2007, 39, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Matoba, S.; Kang, J.G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. P53 regulates mitochondrial respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef] [PubMed]
- Stambolsky, P.; Weisz, L.; Shats, I.; Klein, Y.; Goldfinger, N.; Oren, M.; Rotter, V. Regulation of aif expression by p53. Cell Death Differ. 2006, 13, 2140–2149. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Kachhap, S.; Singh, K.K. Mitochondrial impairment in p53-deficient human cancer cells. Mutagenesis 2003, 18, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Contractor, T.; Harris, C.R. P53 negatively regulates transcription of the pyruvate dehydrogenase kinase pdk2. Cancer Res. 2012, 72, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Boidot, R.; Vegran, F.; Meulle, A.; Le Breton, A.; Dessy, C.; Sonveaux, P.; Lizard-Nacol, S.; Feron, O. Regulation of monocarboxylate transporter mct1 expression by p53 mediates inward and outward lactate fluxes in tumors. Cancer Res. 2012, 72, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Sung, H.J.; Park, J.Y.; Matoba, S.; Hwang, P.M. A pivotal role for p53: Balancing aerobic respiration and glycolysis. J. Bioenerg. Biomembr. 2007, 39, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Tanaka, T.; Poyurovsky, M.V.; Nagano, H.; Mayama, T.; Ohkubo, S.; Lokshin, M.; Hosokawa, H.; Nakayama, T.; Suzuki, Y.; et al. Phosphate-activated glutaminase (gls2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc. Natl. Acad. Sci. USA 2010, 107, 7461–7466. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Xia, Y.; Zweier, J.L.; Kinzler, K.W.; Vogelstein, B. A model for p53-induced apoptosis. Nature 1997, 389, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhang, L.; Hwang, P.M.; Kinzler, K.W.; Vogelstein, B. Puma induces the rapid apoptosis of colorectal cancer cells. Mol. Cell 2001, 7, 673–682. [Google Scholar] [CrossRef]
- Nakano, K.; Vousden, K.H. Puma, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Oda, E.; Ohki, R.; Murasawa, H.; Nemoto, J.; Shibue, T.; Yamashita, T.; Tokino, T.; Taniguchi, T.; Tanaka, N. Noxa, a bh3-only member of the bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000, 288, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Rivera, A.; Maxwell, S.A. The p53-induced gene-6 (proline oxidase) mediates apoptosis through a calcineurin-dependent pathway. J. Biol. Chem. 2005, 280, 29346–29354. [Google Scholar] [CrossRef] [PubMed]
- Forrester, K.; Ambs, S.; Lupold, S.E.; Kapust, R.B.; Spillare, E.A.; Weinberg, W.C.; Felley-Bosco, E.; Wang, X.W.; Geller, D.A.; Tzeng, E.; et al. Nitric oxide-induced p53 accumulation and regulation of inducible nitric oxide synthase expression by wild-type p53. Proc. Natl. Acad. Sci. USA 1996, 93, 2442–2447. [Google Scholar] [CrossRef] [PubMed]
- Subbaramaiah, K.; Chung, W.J.; Michaluart, P.; Telang, N.; Tanabe, T.; Inoue, H.; Jang, M.; Pezzuto, J.M.; Dannenberg, A.J. Resveratrol inhibits cyclooxygenase-2 transcription and activity in phorbol ester-treated human mammary epithelial cells. J. Biol. Chem. 1998, 273, 21875–21882. [Google Scholar] [CrossRef] [PubMed]
- Budanov, A.V.; Sablina, A.A.; Feinstein, E.; Koonin, E.V.; Chumakov, P.M. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial ahpd. Science 2004, 304, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Budanov, A.V.; Karin, M. P53 target genes sestrin1 and sestrin2 connect genotoxic stress and mtor signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Cano, C.E.; Gommeaux, J.; Pietri, S.; Culcasi, M.; Garcia, S.; Seux, M.; Barelier, S.; Vasseur, S.; Spoto, R.P.; Pebusque, M.J.; et al. Tumor protein 53-induced nuclear protein 1 is a major mediator of p53 antioxidant function. Cancer Res. 2009, 69, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Sun, Z.; Wang, X.J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct interaction between nrf2 and p21(cip1/waf1) upregulates the nrf2-mediated antioxidant response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lin, M.; Wu, R.; Wang, X.; Yang, B.; Levine, A.J.; Hu, W.; Feng, Z. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the warburg effect. Proc. Natl. Acad. Sci. USA 2011, 108, 16259–16264. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chaiswing, L.; Velez, J.M.; Batinic-Haberle, I.; Colburn, N.H.; Oberley, T.D.; St Clair, D.K. P53 translocation to mitochondria precedes its nuclear translocation and targets mitochondrial oxidative defense protein-manganese superoxide dismutase. Cancer Res. 2005, 65, 3745–3750. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Mancuso, A.; Wellen, K.E.; Yang, X. Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature 2013, 493, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef] [PubMed]
- Rohrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Levine, A.J. The regulation of energy metabolism and the igf-1/mtor pathways by the p53 protein. Trends Cell Biol. 2010, 20, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hu, W.; de Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The regulation of ampk beta1, tsc2, and pten expression by p53: Stress, cell and tissue specificity, and the role of these gene products in modulating the igf-1-akt-mtor pathways. Cancer Res. 2007, 67, 3043–3053. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. Mtor signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.O.; An, S.; Lee, H.C.; Woo, S.H.; Seo, S.K.; Choe, T.B.; Yoo, D.H.; Lee, S.B.; Um, H.D.; Lee, S.J.; et al. Hypoxic condition- and high cell density-induced expression of redd1 is regulated by activation of hypoxia-inducible factor-1alpha and sp1 through the phosphatidylinositol 3-kinase/akt signaling pathway. Cell Signal 2007, 19, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.L.; Schulze, A. Srebp activity is regulated by mtorc1 and contributes to akt-dependent cell growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Duvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mtor complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Li, E.; Chen, L.; Zhang, Y.; Wei, F.; Liu, J.; Deng, H.; Wang, Y. The creb coactivator crtc2 controls hepatic lipid metabolism by regulating srebp1. Nature 2015, 524, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N.; et al. Mtor complex 1 regulates lipin 1 localization to control the srebp pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef] [PubMed]
- Sundqvist, A.; Bengoechea-Alonso, M.T.; Ye, X.; Lukiyanchuk, V.; Jin, J.; Harper, J.W.; Ericsson, J. Control of lipid metabolism by phosphorylation-dependent degradation of the srebp family of transcription factors by scf(fbw7). Cell Metab. 2005, 1, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Yahagi, N.; Shimano, H.; Matsuzaka, T.; Najima, Y.; Sekiya, M.; Nakagawa, Y.; Ide, T.; Tomita, S.; Okazaki, H.; Tamura, Y.; et al. P53 activation in adipocytes of obese mice. J. Biol. Chem. 2003, 278, 25395–25400. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerod, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [PubMed]
- Buzzai, M.; Jones, R.G.; Amaravadi, R.K.; Lum, J.J.; DeBerardinis, R.J.; Zhao, F.; Viollet, B.; Thompson, C.B. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007, 67, 6745–6752. [Google Scholar] [CrossRef] [PubMed]
- Assaily, W.; Rubinger, D.A.; Wheaton, K.; Lin, Y.; Ma, W.; Xuan, W.; Brown-Endres, L.; Tsuchihara, K.; Mak, T.W.; Benchimol, S. Ros-mediated p53 induction of lpin1 regulates fatty acid oxidation in response to nutritional stress. Mol. Cell 2011, 44, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.; Satija, Y.K.; Das, S. Pgc-1alpha, a key modulator of p53, promotes cell survival upon metabolic stress. Mol. Cell 2011, 44, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lin, J.D. Pgc-1 coactivators in the control of energy metabolism. Acta Biochim. Biophys. Sin. (Shanghai) 2011, 43, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Zaugg, K.; Yao, Y.; Reilly, P.T.; Kannan, K.; Kiarash, R.; Mason, J.; Huang, P.; Sawyer, S.K.; Fuerth, B.; Faubert, B.; et al. Carnitine palmitoyltransferase 1c promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 2011, 25, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Macedo, N.; Feng, J.; Faubert, B.; Chang, N.; Elia, A.; Rushing, E.J.; Tsuchihara, K.; Bungard, D.; Berger, S.L.; Jones, R.G.; et al. Depletion of the novel p53-target gene carnitine palmitoyltransferase 1c delays tumor growth in the neurofibromatosis type i tumor model. Cell Death Differ. 2013, 20, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, I.; Rotter, V. Regulation of lipid metabolism by p53 - fighting two villains with one sword. Trends Endocrinol. Metab. 2012, 23, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A.; Akkari, L.; Simon, J.; Grace, D.; Tschaharganeh, D.F.; Bolden, J.E.; Zhao, Z.; Thapar, V.; Joyce, J.A.; Krizhanovsky, V.; et al. Non-cell-autonomous tumor suppression by p53. Cell 2013, 153, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Schwitalla, S.; Ziegler, P.K.; Horst, D.; Becker, V.; Kerle, I.; Begus-Nahrmann, Y.; Lechel, A.; Rudolph, K.L.; Langer, R.; Slotta-Huspenina, J.; et al. Loss of p53 in enterocytes generates an inflammatory microenvironment enabling invasion and lymph node metastasis of carcinogen-induced colorectal tumors. Cancer Cell 2013, 23, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.P.; Wang, W.L.; Chang, Y.L.; Wu, C.T.; Chao, Y.C.; Kao, S.H.; Yuan, A.; Lin, C.W.; Yang, S.C.; Chan, W.K.; et al. P53 controls cancer cell invasion by inducing the mdm2-mediated degradation of slug. Nat. Cell Biol. 2009, 11, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P.; et al. Mutant p53 drives invasion by promoting integrin recycling. Cell 2009, 139, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. Amp-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Dameron, K.M.; Volpert, O.V.; Tainsky, M.A.; Bouck, N. Control of angiogenesis in fibroblasts by p53 regulation of thrombospondin-1. Science 1994, 265, 1582–1584. [Google Scholar] [CrossRef] [PubMed]
- Futamura, M.; Kamino, H.; Miyamoto, Y.; Kitamura, N.; Nakamura, Y.; Ohnishi, S.; Masuda, Y.; Arakawa, H. Possible role of semaphorin 3f, a candidate tumor suppressor gene at 3p21.3, in p53-regulated tumor angiogenesis suppression. Cancer Res. 2007, 67, 1451–1460. [Google Scholar] [CrossRef] [PubMed]
- Farhang Ghahremani, M.; Goossens, S.; Nittner, D.; Bisteau, X.; Bartunkova, S.; Zwolinska, A.; Hulpiau, P.; Haigh, K.; Haenebalcke, L.; Drogat, B.; et al. P53 promotes vegf expression and angiogenesis in the absence of an intact p21-rb pathway. Cell Death Differ. 2013, 20, 888–897. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Marrero, L.; Rodriguez, P.; Del Valle, L.; Ochoa, A.; Cui, Y. Trp53 inactivation in the tumor microenvironment promotes tumor progression by expanding the immunosuppressive lymphoid-like stromal network. Cancer Res. 2013, 73, 1668–1675. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zheng, M.; Kibe, R.; Huang, Y.; Marrero, L.; Warren, S.; Zieske, A.W.; Iwakuma, T.; Kolls, J.K.; Cui, Y. Trp53 negatively regulates autoimmunity via the stat3-th17 axis. FASEB J. 2011, 25, 2387–2398. [Google Scholar] [CrossRef] [PubMed]
- Menendez, D.; Shatz, M.; Resnick, M.A. Interactions between the tumor suppressor p53 and immune responses. Curr. Opin. Oncol. 2013, 25, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Komarova, E.A.; Krivokrysenko, V.; Wang, K.; Neznanov, N.; Chernov, M.V.; Komarov, P.G.; Brennan, M.L.; Golovkina, T.V.; Rokhlin, O.W.; Kuprash, D.V.; et al. P53 is a suppressor of inflammatory response in mice. FASEB J. 2005, 19, 1030–1032. [Google Scholar] [CrossRef] [PubMed]
- Martins, C.P.; Brown-Swigart, L.; Evan, G.I. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell 2006, 127, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Kaplon, J.; Zheng, L.; Meissl, K.; Chaneton, B.; Selivanov, V.A.; Mackay, G.; van der Burg, S.H.; Verdegaal, E.M.; Cascante, M.; Shlomi, T.; et al. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 2013, 498, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.L.; Poffenberger, M.C.; Chang, C.H.; Jones, R.G. Fueling immunity: Insights into metabolism and lymphocyte function. Science 2013, 342, 1242454. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Khoo, K.H.; Verma, C.S.; Lane, D.P. Drugging the p53 pathway: Understanding the route to clinical efficacy. Nat. Rev. Drug Discov. 2014, 13, 217–236. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of mdm2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Zache, N.; Lambert, J.M.; Wiman, K.G.; Bykov, V.J. Prima-1met inhibits growth of mouse tumors carrying mutant p53. Cell Oncol. 2008, 30, 411–418. [Google Scholar] [PubMed]
- Cesar-Razquin, A.; Snijder, B.; Frappier-Brinton, T.; Isserlin, R.; Gyimesi, G.; Bai, X.; Reithmeier, R.A.; Hepworth, D.; Hediger, M.A.; Edwards, A.M.; et al. A call for systematic research on solute carriers. Cell 2015, 162, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Oronsky, B.; Scicinski, J.; Reid, T.; Oronsky, A.; Carter, C.; Oronsky, N.; Cabrales, P. Rrx-001, a novel clinical-stage chemosensitizer, radiosensitizer, and immunosensitizer, inhibits glucose 6-phosphate dehydrogenase in human tumor cells. Discov. Med. 2016, 21, 251–265. [Google Scholar] [PubMed]
- Thurnher, M.; Nussbaumer, O.; Gruenbacher, G. Novel aspects of mevalonate pathway inhibitors as antitumor agents. Clin. Cancer Res. 2012, 18, 3524–3531. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flöter, J.; Kaymak, I.; Schulze, A. Regulation of Metabolic Activity by p53. Metabolites 2017, 7, 21. https://doi.org/10.3390/metabo7020021
Flöter J, Kaymak I, Schulze A. Regulation of Metabolic Activity by p53. Metabolites. 2017; 7(2):21. https://doi.org/10.3390/metabo7020021
Chicago/Turabian StyleFlöter, Jessica, Irem Kaymak, and Almut Schulze. 2017. "Regulation of Metabolic Activity by p53" Metabolites 7, no. 2: 21. https://doi.org/10.3390/metabo7020021
APA StyleFlöter, J., Kaymak, I., & Schulze, A. (2017). Regulation of Metabolic Activity by p53. Metabolites, 7(2), 21. https://doi.org/10.3390/metabo7020021