Recent Advances in Understanding the Complexity of Alcohol-Induced Pancreatic Dysfunction and Pancreatitis Development

 , , and
, , and 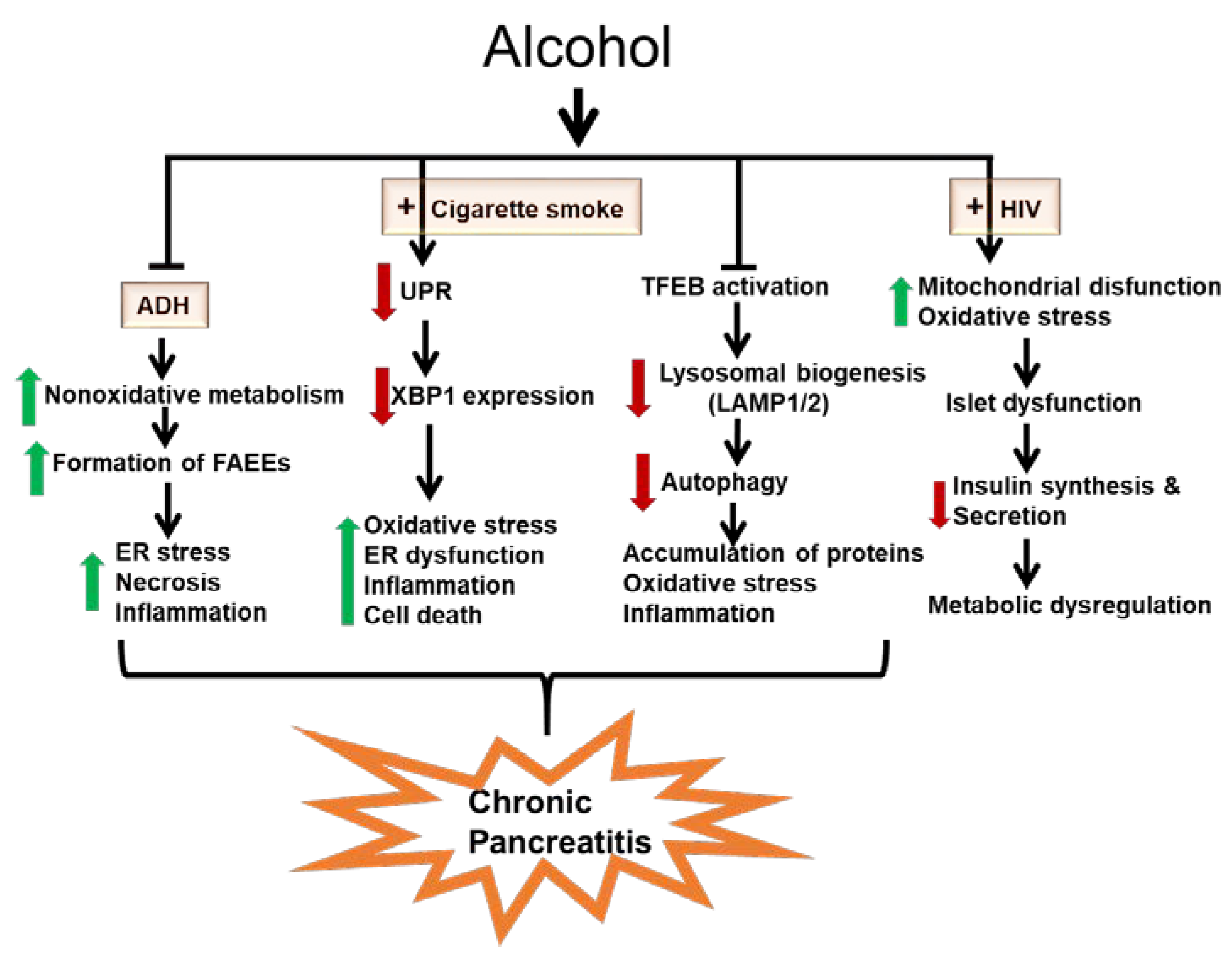{kind=link}
Abstract
:1. Introduction
- (i)
- Metabolic basis for alcoholic pancreatitis
- (ii)
- Role of the UPR in the development of alcoholic pancreatitis
- (iii)
- Role of impaired autophagy in the pathogenesis of alcoholic pancreatitis
- (iv)
- Chronic alcohol consumption dysregulates pancreatic endocrine function and exacerbates metabolic alterations in people living with human immunodeficiency virus (HIV)
2. Metabolic Basis for Alcoholic Pancreatitis
3. Unfolded Protein Response in Pancreatitis Development
4. Impaired Autophagy-Lysosomal Pathway in the Pathogenesis of Pancreatitis
5. Chronic Alcohol Consumption Dysregulates Pancreatic Endocrine Function and Exacerbates Metabolic Alterations in People Living with HIV
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Vladimir, P.; Dag, R. Global Status Report on Alcohol and Health 2018; Licence: CC BY-NC-SA 3.0 IGO; World Health Organization: Geneva, Switzerland, 2018; ISBN 978-92-4-156563-9. [Google Scholar]
- Sacks, J.J.; Gonzales, K.R.; Bouchery, E.E.; Tomedi, L.E.; Brewer, R.D. 2010 National and State Costs of Excessive Alcohol Consumption. Am. J. Prev. Med. 2015, 49, e73–e79. [Google Scholar] [CrossRef] [PubMed]
- Laposata, E.; Lange, L. Presence of nonoxidative ethanol metabolism in human organs commonly damaged by ethanol abuse. Science 1986, 231, 497–499. [Google Scholar] [CrossRef] [PubMed]
- Lankisch, P.G. Diagnosis of chronic pancreatitis. Lancet 1998, 351, 599. [Google Scholar] [CrossRef]
- Kim, J.Y.; Song, E.H.; Lee, H.J.; Oh, Y.K.; Park, Y.S.; Park, J.W.; Kim, B.J.; Kim, D.J.; Lee, I.; Song, J.; et al. Chronic ethanol consumption-induced pancreatic {Al-Daghri, #37}-cell dysfunction and apoptosis through glucokinase nitration and its down-regulation. J. Biol. Chem. 2010, 285, 37251–37262. [Google Scholar] [PubMed] [Green Version]
- Dembele, K.; Nguyen, K.H.; Hernandez, T.A.; Nyomba, B.L.G. Effects of ethanol on pancreatic beta-cell death: Interaction with glucose and fatty acids. Cell Boil. Toxicol. 2008, 25, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Gukovskaya, A.; Pandol, S.J.; Gukovsky, I. New insights into the pathways initiating and driving pancreatitis. Curr. Opin. Gastroenterol. 2016, 32, 429–435. [Google Scholar] [CrossRef]
- Gukovskaya, A.; Gorelick, F.S.; Groblewski, G.E.; Mareninova, O.A.; Lugea, A.; Antonucci, L.; Waldron, R.T.; Habtezion, A.; Karin, M.; Pandol, S.J.; et al. Recent Insights into the Pathogenic Mechanism of Pancreatitis. Pancreas 2019, 48, 459–470. [Google Scholar] [CrossRef]
- Bonnet, F.; Disse, E.; Laville, M.; Mari, A.; Hojlund, K.; Anderwald, C.H.; Piatti, P.; Balkau, B.; RISC Study Group. Moderate alcohol consumption is associated with improved insulin sensitivity, reduced basal insulin secretion rate and lower fasting glucagon concentration in healthy women. Diabetologia 2012, 55, 3228–3237. [Google Scholar] [CrossRef] [Green Version]
- Patto, R.J.; Russo, E.K.; Borges, D.R.; Neves, M.M. The enteroinsular axis and endocrine pancreatic function in chronic alcohol consumers: Evidence for early beta-cell hypofunction. Mt. Sinai J. Med. A J. Transl. Pers. Med. 1993, 60, 317–320. [Google Scholar]
- Rasineni, K.; Thomes, P.G.; Kubik, J.L.; Harris, E.N.; Kharbanda, K.K.; Casey, C.A. Chronic alcohol exposure alters circulating insulin and ghrelin levels: Role of ghrelin in hepatic steatosis. Am. J. Physiol. Liver Physiol. 2019, 316, G453–G461. [Google Scholar] [CrossRef]
- Steiner, J.L.; Crowell, K.; Lang, C.H. Impact of Alcohol on Glycemic Control and Insulin Action. Biomolecules 2015, 5, 2223–2246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasineni, K.; Kubik, J.L.; Casey, C.A.; Kharbanda, K.K. Inhibition of Ghrelin Activity by Receptor Antagonist [d-Lys-3] GHRP-6 Attenuates Alcohol-Induced Hepatic Steatosis by Regulating Hepatic Lipid Metabolism. Biomolecules 2019, 9, 517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Luo, Y.; Feng, A.; Li, T.; Yang, X.; Nofech-Mozes, R.; Yu, M.; Wang, C.; Li, Z.; Yi, F.; et al. Ethanol induced impairment of glucose metabolism involves alterations of gabaergic signaling in pancreatic beta-cells. Toxicology 2014, 326, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, D.; Micic, A.; Dimitrijevic-Sreckovic, V.; Kerkez, M.; Nikolic, B. Effect of alcohol on insulin secretion and viability of human pancreatic islets. Srp. Arh. za Celok. Lek. 2017, 145, 159–164. [Google Scholar] [CrossRef]
- Nguyen, K.H.; Lee, J.H.; Nyomba, B.L. Ethanol causes endoplasmic reticulum stress and impairment of insulin secretion in pancreatic beta-cells. Alcohol 2012, 46, 89–99. [Google Scholar] [CrossRef]
- Apte, M.V.; Wilson, J.; A Korsten, M.; McCaughan, G.W.; Haber, P.S.; Pirola, R.C. Effects of ethanol and protein deficiency on pancreatic digestive and lysosomal enzymes. Gut 1995, 36, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Cosen-Binker, L.I.; Binker, M.G.; Wang, C.-C.; Hong, W.; Gaisano, H.Y. VAMP8 is the v-SNARE that mediates basolateral exocytosis in a mouse model of alcoholic pancreatitis. J. Clin. Investig. 2008, 118, 2535–2551. [Google Scholar] [CrossRef] [Green Version]
- Lugea, A.; Tischler, D.; Nguyen, J.; Gong, J.; Gukovsky, I.; French, S.W.; Gorelick, F.S.; Pandol, S.J. Adaptive unfolded protein response attenuates alcohol-induced pancreatic damage. Gastroenterology 2010, 140, 987–997. [Google Scholar] [CrossRef] [Green Version]
- Lugea, A.; Waldron, R.T.; Mareninova, O.A.; Shalbueva, N.; Deng, N.; Su, H.-Y.; Thomas, D.D.; Jones, E.; Messenger, S.W.; Yang, J.; et al. Human Pancreatic Acinar Cells. Am. J. Pathol. 2017, 187, 2726–2743. [Google Scholar] [CrossRef] [Green Version]
- Orabi, A.I.; Shah, A.U.; Muili, K.; Luo, Y.; Mahmood, S.M.; Ahmad, A.; Reed, A.; Husain, S.Z. Ethanol Enhances Carbachol-induced Protease Activation and Accelerates Ca2+ Waves in Isolated Rat Pancreatic Acini*. J. Boil. Chem. 2011, 286, 14090–14097. [Google Scholar] [CrossRef] [Green Version]
- Haber, P.S.; Wilson, J.; Apte, M.V.; A Korsten, M.; Pirola, R.C. Chronic ethanol consumption increases the fragility of rat pancreatic zymogen granules. Gut 1994, 35, 1474–1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gukovskaya, A.; Gukovsky, I.; Algül, H.; Habtezion, A. Autophagy, Inflammation, and Immune Dysfunction in the Pathogenesis of Pancreatitis. Gastroenterology 2017, 153, 1212–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerch, M.M.; Gorelick, F.S. Models of Acute and Chronic Pancreatitis. Gastroenterology 2013, 144, 1180–1193. [Google Scholar] [CrossRef] [PubMed]
- Kaphalia, B.S.; Bhopale, K.K.; Kondraganti, S.; Wu, H.; Boor, P.J.; Ansari, G.S. Pancreatic injury in hepatic alcohol dehydrogenase-deficient deer mice after subchronic exposure to ethanol. Toxicol. Appl. Pharmacol. 2010, 246, 154–162. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Forsmark, C.E. Chronic pancreatitis. Curr. Opin. Gastroenterol. 2017, 33, 396–403. [Google Scholar] [CrossRef]
- Lew, D.; Afghani, E.; Pandol, S. Chronic Pancreatitis: Current Status and Challenges for Prevention and Treatment. Dig. Dis. Sci. 2017, 62, 1702–1712. [Google Scholar] [CrossRef]
- Irving, H.M.; Samokhvalov, A.V.; Rehm, J. Alcohol as a risk factor for pancreatitis. A systematic review and meta-analysis. JOP J. pancreas 2009, 10, 387–392. [Google Scholar]
- Setiawan, V.W.; Monroe, K.; Lugea, A.; Yadav, D.; Pandol, S. Uniting Epidemiology and Experimental Disease Models for Alcohol-Related Pancreatic Disease. Alcohol Res. Curr. Rev. 2017, 38, 173–182. [Google Scholar]
- Yadav, D.; Lowenfels, A.B. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology 2013, 144, 1252–1261. [Google Scholar] [CrossRef] [Green Version]
- Yadav, D.; Hawes, R.H.; E Brand, R.; Anderson, M.A.; Money, M.E.; Banks, P.A.; Bishop, M.D.; Baillie, J.; Sherman, S.; DiSario, J.; et al. Alcohol consumption, cigarette smoking, and the risk of recurrent acute and chronic pancreatitis. Arch. Intern. Med. 2009, 169, 1035–1045. [Google Scholar] [CrossRef] [Green Version]
- Jalaly, N.Y.; A Moran, R.; Fargahi, F.; A Khashab, M.; Kamal, A.; Lennon, A.M.; Walsh, C.; A Makary, M.; Whitcomb, D.C.; Yadav, D.; et al. An Evaluation of Factors Associated With Pathogenic PRSS1, SPINK1, CTFR, and/or CTRC Genetic Variants in Patients With Idiopathic Pancreatitis. Am. J. Gastroenterol. 2017, 112, 1320–1329. [Google Scholar] [CrossRef] [PubMed]
- Whitcomb, D.C. Genetic risk factors for pancreatic disorders. Gastroenterology 2013, 144, 1292–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitcomb, D.C.; Alzheimer’s Disease Genetics Consortium; LaRusch, J.; Krasinskas, A.M.; Klei, L.; Smith, J.P.; Brand, R.E.; Neoptolemos, J.P.; Lerch, M.M.; Tector, M.; et al. Common genetic variants in the CLDN2 and PRSS1-PRSS2 loci alter risk for alcohol-related and sporadic pancreatitis. Nat. Genet. 2012, 44, 1349–1354. [Google Scholar] [CrossRef] [Green Version]
- Yadav, D.; Park, W.G.; Fogel, E.L.; Li, L.; Chari, S.T.; Feng, Z.; Fisher, W.E.; Forsmark, C.E.; Jeon, C.Y.; Habtezion, A.; et al. PROspective Evaluation of Chronic Pancreatitis for EpidEmiologic and Translational StuDies. Pancreas 2018, 47, 1229–1238. [Google Scholar] [CrossRef] [PubMed]
- Vonlaufen, A.; Wilson, J.S.; Pirola, R.C.; Apte, M.V. Role of Alcohol Metabolism in Chronic Pancreatitis. Alcohol Res. Heal. J. Natl. Inst. Alcohol Abus. Alcohol. 2007, 30, 48–54. [Google Scholar]
- Alsamarrai, A.; Das, S.L.; Windsor, J.; Petrov, M.S. Factors That Affect Risk for Pancreatic Disease in the General Population: A Systematic Review and Meta-analysis of Prospective Cohort Studies. Clin. Gastroenterol. Hepatol. 2014, 12, 1635–1644. [Google Scholar] [CrossRef]
- Nuutinen, H.; Lindros, K.O.; Salaspuro, M. Determinants of Blood Acetaldehyde Level during Ethanol Oxidation in Chronic Alcoholics. Alcohol. Clin. Exp. Res. 1983, 7, 163–168. [Google Scholar] [CrossRef]
- Panés, J.; Caballería, J.; Guitart, R.; Pares, A.; Soler, X.; Rodamilans, M.; Navasa, M.; Pares, X.; Bosch, J.; Rodés, J. Determinants of Ethanol and Acetaldehyde Metabolism in Chronic Alcoholics. Alcohol. Clin. Exp. Res. 1993, 17, 48–53. [Google Scholar] [CrossRef]
- Kaphalia, B.S.; Khan, M.F.; Caroll, R.M.; Aronson, J.; Ansari, G.A. Subchronic toxicity of 2-chloroethanol and 2-bromoethanol in rats. Res. Commun. Pharmacol. Toxicol. 1996, 1, 173–186. [Google Scholar]
- Haber, P.; Apte, M.V.; Applegate, T.L.; Norton, I.D.; Korsten, M.A.; Pirola, R.C.; Wilson, J. Metabolism of ethanol by rat pancreatic acinar cells. J. Lab. Clin. Med. 1998, 132, 294–302. [Google Scholar] [CrossRef]
- Gukovskaya, A.S.; Mouria, M.; Gukovsky, I.; Reyes, C.N.; Kasho, V.N.; Faller, L.D.; Pandol, S.J. Ethanol metabolism and transcription factor activation in pancreatic acinar cells in rats. Gastroenterology 2002, 122, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Werner, J.; Saghir, M.; Warshaw, A.L.; Lewandrowski, K.B.; Laposata, M.; Iozzo, R.V.; Carter, E.A.; Schatz, R.J.; Castillo, C.F.-D. Alcoholic pancreatitis in rats: Injury from nonoxidative metabolites of ethanol. Am. J. Physiol. Liver Physiol. 2002, 283, G65–G73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyasaka, K.; Ohta, M.; Takano, S.; Hayashi, H.; Higuchi, S.; Maruyama, K.; Tando, Y.; Nakamura, T.; Takata, Y.; Funakoshi, A. Carboxylester lipase gene polymorphism as a risk of alcohol-induced pancreatitis. Pancreas 2005, 30, e87–e91. [Google Scholar] [CrossRef] [PubMed]
- Kaphalia, B.S.; Cai, P.; Khan, M.F.; Okorodudu, A.O.; Ansari, G. Fatty acid ethyl esters: Markers of alcohol abuse and alcoholism. Alcohol 2004, 34, 151–158. [Google Scholar] [CrossRef]
- Shalbueva, N.; Mareninova, O.A.; Gerloff, A.; Yuan, J.; Waldron, R.T.; Pandol, S.J.; Gukovskaya, A. Effects of oxidative alcohol metabolism on the mitochondrial permeability transition pore and necrosis in a mouse model of alcoholic pancreatitis. Gastroenterology 2012, 144, 437–446.e6. [Google Scholar] [CrossRef] [Green Version]
- Dolai, S.; Liang, T.; Lam, P.P.; Fernandez, N.; Chidambaram, S.; Gaisano, H.Y. Effects of Ethanol Metabolites on Exocytosis of Pancreatic Acinar Cells in Rats. Gastroenterology 2012, 143, 832–843.e7. [Google Scholar] [CrossRef]
- Apte, M.V.; Phillips, P.A.; Fahmy, R.G.; Darby, S.J.; Rodgers, S.C.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Naidoo, D.; Wilson, J. Does alcohol directly stimulate pancreatic fibrogenesis? Studies with rat pancreatic stellate cells. Gastroenterology 2000, 118, 780–794. [Google Scholar] [CrossRef]
- Chiang, C.-P.; Wu, C.-W.; Lee, S.-P.; Chung, C.-C.; Wang, C.-W.; Lee, S.-L.; Nieh, S.; Yin, S.-J. Expression Pattern, Ethanol-Metabolizing Activities, and Cellular Localization of Alcohol and Aldehyde Dehydrogenases in Human Pancreas: Implications for Pathogenesis of Alcohol-Induced Pancreatic Injury. Alcohol. Clin. Exp. Res. 2009, 33, 1059–1068. [Google Scholar] [CrossRef]
- Vonlaufen, A.; Xu, Z.; Daniel, B.; Kumar, R.; Pirola, R.; Wilson, J.; Apte, M.V. Bacterial Endotoxin: A Trigger Factor for Alcoholic Pancreatitis? Evidence from a Novel, Physiologically Relevant Animal Model. Gastroenterology 2007, 133, 1293–1303. [Google Scholar] [CrossRef]
- Lee, A.T.K.; Xu, Z.; Pothula, S.; Patel, M.B.; Pirola, R.C.; Wilson, J.; Apte, M.V. Alcohol and Cigarette Smoke Components Activate Human Pancreatic Stellate Cells: Implications for the Progression of Chronic Pancreatitis. Alcohol. Clin. Exp. Res. 2015, 39, 2123–2133. [Google Scholar] [CrossRef]
- Hu, R.; Wang, Y.-L.; Edderkaoui, M.; Lugea, A.; Apte, M.V.; Pandol, S.J. Ethanol augments PDGF-induced NADPH oxidase activity and proliferation in rat pancreatic stellate cells. Pancreatology 2007, 7, 332–340. [Google Scholar] [CrossRef] [Green Version]
- Apte, M.V.; Wilson, J.; Lugea, A.; Pandol, S. A starring role for stellate cells in the pancreatic cancer microenvironment. Gastroenterology 2013, 144, 1210–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omary, B.; Lugea, A.; Lowe, A.W.; Pandol, S.J. The pancreatic stellate cell: A star on the rise in pancreatic diseases. J. Clin. Investig. 2007, 117, 50–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfützer, R.H.; Tadic, S.D.; Li, H.-S.; Thompson, B.S.; Zhang, J.-Y.; Ford, M.E.; Eagon, P.K.; Whitcomb, D.C. Pancreatic Cholesterol Esterase, ES-10, and Fatty Acid Ethyl Ester Synthase III Gene Expression are Increased in the Pancreas and Liver but Not in the Brain or Heart with Long-term Ethanol Feeding in Rats. Pancreas 2002, 25, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Booth, D.M.; Cane, M.; Chvanov, M.; A Javed, M.; Elliott, V.L.; A Armstrong, J.; Dingsdale, H.; Cash, N.; Li, Y.; et al. Fatty acid ethyl ester synthase inhibition ameliorates ethanol-induced Ca2+-dependent mitochondrial dysfunction and acute pancreatitis. Gut 2013, 63, 1313–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amer, S.M.; Bhopale, K.K.; Kakumanu, R.D.; Popov, V.L.; Rampy, B.A.; El-Mehallawi, I.H.; Ashmawy, M.M.; Ansari, G.A.S.; Kaphalia, B.S. Hepatic alcohol dehydrogenase deficiency induces pancreatic injury in chronic ethanol feeding model of deer mice. Exp. Mol. Pathol. 2018, 104, 89–97. [Google Scholar] [CrossRef]
- Wu, H.; Bhopale, K.K.; Ansari, G.A.S.; Kaphalia, B.S. Ethanol-induced cytotoxicity in rat pancreatic acinar AR42J cells: Role of fatty acid ethyl esters. Alcohol Alcohol. 2007, 43, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Bhopale, K.K.; Falzon, M.; Ansari, G.A.S.; Kaphalia, B.S. Alcohol oxidizing enzymes and ethanol-induced cytotoxicity in rat pancreatic acinar AR42J cells. Vitr. Cell. Dev. Boil. - Anim. 2013, 50, 373–380. [Google Scholar] [CrossRef]
- Pandol, S.; Gorelick, F.S.; Lugea, A.P. Environmental and Genetic Stressors and the Unfolded Protein Response in Exocrine Pancreatic Function – A Hypothesis. Front. Physiol. 2011, 2, 8. [Google Scholar] [CrossRef] [Green Version]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Tirasophon, W.; Shen, X.; Michalak, M.; Prywes, R.; Okada, T.; Yoshida, H.; Mori, K.; Kaufman, R.J. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genome Res. 2002, 16, 452–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA Is Induced by ATF6 and Spliced by IRE1 in Response to ER Stress to Produce a Highly Active Transcription Factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Matsui, T.; Hosokawa, N.; Kaufman, R.J.; Nagata, K.; Mori, K. A time-dependent phase shift in the mammalian unfolded protein response. Dev. Cell 2003, 4, 265–271. [Google Scholar] [CrossRef] [Green Version]
- Acosta-Alvear, D.; Zhou, Y.; Blais, A.; Tsikitis, M.; Lents, N.H.; Arias, C.; Lennon, C.J.; Kluger, Y.; Dynlacht, B.D. XBP1 Controls Diverse Cell Type- and Condition-Specific Transcriptional Regulatory Networks. Mol. Cell 2007, 27, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Scheuner, N.; Ron, D.; Pennathur, S.; Kaufman, R.J. Chop deletion reduces oxidative stress, improves β cell function, and promotes cell survival in multiple mouse models of diabetes. J. Clin. Investig. 2008, 118, 3378–3389. [Google Scholar] [CrossRef] [Green Version]
- Zinszner, H.; Kuroda, M.; Wang, X.; Batchvarova, N.; Lightfoot, R.T.; Remotti, H.; Stevens, J.L.; Ron, D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genome Res. 1998, 12, 982–995. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Proics, E.; De Bieville, C.H.D.; Rousseau, D.; Bonnafous, S.; Patouraux, S.; Adam, G.; Lavallard, V.; Rovere, C.; Le Thuc, O.; et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. 2015, 6, e1879. [Google Scholar] [CrossRef] [Green Version]
- Lei, Y.; Wang, S.; Ren, B.; Wang, J.; Chen, J.; Lu, J.; Zhan, S.; Fu, Y.; Huang, L.; Tan, J. CHOP favors endoplasmic reticulum stress-induced apoptosis in hepatocellular carcinoma cells via inhibition of autophagy. PLoS ONE 2017, 12, e0183680. [Google Scholar] [CrossRef] [Green Version]
- Su, H.-Y.; Waldron, R.T.; Deng, N.; Liu, Z.; Pandol, S.; Lugea, A. 317 - Deficient Unfolded Protein Response (UPR) in Adult Pancreatic Acinar Cells Results in Significant Reprogramming in Genes Related with Mitochondrial Function. Gastroenterology 2018, 154. [Google Scholar] [CrossRef]
- Waldron, R.T.; Su, H.-Y.; Piplani, H.; Capri, J.; Cohn, W.; Whitelegge, J.P.; Faull, K.F.; Sakkiah, S.; Abrol, R.; Yang, W.; et al. Ethanol Induced Disordering of Pancreatic Acinar Cell Endoplasmic Reticulum: An ER Stress/Defective Unfolded Protein Response Model. Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 479–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lugea, A.; Gerloff, A.; Su, H.-Y.; Xu, Z.; Go, A.; Hu, C.; French, S.W.; Wilson, J.S.; Apte, M.V.; Waldron, R.T.; et al. The Combination of Alcohol and Cigarette Smoke Induces Endoplasmic Reticulum Stress and Cell Death in Pancreatic Acinar Cells. Gastroenterology 2017, 153, 1674–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steingrimsson, E.; Copeland, N.G.; Jenkins, N.A. Melanocytes and theMicrophthalmiaTranscription Factor Network. Annu. Rev. Genet. 2004, 38, 365–411. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef] [Green Version]
- Sardiello, M.; Palmieri, M.; Di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A Gene Network Regulating Lysosomal Biogenesis and Function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia-Arencibia, M.; Vetrini, F.; Erdin, S.; Huynh, T.; Medina, D.L.; Colella, P.; Sardiello, M.; et al. TFEB Links Autophagy to Lysosomal Biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Boil. 2013, 14, 283–296. [Google Scholar] [CrossRef] [Green Version]
- Vega-Rubin-De-Celis, S.; Peña-Llopis, S.; Konda, M.; Brugarolas, J. Multistep regulation of TFEB by MTORC1. Autophagy 2017, 13, 464–472. [Google Scholar] [CrossRef] [Green Version]
- Ferron, M.; Settembre, C.; Shimazu, J.; Lacombe, J.; Kato, S.; Rawlings, D.J.; Ballabio, A.; Karsenty, G. A RANKL-PKCBETA-TFEB signaling cascade is necessary for lysosomal biogenesis in osteoclasts. Genes Dev. 2013, 27, 955–969. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Ni, H.-M.; Chao, X.; Wang, H.; Bridges, B.; Kumer, S.; Schmitt, T.; Mareninova, O.; Gukovskaya, A.; De Lisle, R.C.; et al. Impaired TFEB-mediated lysosomal biogenesis promotes the development of pancreatitis in mice and is associated with human pancreatitis. Autophagy 2019, 15, 1954–1969. [Google Scholar] [CrossRef]
- Wang, S.; Ding, W.-X. Does Autophagy Promote or Protect Against the Pathogenesis of Pancreatitis? Gastroenterology 2018, 155, 1273–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gukovskaya, A.; Gukovsky, I. Autophagy and pancreatitis. Am. J. Physiol. Liver Physiol. 2012, 303, G993–G1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lugea, A.; Gong, J.; Nguyen, J.; Nieto, J.; French, S.W.; Pandol, S.J. Cholinergic mediation of alcohol-induced experimental pancreatitis. Alcohol. Clin. Exp. Res. 2010, 34, 1768–1781. [Google Scholar] [CrossRef] [Green Version]
- Mareninova, O.A.; Sendler, M.; Malla, S.R.; Yakubov, I.; French, S.W.; Tokhtaeva, E.; Vagin, O.; Oorschot, V.; Lüllmann-Rauch, R.; Blanz, J.; et al. Lysosome-Associated Membrane Proteins (LAMP) Maintain Pancreatic Acinar Cell Homeostasis: LAMP-2–Deficient Mice Develop Pancreatitis. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 678–694. [Google Scholar] [CrossRef] [Green Version]
- Fortunato, F.; Bürgers, H.; Bergmann, F.; Rieger, P.; Büchler, M.W.; Kroemer, G.; Werner, J. Impaired Autolysosome Formation Correlates With Lamp-2 Depletion: Role of Apoptosis, Autophagy, and Necrosis in Pancreatitis. Gastroenterology 2009, 137, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Diakopoulos, K.N.; Lesina, M.; Wörmann, S.; Song, L.; Aichler, M.; Schild, L.; Artati, A.; Römisch-Margl, W.; Wartmann, T.; Fischer, R.; et al. Impaired Autophagy Induces Chronic Atrophic Pancreatitis in Mice via Sex- and Nutrition-Dependent Processes. Gastroenterology 2015, 148, 626–638.e17. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Yang, F.; Wang, X.; Wang, Y.; Xu, M.; Frank, J.A.; Ke, Z.; Zhang, Z.; Shi, X.; Luo, J. Chronic plus binge ethanol exposure causes more severe pancreatic injury and inflammation. Toxicol. Appl. Pharmacol. 2016, 308, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Ni, H.-M.; Chao, X.; Ma, X.; Kolodecik, T.; De Lisle, R.; Ballabio, A.; Pacher, P.; Ding, W.-X. Critical Role of TFEB-Mediated Lysosomal Biogenesis in Alcohol-Induced Pancreatitis in Mice and Humans. Cell. Mol. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Grasso, D.; Ropolo, A.; Ré, A.L.; Boggio, V.; Molejón, M.I.; Iovanna, J.L.; Gonzalez, C.; Urrutia, R.; Vaccaro, M. Zymophagy, a Novel Selective Autophagy Pathway Mediated by VMP1-USP9x-p62, Prevents Pancreatic Cell Death. J. Boil. Chem. 2010, 286, 8308–8324. [Google Scholar] [CrossRef] [Green Version]
- Halangk, W.; Lerch, M.M.; Brandt-Nedelev, B.; Roth, W.; Ruthenbuerger, M.; Reinheckel, T.; Domschke, W.; Lippert, H.; Peters, C.; Deussing, J.M. Role of cathepsin B in intracellular trypsinogen activation and the onset of acute pancreatitis. J. Clin. Investig. 2000, 106, 773–781. [Google Scholar] [CrossRef] [Green Version]
- Andersen, B.; Hagen, C.; Faber, O.; Lindholm, J.; Boisen, P.; Worning, H. Glucose tolerance and B cell function in chronic alcoholism: Its relation to hepatic histology and exocrine pancreatic function. Metabolism 1983, 32, 1029–1032. [Google Scholar] [CrossRef]
- A Hätönen, K.; Virtamo, J.; Eriksson, J.; Perälä, M.-M.; Sinkko, H.K.; Leiviskä, J.; Valsta, L.M. Modifying effects of alcohol on the postprandial glucose and insulin responses in healthy subjects. Am. J. Clin. Nutr. 2012, 96, 44–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pezzarossa, A.; Cervigni, C.; Ghinelli, F.; Molina, E.; Gnudi, A. Glucose tolerance in chronic alcoholics after alcohol withdrawal: Effect of accompanying diet. Metabolism 1986, 35, 984–988. [Google Scholar] [CrossRef]
- Kim, J.Y.; Hwang, J.-Y.; Lee, D.Y.; Song, E.H.; Park, K.J.; Kim, G.H.; Jeong, E.A.; Lee, Y.J.; Go, M.J.; Kim, D.J.; et al. Chronic ethanol consumption inhibits glucokinase transcriptional activity by Atf3 and triggers metabolic syndrome in vivo. J. Boil. Chem. 2014, 289, 27065–27079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-J.; Ju, A.; Lim, S.-G.; Kim, D.-J. Chronic alcohol consumption, type 2 diabetes mellitus, insulin-like growth factor-I (IGF-I), and growth hormone (GH) in ethanol-treated diabetic rats. Life Sci. 2013, 93, 778–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, N.G.; Suhaidi, F.A.; Law, W.X.; Liang, N.-C. Chronic moderate alcohol drinking alters insulin release without affecting cognitive and emotion-like behaviors in rats. Alcohol (Fayetteville N.Y.) 2017, 70, 11–22. [Google Scholar] [CrossRef]
- Hodge, A.M.; Dowse, G.K.; Collins, V.R.; Zimmet, P.Z. Abnormal Glucose Tolerance and Alcohol Consumption in Three Populations at High Risk of Non-lnsulin-dependent Diabetes Mellitus. Am. J. Epidemiology 1993, 137, 178–189. [Google Scholar] [CrossRef]
- Wei, M.; Gibbons, L.W.; Mitchell, T.L.; Kampert, J.B.; Blair, S.N. Alcohol intake and incidence of type 2 diabetes in men. Diabetes Care 2000, 23, 18–22. [Google Scholar] [CrossRef] [Green Version]
- Huh, J.H.; Jeon, H.; Park, S.M.; Choi, E.; Lee, G.S.; Kim, J.W.; Lee, K.J. Diabetes Mellitus is Associated With Mortality in Acute Pancreatitis. J. Clin. Gastroenterol. 2018, 52, 178–183. [Google Scholar] [CrossRef]
- Andrali, S.S.; Sampley, M.L.; Vanderford, N.L.; Özcan, S. Glucose regulation of insulin gene expression in pancreatic β-cells. Biochem. J. 2008, 415, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.S.; Hebrok, M. All mixed up: Defining roles for beta-cell subtypes in mature islets. Genes Dev. 2017, 31, 228–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneto, H.; Miyatsuka, T.; Fujitani, Y.; Noguchi, H.; Song, K.-H.; Yoon, K.-H.; Matsuoka, T.-A. Role of PDX-1 and MafA as a potential therapeutic target for diabetes. Diabetes Res. Clin. Pr. 2007, 77, S127–S137. [Google Scholar] [CrossRef] [PubMed]
- Sasikala, M.; Talukdar, R.; Pavan kumar, P.; Radhika, G.; Rao, G.V.; Pradeep, R.; Subramanyam, C.; Nageshwar Reddy, D. Beta-cell dysfunction in chronic pancreatitis. Dig. Dis. Sci. 2012, 57, 1764–1772. [Google Scholar] [CrossRef] [PubMed]
- Kaneto, H.; Miyatsuka, T.; Kawamori, D.; Shiraiwa, T.; Fujitani, Y.; Matsuoka, T.A. Pdx-1 and mafa in beta-cell differentiation and dysfunction. Expert Rev Endocrinol Metab. 2006, 1, 587–600. [Google Scholar] [CrossRef]
- Robertson, R.P.; Harmon, J.S. Diabetes, glucose toxicity, and oxidative stress: A case of double jeopardy for the pancreatic islet β cell. Free. Radic. Boil. Med. 2006, 41, 177–184. [Google Scholar] [CrossRef]
- Dressel, U.; Bailey, P.; Wang, S.-C.; Downes, M.; Evans, R.M.; Muscat, G. A Dynamic Role for HDAC7 in MEF2-mediated Muscle Differentiation. J. Boil. Chem. 2001, 276, 17007–17013. [Google Scholar] [CrossRef] [Green Version]
- Meroni, M.; Longo, M.; Rametta, R.; Dongiovanni, P. Genetic and Epigenetic Modifiers of Alcoholic Liver Disease. Int. J. Mol. Sci. 2018, 19, 3857. [Google Scholar] [CrossRef] [Green Version]
- Miska, E.A.; Karlsson, C.; Langley, E.; Nielsen, S.J.; Pines, J.; Kouzarides, T. HDAC4 deacetylase associates with and represses the MEF2 transcription factor. EMBO J. 1999, 18, 5099–5107. [Google Scholar] [CrossRef]
- Palmisano, M.; Pandey, S.C. Epigenetic mechanisms of alcoholism and stress-related disorders. Alcohol 2017, 60, 7–18. [Google Scholar] [CrossRef]
- Pandey, S.C.; Kyzar, E.J.; Zhang, H. Epigenetic basis of the dark side of alcohol addiction. Neuropharmacology 2017, 122, 74–84. [Google Scholar] [CrossRef]
- Par, A.; Par, G. Alcoholic liver disease: The roles of genetic-epigenetic factors and the effect of abstinence. Orv. Hetil. 2019, 160, 524–532. [Google Scholar] [PubMed]
- Simon, L.; Ford, S.M.; Song, K.; Berner, P.; Stouwe, C.V.; Nelson, S.; Bagby, G.J.; Molina, P.E. Decreased myoblast differentiation in chronic binge alcohol-administered simian immunodeficiency virus-infected male macaques: Role of decreased miR-206. Am. J. Physiol. Integr. Comp. Physiol. 2017, 313, R240–R250. [Google Scholar] [CrossRef] [PubMed]
- Simon, L.; Hollenbach, A.D.; Zabaleta, J.; Molina, P.E. Chronic binge alcohol administration dysregulates global regulatory gene networks associated with skeletal muscle wasting in simian immunodeficiency virus-infected macaques. BMC Genom. 2015, 16, 1097. [Google Scholar] [CrossRef] [Green Version]
- Adler, K.; Molina, P.E.; Simon, L. Epigenomic mechanisms of alcohol-induced impaired differentiation of skeletal muscle stem cells; role of Class IIA histone deacetylases. Physiol. Genom. 2019, 51, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Galvan, F.H.; Bing, E.G.; A Fleishman, J.; London, A.S.; Caetano, R.; Burnam, M.A.; Longshore, D.; Morton, S.C.; Orlando, M.; Shapiro, M. The prevalence of alcohol consumption and heavy drinking among people with HIV in the United States: Results from the HIV Cost and Services Utilization Study. J. Stud. Alcohol 2002, 63, 179–186. [Google Scholar] [CrossRef]
- Grant, B.F.; A Dawson, D.; Stinson, F.S.; Chou, S.; Dufour, M.C.; Pickering, R.P. The 12-month prevalence and trends in DSM-IV alcohol abuse and dependence: United States, 1991–1992 and 2001–2002. Drug Alcohol Depend. 2004, 74, 223–234. [Google Scholar] [CrossRef]
- LeCapitaine, N.J.; Wang, Z.Q.; Dufour, J.P.; Potter, B.J.; Bagby, G.J.; Nelson, S.; Cefalu, W.T.; Molina, P.E. Disrupted Anabolic and Catabolic Processes May Contribute to Alcohol-Accentuated SAIDS-Associated Wasting. J. Infect. Dis. 2011, 204, 1246–1255. [Google Scholar] [CrossRef]
- Ford, S.M., Jr.; Peter, L.S.; Berner, P.; Cook, G.; Stouwe, C.V.; Dufour, J.P.; Bagby, G.; Nelson, S.; Molina, P.E. Differential contribution of chronic binge alcohol and antiretroviral therapy to metabolic dysregulation in SIV-infected male macaques. Am. J. Physiol. Metab. 2018, 315, E892–E903. [Google Scholar] [CrossRef]
- Ford, S.M.; Simon, L.; Stouwe, C.V.; Allerton, T.; Mercante, D.E.; Byerley, L.O.; Dufour, J.P.; Bagby, G.J.; Nelson, S.; Molina, P.E. Chronic binge alcohol administration impairs glucose-insulin dynamics and decreases adiponectin in asymptomatic simian immunodeficiency virus-infected macaques. Am. J. Physiol. Integr. Comp. Physiol. 2016, 311, R888–R897. [Google Scholar] [CrossRef]
- Chen, X.; Sebastian, B.M.; Nagy, L.E. Chronic ethanol feeding to rats decreases adiponectin secretion by subcutaneous adipocytes. Am. J. Physiol. Metab. 2006, 292, E621–E628. [Google Scholar] [CrossRef]
- Jung, S.; Kim, M.K.; Shin, J.; Choi, B.Y. A cross-sectional analysis of the relationship between daily alcohol consumption and serum adiponectin levels among adults aged 40 years or more in a rural area of Korea. Eur. J. Clin. Nutr. 2013, 67, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Makita, S.; Abiko, A.; Nagai, M.; Yonezawa, S.; Koshiyama, M.; Ohta, M.; Nakamura, M. Influence of daily alcohol consumption on serum adiponectin levels in men. Metabolism 2013, 62, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Rasineni, K.; Kubik, J.L.; Knight, K.L.; Hall, L.; Casey, C.A.; Kharbanda, K.K. Ghrelin regulates adipose tissue metabolism: Role in hepatic steatosis. Chem. Biol. Interact. 2020, 322, 109059. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Jin, X.; Ye, X.; Wu, H.; Ren, W.; Zhang, R.; Long, J.; Ying, C. Long term intake of 0.1% ethanol decreases serum adiponectin by suppressing ppargamma expression via p38 mapk pathway. Food Chem. Toxicol. 2014, 65, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Ravazzola, M.; Park, B.H.; Bashmakov, Y.K.; Orci, L.; Unger, R.H. Metabolic mechanisms of failure of intraportally transplanted pancreatic beta-cells in rats: Role of lipotoxicity and prevention by leptin. Diabetes 2007, 56, 2295–2301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, W.L.; Miller, R.A.; Wang, Z.V.; Sun, K.; Barth, B.M.; Bui, H.H.; Davis, K.E.; Bikman, B.T.; Halberg, N.; Rutkowski, J.M.; et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat. Med. 2010, 17, 55–63. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rasineni, K.; Srinivasan, M.P.; Balamurugan, A.N.; Kaphalia, B.S.; Wang, S.; Ding, W.-X.; Pandol, S.J.; Lugea, A.; Simon, L.; Molina, P.E.; et al. Recent Advances in Understanding the Complexity of Alcohol-Induced Pancreatic Dysfunction and Pancreatitis Development. Biomolecules 2020, 10, 669. https://doi.org/10.3390/biom10050669
Rasineni K, Srinivasan MP, Balamurugan AN, Kaphalia BS, Wang S, Ding W-X, Pandol SJ, Lugea A, Simon L, Molina PE, et al. Recent Advances in Understanding the Complexity of Alcohol-Induced Pancreatic Dysfunction and Pancreatitis Development. Biomolecules. 2020; 10(5):669. https://doi.org/10.3390/biom10050669
Chicago/Turabian StyleRasineni, Karuna, Mukund P. Srinivasan, Appakalai N. Balamurugan, Bhupendra S. Kaphalia, Shaogui Wang, Wen-Xing Ding, Stephen J. Pandol, Aurelia Lugea, Liz Simon, Patricia E. Molina, and et al. 2020. "Recent Advances in Understanding the Complexity of Alcohol-Induced Pancreatic Dysfunction and Pancreatitis Development" Biomolecules 10, no. 5: 669. https://doi.org/10.3390/biom10050669