
Aldehyde Trapping by ADX-102 Is Protective against Cigarette Smoke and Alcohol Mediated Lung Cell Injury

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Treatments
2.3. Cigarette Smoke Extract Preparation
2.4. In Vitro Wound Closure (Migration) Assay
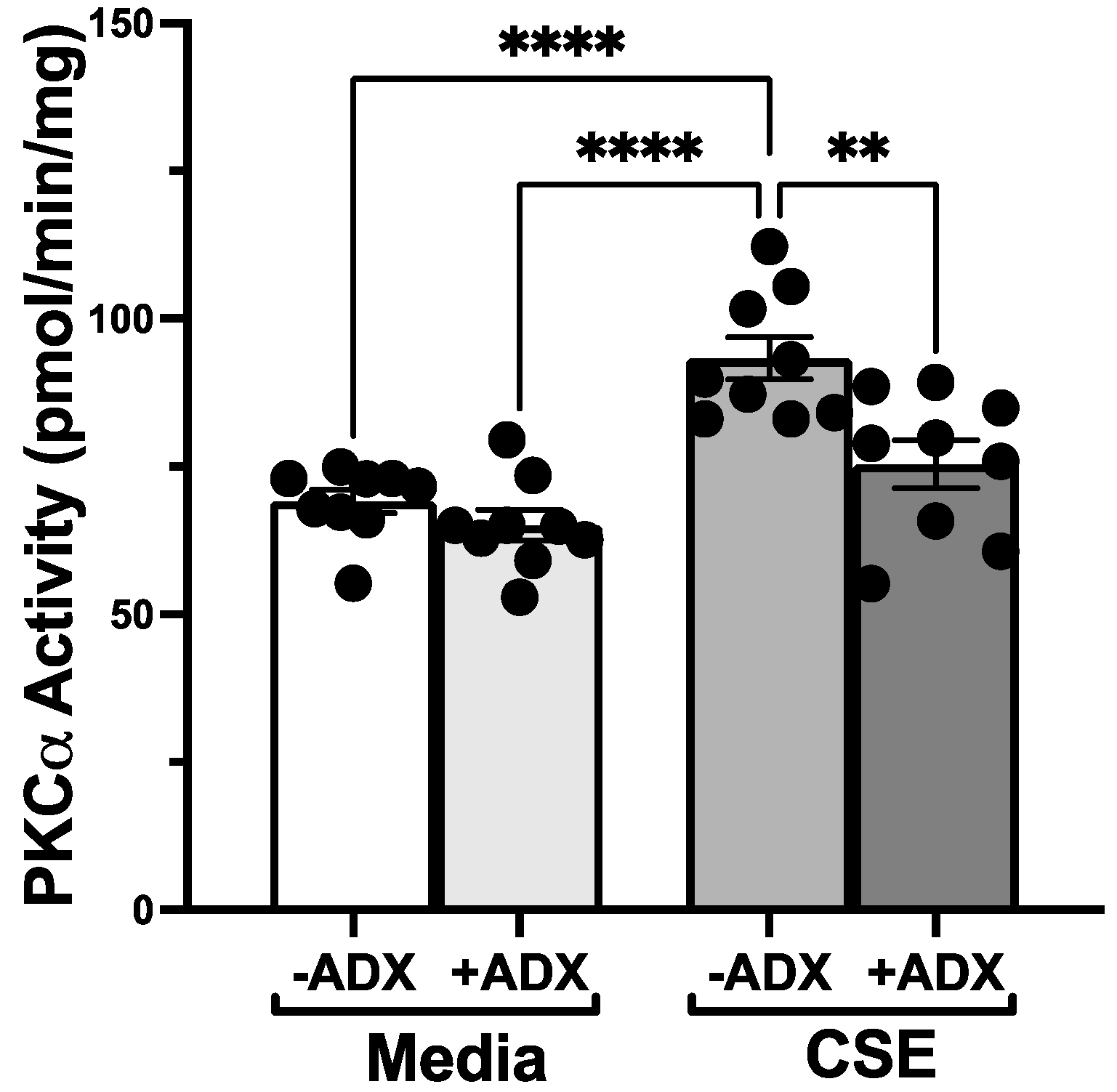
2.5. PKCα and PKCε Activity Assays
2.6. Cilia Beating Assay (SAVA)
2.7. Transepithelial Resistance Assay
2.8. Cell Viability Assay
2.9. ELISA for MAA Adducts
2.10. Statistical Analysis
3. Results
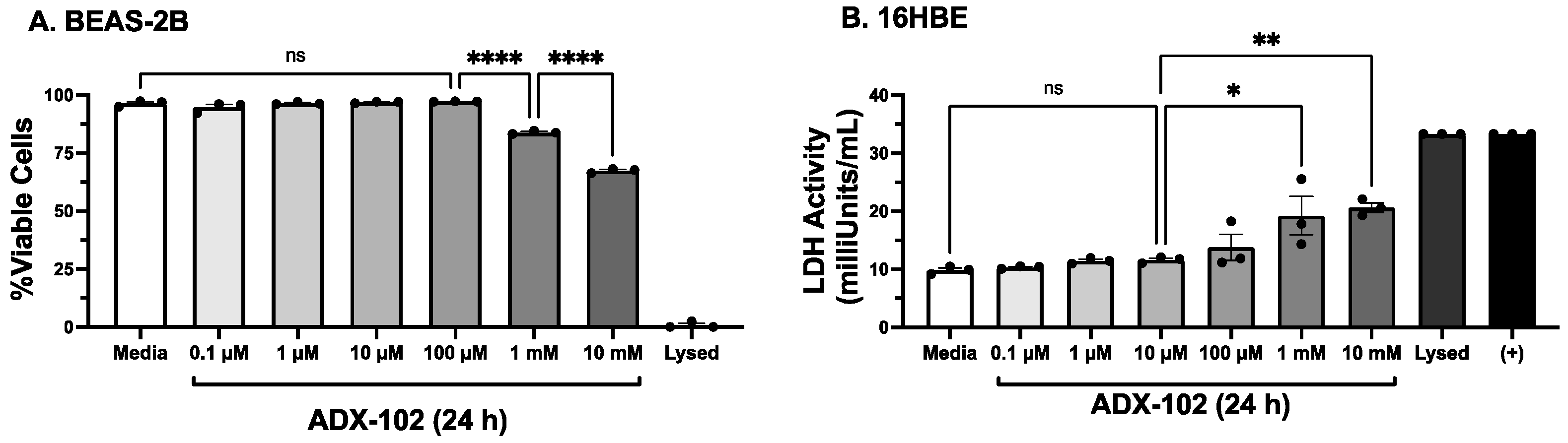
3.1. Only High Concentrations of ADX-102 Affect Cell Viability
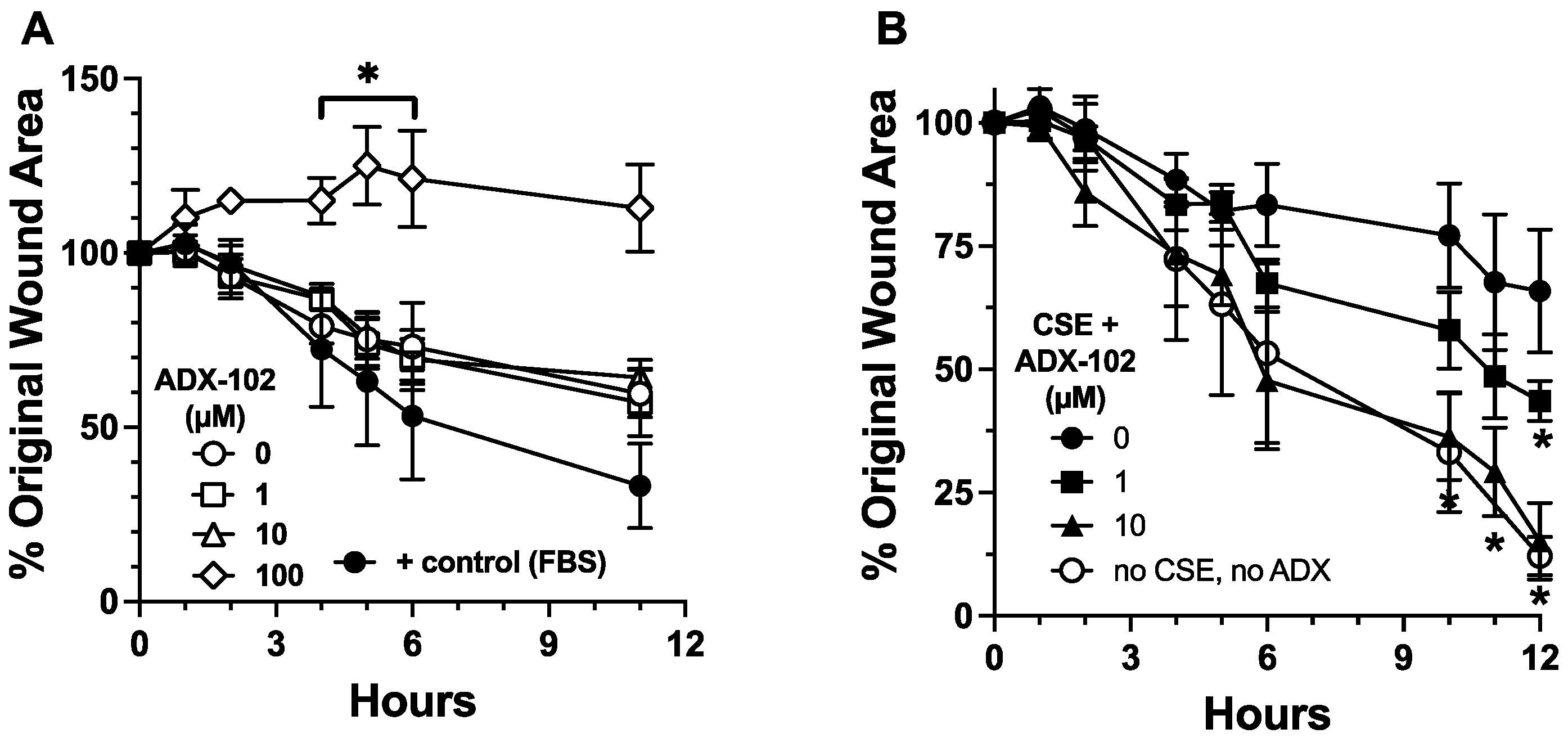
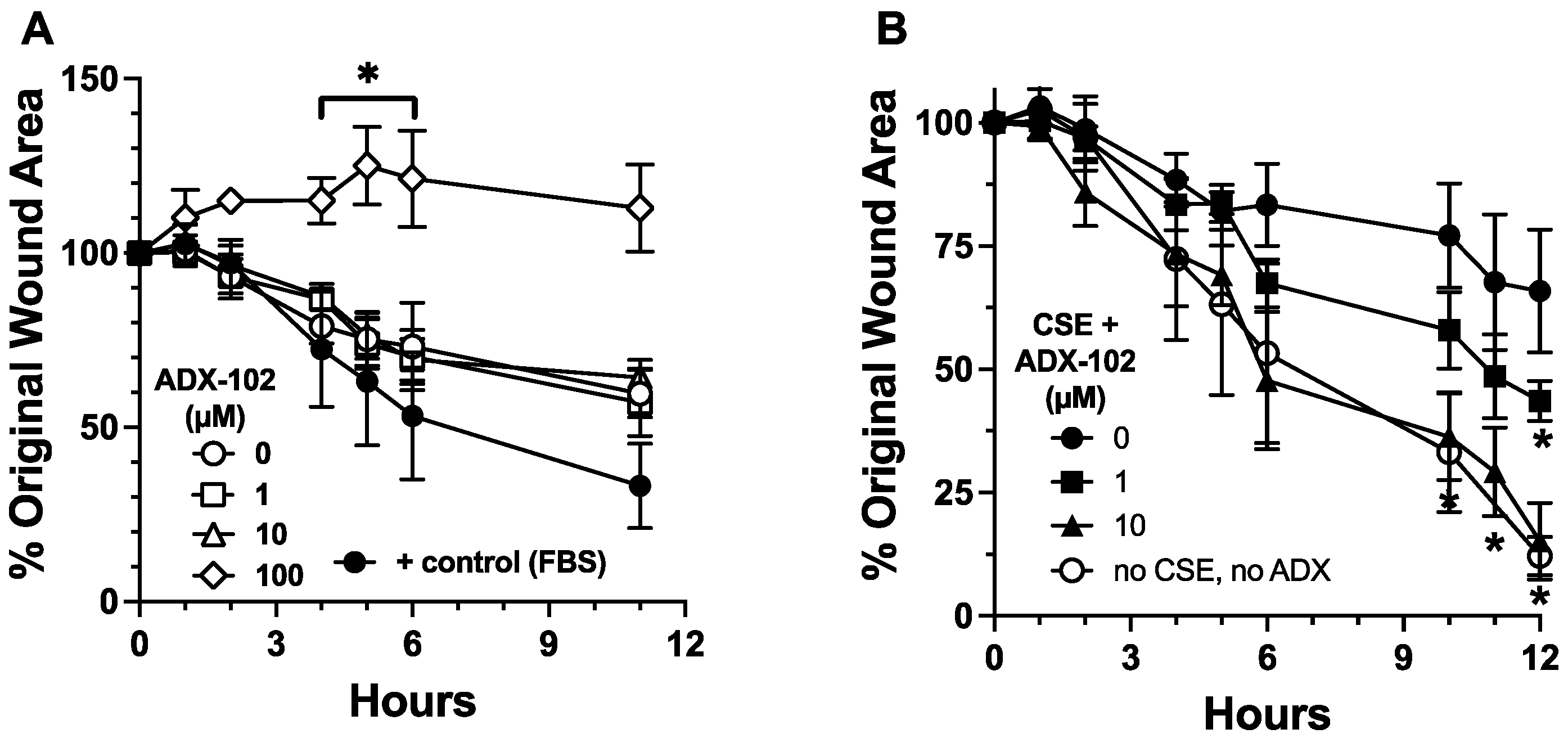
3.2. ADX-102 and Wound Repair
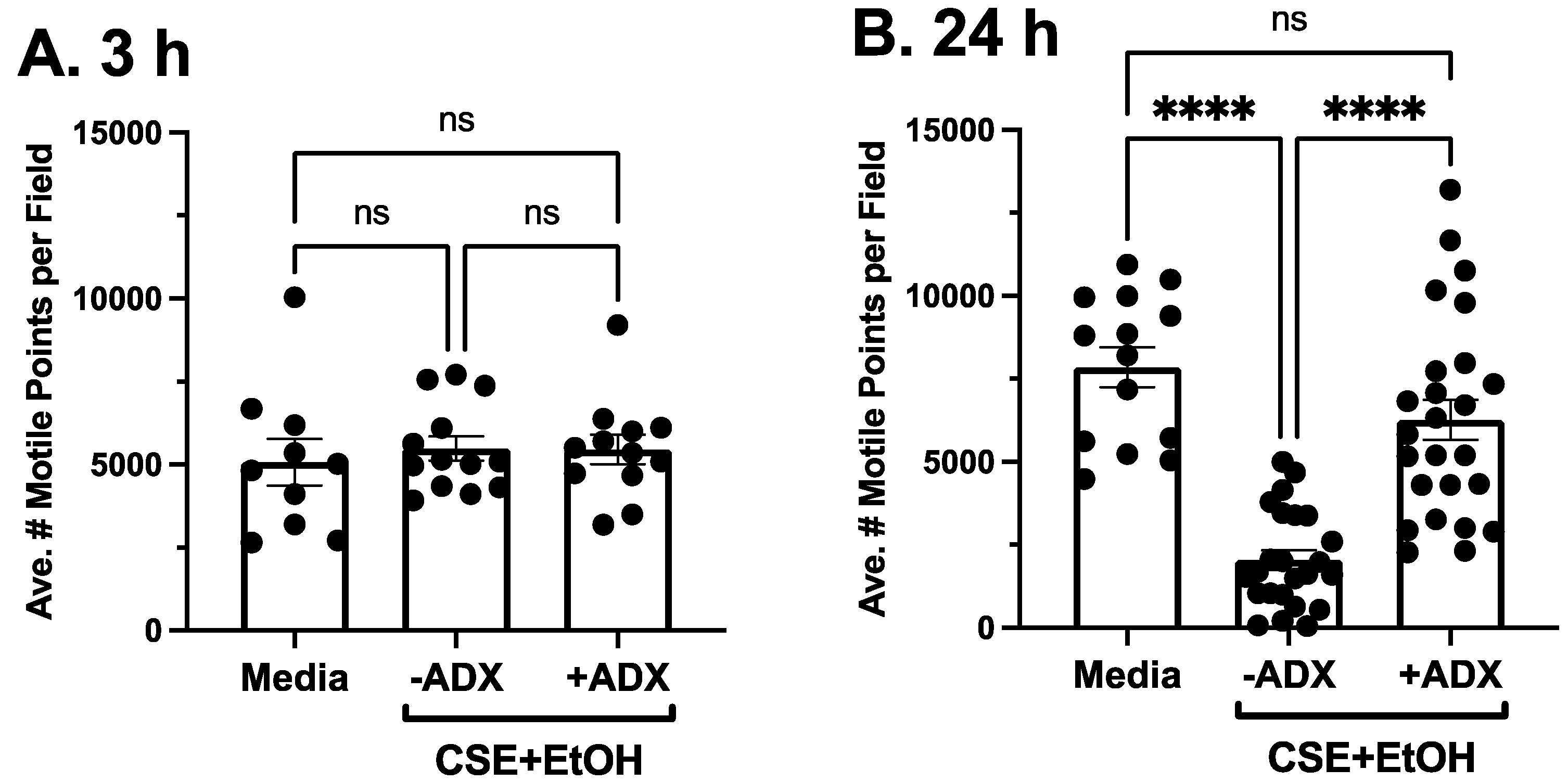
3.3. ADX-102 and Cilia Beating
3.4. ADX-102 and Cilia Loss
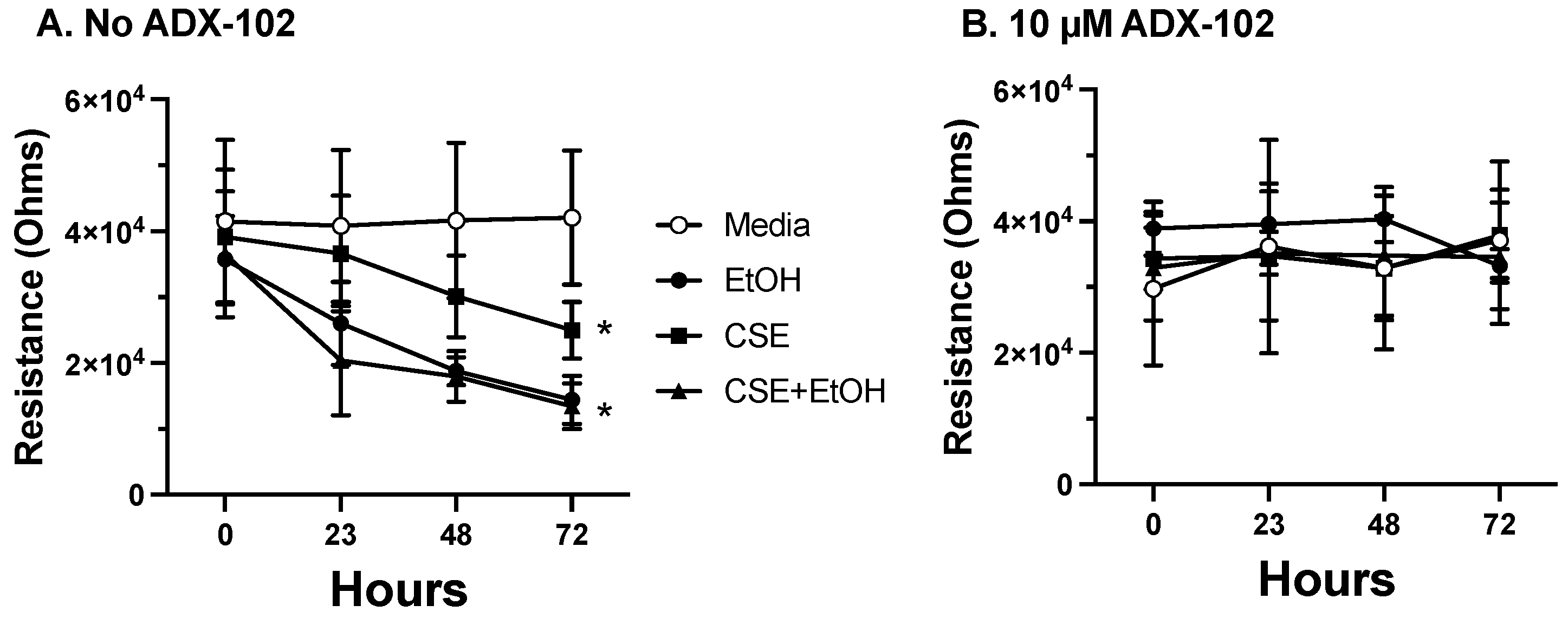
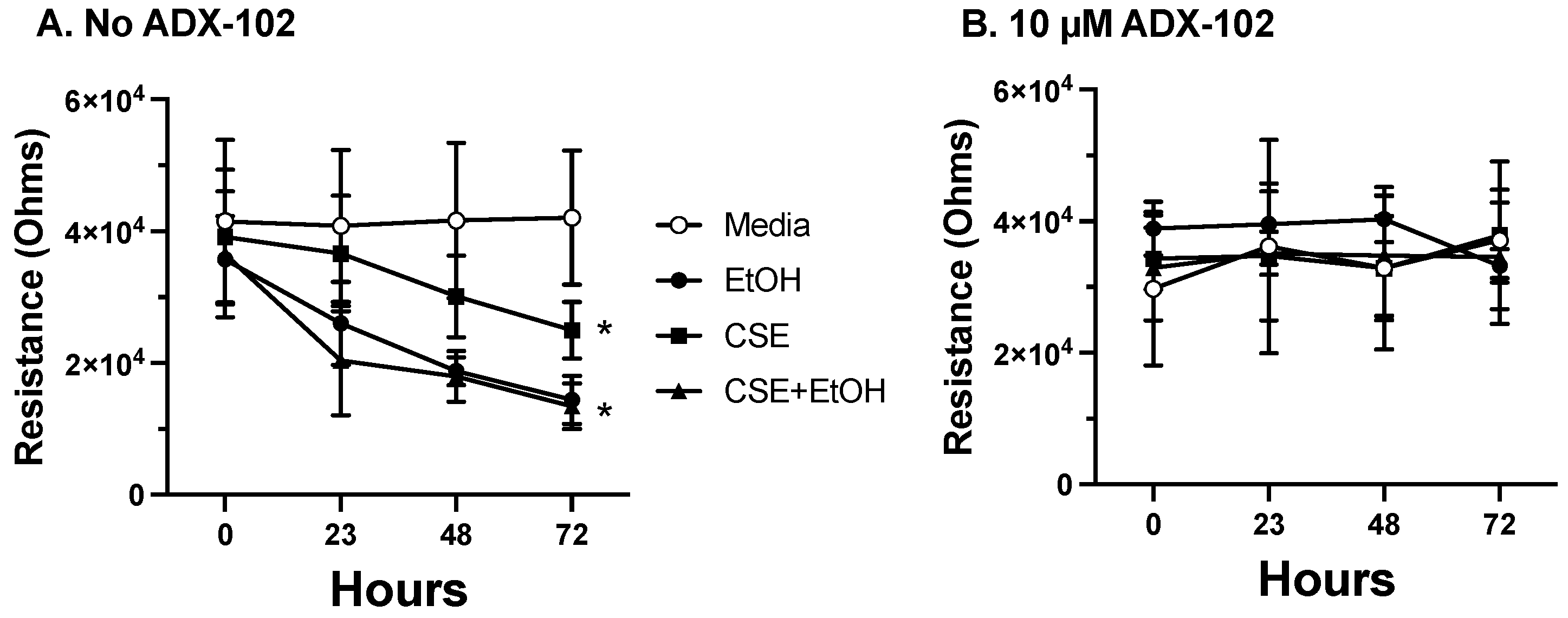
3.5. ADX-102 and Barrier Function
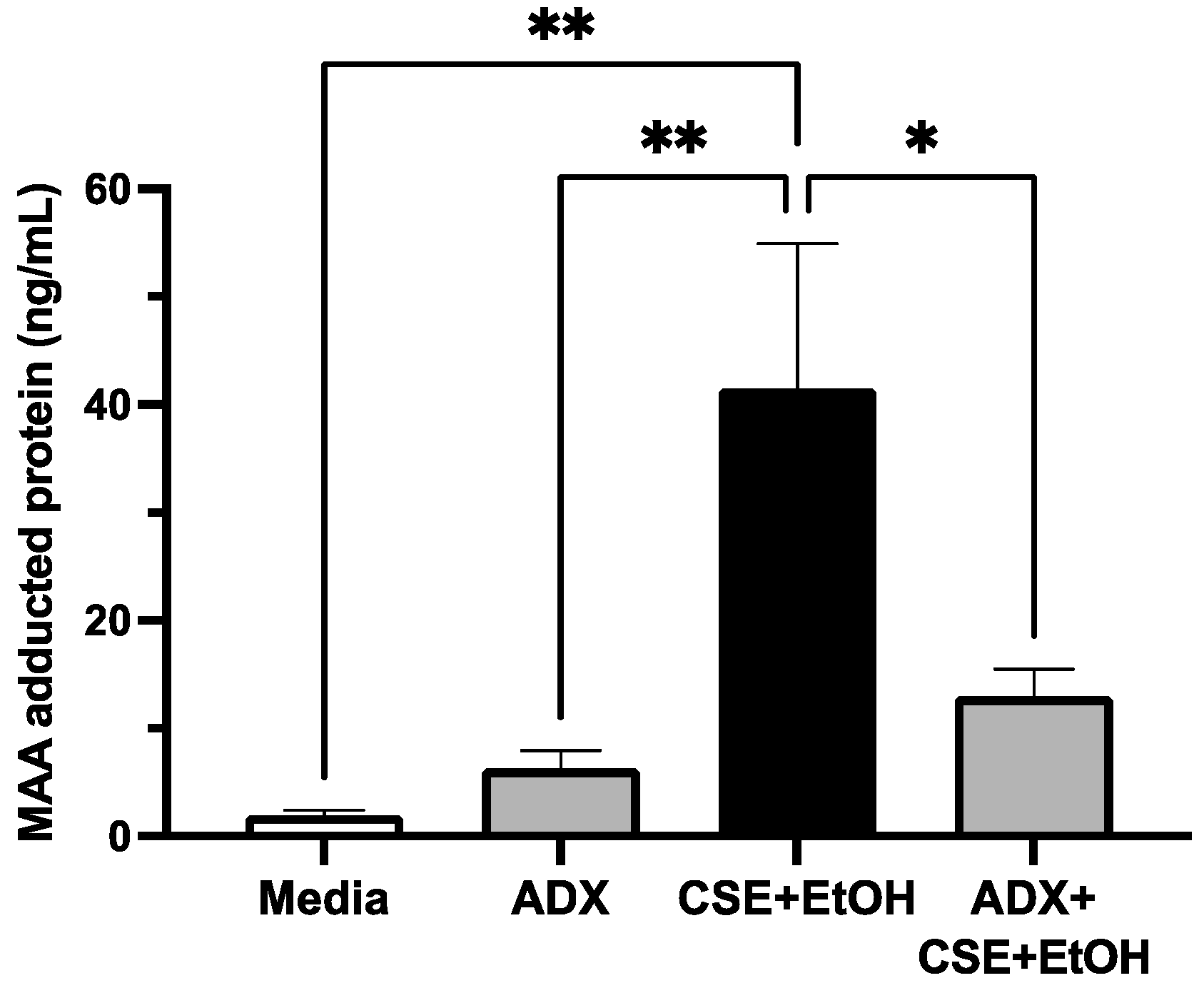
3.6. ADX-102 and MAA Adduct Formation
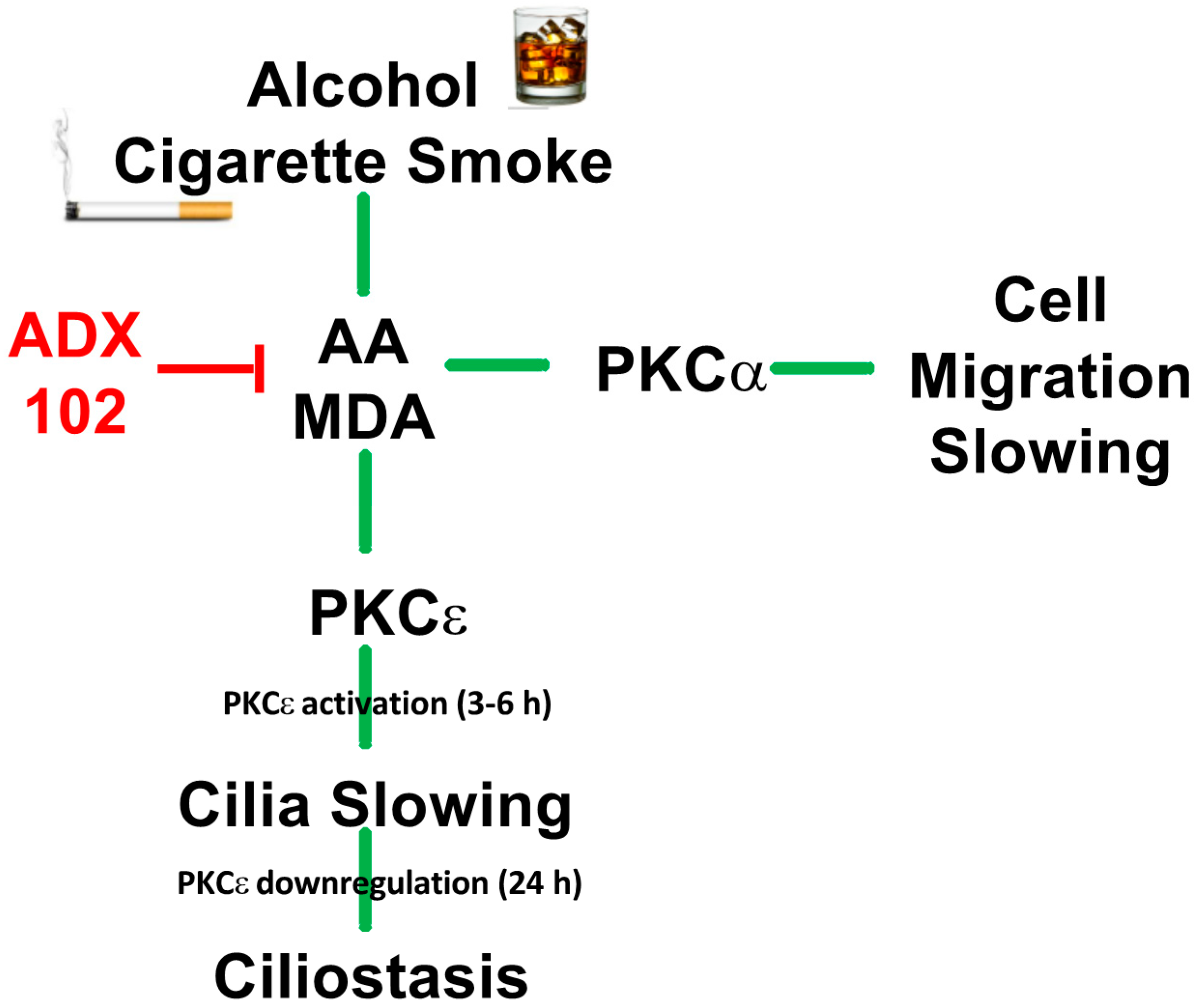
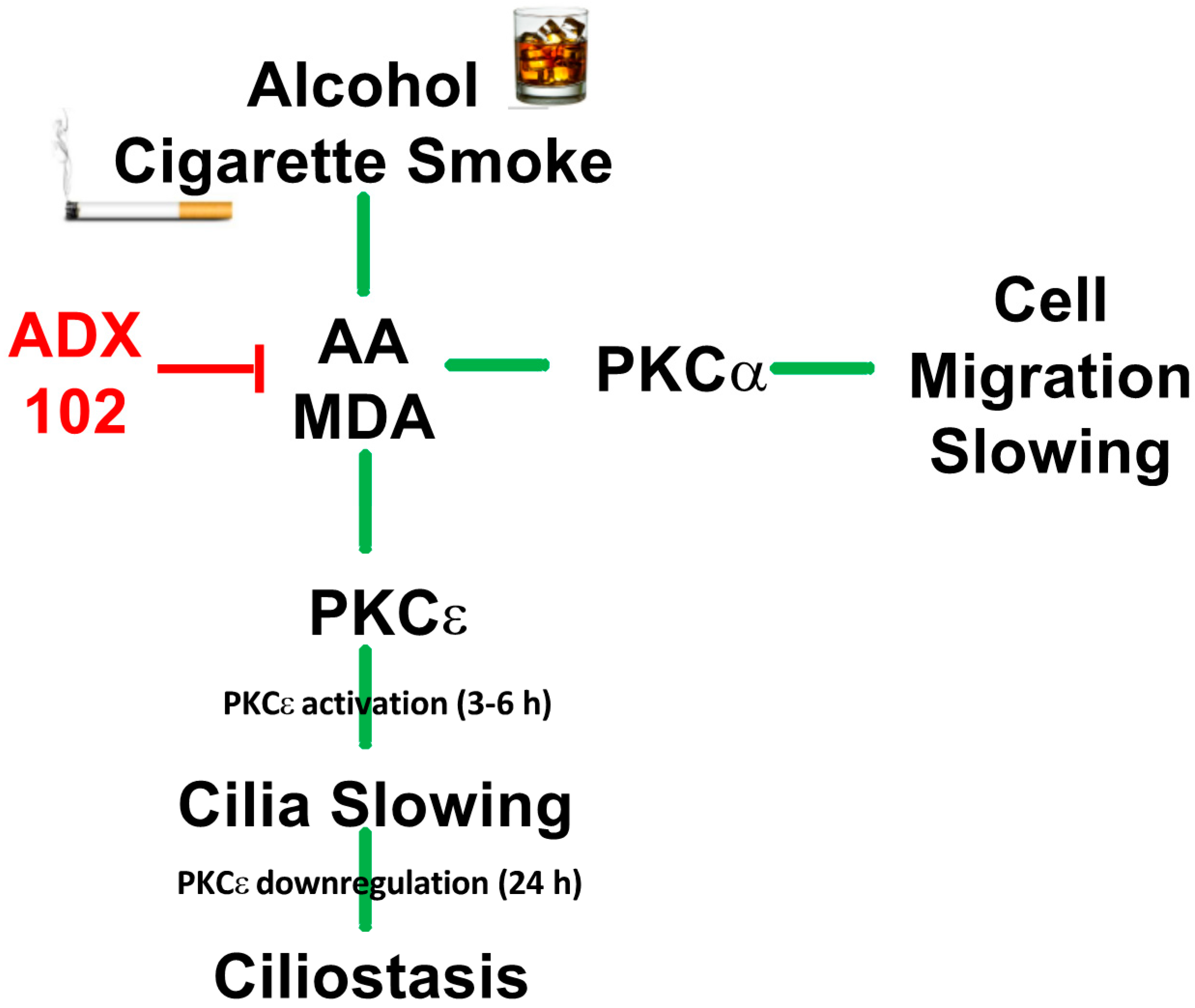
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Office on Smoking and Health, Centers for Disease Control and Prevention, Smoking & Tobacco Use: Fast Facts. Available online: https://www.cdc.gov/tobacco/data_statistics/fact_sheets/fast_facts/index.htm#beginning (accessed on 5 October 2021).
- Romberger, D.J.; Grant, K. Alcohol consumption and smoking status: The role of smoking cessation. Biomed. Pharmacother. 2004, 58, 77–83. [Google Scholar] [CrossRef]
- Chudomelka, L.; Wyatt, T.A. Cross-fading: The importance of tissue injury research on dual misuse of alcohol and JUUL. Alcohol 2020, 86, 43–44. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.C.; Applewhite, L.; Ritzenthaler, J.D.; Roman, J.; Fernandez, A.L.; Eaton, D.C.; Brown, L.A.; Guidot, D.M. Chronic ethanol ingestion in rats decreases granulocyte-macrophage colony-stimulating factor receptor expression and downstream signaling in the alveolar macrophage. J. Immunol. 2005, 175, 6837–6845. [Google Scholar] [CrossRef] [Green Version]
- Simet, S.M.; Sisson, J.H. Alcohol’s Effects on Lung Health and Immunity. Alcohol Res. 2015, 37, 199–208. [Google Scholar] [PubMed]
- Boé, D.M.; Vandivier, R.W.; Burnham, E.L.; Moss, M. Alcohol abuse and pulmonary disease. J. Leukoc. Biol. 2009, 86, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Kaphalia, L.; Calhoun, W.J. Alcoholic lung injury: Metabolic, biochemical and immunological aspects. Toxicol. Lett. 2013, 222, 171–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cederbaum, A.I. Role of CYP2E1 in ethanol-induced oxidant stress, fatty liver and hepatotoxicity. Dig. Dis. 2010, 28, 802–811. [Google Scholar] [CrossRef] [Green Version]
- National Institute on Alcohol Abuse and Alcoholism, Alcohol Facts and Statistics. Available online: https://www.niaaa.nih.gov/publications/brochures-and-fact-sheets/alcohol-facts-and-statistics (accessed on 29 March 2021).
- Fujioka, K.; Shibamoto, T. Determination of toxic carbonyl compounds in cigarette smoke. Environ. Toxicol. 2006, 21, 47–54. [Google Scholar] [CrossRef]
- Weng, M.W.; Lee, H.W.; Park, S.H.; Hu, Y.; Wang, H.T.; Chen, L.C.; Rom, W.N.; Huang, W.C.; Lepor, H.; Wu, X.R.; et al. Aldehydes are the predominant forces inducing DNA damage and inhibiting DNA repair in tobacco smoke carcinogenesis. Proc. Natl. Acad. Sci. USA 2018, 115, E6152–E6161. [Google Scholar] [CrossRef] [Green Version]
- McCaskill, M.L.; Kharbanda, K.K.; Tuma, D.J.; Reynolds, J.D.; DeVasure, J.M.; Sisson, J.H.; Wyatt, T.A. Hybrid malondialdehyde and acetaldehyde protein adducts form in the lungs of mice exposed to alcohol and cigarette smoke. Alcohol Clin. Exp. Res. 2011, 35, 1106–1113. [Google Scholar] [CrossRef] [Green Version]
- Wyatt, T.A.; Kharbanda, K.K.; Tuma, D.J.; Sisson, J.H.; Spurzem, J.R. Malondialdehyde-acetaldehyde adducts decrease bronchial epithelial wound repair. Alcohol 2005, 36, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, T.A.; Sisson, J.H.; Allen-Gipson, D.S.; McCaskill, M.L.; Boten, J.A.; DeVasure, J.M.; Bailey, K.L.; Poole, J.A. Co-exposure to cigarette smoke and alcohol decreases airway epithelial cell cilia beating in a protein kinase Cepsilon-dependent manner. Am. J. Pathol. 2012, 181, 431–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldeyra Therapeutics, Inc. Dry Eye Disease. Available online: https://www.aldeyra.com/pipeline-disease-areas/ocular-diseases/dry-eye-disease/ (accessed on 8 September 2021).
- Dossou, S.J.; Bré, M.H.; Hallworth, R. Mammalian cilia function is independent of the polymeric state of tubulin glycylation. Cell Motil. Cytoskelet 2007, 64, 847–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantral, D.E.; Sisson, J.H.; Veys, T.; Rennard, S.I.; Spurzem, J.R. Effects of cigarette smoke extract on bovine bronchial epithelial cell attachment and migration. Am. J. Physiol.-Lung Cell. Mol. Physiol. 1995, 268, L723–L728. [Google Scholar] [CrossRef]
- Allen-Gipson, D.S.; Wong, J.; Spurzem, J.R.; Sisson, J.H.; Wyatt, T.A. Adenosine A2A receptors promote adenosine-stimulated wound healing in bronchial epithelial cells. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2006, 290, L849–L855. [Google Scholar] [CrossRef]
- Spurzem, J.R.; Gupta, J.; Veys, T.; Kneifl, K.R.; Rennard, S.I.; Wyatt, T.A. Activation of protein kinase A accelerates bovine bronchial epithelial cell migration. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2002, 282, L1108–L1116. [Google Scholar] [CrossRef] [Green Version]
- Elliott, M.K.; Sisson, J.H.; Wyatt, T.A. Effects of cigarette smoke and alcohol on ciliated tracheal epithelium and inflammatory cell recruitment. Am. J. Respir. Cell Mol. Biol. 2007, 36, 452–459. [Google Scholar] [CrossRef]
- Sisson, J.H.; Stoner, J.A.; Ammons, B.A.; Wyatt, T.A. All-digital image capture and whole-field analysis of ciliary beat frequency. J. Microsc. 2003, 211, 103–111. [Google Scholar] [CrossRef]
- Wyatt, T.A.; Schmidt, S.C.; Rennard, S.I.; Tuma, D.J.; Sisson, J.H. Acetaldehyde-stimulated PKC activity in airway epithelial cells treated with smoke extract from normal and smokeless cigarettes. Proc. Soc. Exp. Biol. Med. 2000, 225, 91–97. [Google Scholar] [CrossRef]
- Olivera, D.S.; Boggs, S.E.; Beenhouwer, C.; Aden, J.; Knall, C. Cellular mechanisms of mainstream cigarette smoke-induced lung epithelial tight junction permeability changes in vitro. Inhal. Toxicol. 2007, 19, 13–22. [Google Scholar] [CrossRef]
- Schlingmann, B.; Overgaard, C.E.; Molina, S.A.; Lynn, K.S.; Mitchell, L.A.; Dorsainvil White, S.; Mattheyses, A.L.; Guidot, D.M.; Capaldo, C.T.; Koval, M. Regulation of claudin/zonula occludens-1 complexes by hetero-claudin interactions. Nat. Commun. 2016, 7, 12276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sapkota, M.; Burnham, E.L.; DeVasure, J.M.; Sweeter, J.M.; Hunter, C.D.; Duryee, M.J.; Klassen, L.W.; Kharbanda, K.K.; Sisson, J.H.; Thiele, G.M.; et al. Malondialdehyde-Acetaldehyde (MAA) Protein Adducts Are Found Exclusively in the Lungs of Smokers with Alcohol Use Disorders and Are Associated with Systemic Anti-MAA Antibodies. Alcohol Clin. Exp. Res. 2017, 41, 2093–2099. [Google Scholar] [CrossRef] [PubMed]
- Altuntaş, I.; Dane, S.; Gümüştekin, K. Effects of cigarette smoking on lipid peroxidation. J. Basic Clin. Physiol. Pharmacol. 2002, 13, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Runge, D.M.; Stock, T.W.; Lehmann, T.; Taege, C.; Bernauer, U.; Stolz, D.B.; Hofmann, S.; Foth, H. Expression of cytochrome P450 2E1 in normal human bronchial epithelial cells and activation by ethanol in culture. Arch. Toxicol. 2001, 75, 335–345. [Google Scholar] [CrossRef]
- van der Vliet, A.; Bove, P.F. Purinergic signaling in wound healing and airway remodeling. Subcell. Biochem. 2011, 55, 139–157. [Google Scholar] [CrossRef]
- Thiele, G.M.; Worrall, S.; Tuma, D.J.; Klassen, L.W.; Wyatt, T.A.; Nagata, N. The chemistry and biological effects of malondialdehyde-acetaldehyde adducts. Alcohol Clin. Exp. Res. 2001, 25, 218S–224S. [Google Scholar] [CrossRef]
- Berger, J.P.; Simet, S.M.; DeVasure, J.M.; Boten, J.A.; Sweeter, J.M.; Kharbanda, K.K.; Sisson, J.H.; Wyatt, T.A. Malondialdehyde-acetaldehyde (MAA) adducted proteins bind to scavenger receptor A in airway epithelial cells. Alcohol 2014, 48, 493–500. [Google Scholar] [CrossRef] [Green Version]
- Rosenblatt, M.B.; Trinidad, S.; Lisa, J.R.; Tchertkoff, V. Specific epithelial degeneration (ciliocytophthoria) in inflammatory and malignant respiratory disease. Dis. Chest. 1963, 43, 605–612. [Google Scholar] [CrossRef] [Green Version]
- Sapkota, M.; Wyatt, T.A. Alcohol, Aldehydes, Adducts and Airways. Biomolecules 2015, 5, 2987–3008. [Google Scholar] [CrossRef] [Green Version]
- Slager, R.E.; Devasure, J.M.; Pavlik, J.A.; Sisson, J.H.; Wyatt, T.A. RACK1, a PKC targeting protein, is exclusively localized to basal airway epithelial cells. J. Histochem. Cytochem. 2008, 56, 7–14. [Google Scholar] [CrossRef]
- National Institute on Alcohol Abuse and Alcoholism, Understanding Alcohol Use Disorders. Available online: https://www.niaaa.nih.gov/publications/brochures-and-fact-sheets/understanding-alcohol-use-disorder (accessed on 8 September 2021).
- National Institue on Drug Abuse, AUDIT. Available online: https://www.drugabuse.gov/sites/default/files/audit.pdf (accessed on 8 September 2021).
- Substance Abuse and Mental Health Services Administration, U.S. Department of Health and Human Services. 2019 NSDUH Detailed Tables. Available online: https://www.samhsa.gov/data/release/2019-national-survey-drug-use-and-health-nsduh-releases (accessed on 8 September 2021).
- Gentry, M.J. Pneumococcal pneumonia in alcoholism and HIV infection. Alcohol Clin. Exp. Res. 1998, 22, 201s–203s. [Google Scholar] [CrossRef] [PubMed]
- Tabak, C.; Smit, H.A.; Rasanen, L.; Fidanza, F.; Menotti, A.; Nissinen, A.; Feskens, E.J.; Heederik, D.; Kromhout, D. Alcohol consumption in relation to 20-year COPD mortality and pulmonary function in middle-aged men from three European countries. Epidemiology 2001, 12, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Bailey, K.L.; Samuelson, D.R.; Wyatt, T.A. Alcohol use disorder: A pre-existing condition for COVID-19? Alcohol 2021, 90, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.; Cavanagh, B.; Shields, A.L.; Karpecki, P.; Sheppard, J.; Brady, T.C. Clinically Relevant Activity of the Novel RASP Inhibitor Reproxalap in Allergic Conjunctivitis: The Phase 3 ALLEVIATE Trial. Am. J. Ophthalmol. 2021, 230, 60–67. [Google Scholar] [CrossRef]
- Clark, D.; Sheppard, J.; Brady, T.C. A Randomized Double-Masked Phase 2a Trial to Evaluate Activity and Safety of Topical Ocular Reproxalap, a Novel RASP Inhibitor, in Dry Eye Disease. J. Ocul. Pharmacol. Ther. 2021, 37, 193–199. [Google Scholar] [CrossRef]
- MacDonald, S.; Halilovic, A.; Brady, T. Novel Small Molecule Aldehyde Sequestering Agents Demonstrate Broad Therapeutic Potential for Ocular Inflammation. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2663. [Google Scholar]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ochoa, C.A.; Nissen, C.G.; Mosley, D.D.; Bauer, C.D.; Jordan, D.L.; Bailey, K.L.; Wyatt, T.A. Aldehyde Trapping by ADX-102 Is Protective against Cigarette Smoke and Alcohol Mediated Lung Cell Injury. Biomolecules 2022, 12, 393. https://doi.org/10.3390/biom12030393
Ochoa CA, Nissen CG, Mosley DD, Bauer CD, Jordan DL, Bailey KL, Wyatt TA. Aldehyde Trapping by ADX-102 Is Protective against Cigarette Smoke and Alcohol Mediated Lung Cell Injury. Biomolecules. 2022; 12(3):393. https://doi.org/10.3390/biom12030393
Chicago/Turabian StyleOchoa, Carmen A., Claire G. Nissen, Deanna D. Mosley, Christopher D. Bauer, Destiny L. Jordan, Kristina L. Bailey, and Todd A. Wyatt. 2022. "Aldehyde Trapping by ADX-102 Is Protective against Cigarette Smoke and Alcohol Mediated Lung Cell Injury" Biomolecules 12, no. 3: 393. https://doi.org/10.3390/biom12030393