
3-Chloro-3-methyl-2,6-diarylpiperidin-4-ones as Anti-Cancer Agents: Synthesis, Biological Evaluation, Molecular Docking, and In Silico ADMET Prediction

 ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis of 3-Chloro-3-methyl-2,6-diarylpiperidin-4-ones [(I)–(V)]
2.2. Analytical Data
2.2.1. 3-Chloro-3-methyl-2,6-diphenylpiperidin-4-one [Compound (I)]
2.2.2. 3-Chloro-2,6-bis(4-chlorophenyl)-3-methylpiperidin-4-one [Compound (II)]
2.2.3. 3-Chloro-2,6-bis(4-fluorophenyl)-3-methylpiperidin-4-one [Compound (III)]
2.2.4. 3-Chloro-3-methyl-2,6-di-p-tolylpiperidin-4-one [Compound (IV)]
2.2.5. 3-Chloro-2,6-bis(4-methoxyphenyl)-3-methylpiperidin-4-one [Compound (V)]
2.3. Spectral Characterization
2.4. Biological Study
2.4.1. Cell Culture and Cell Treatment
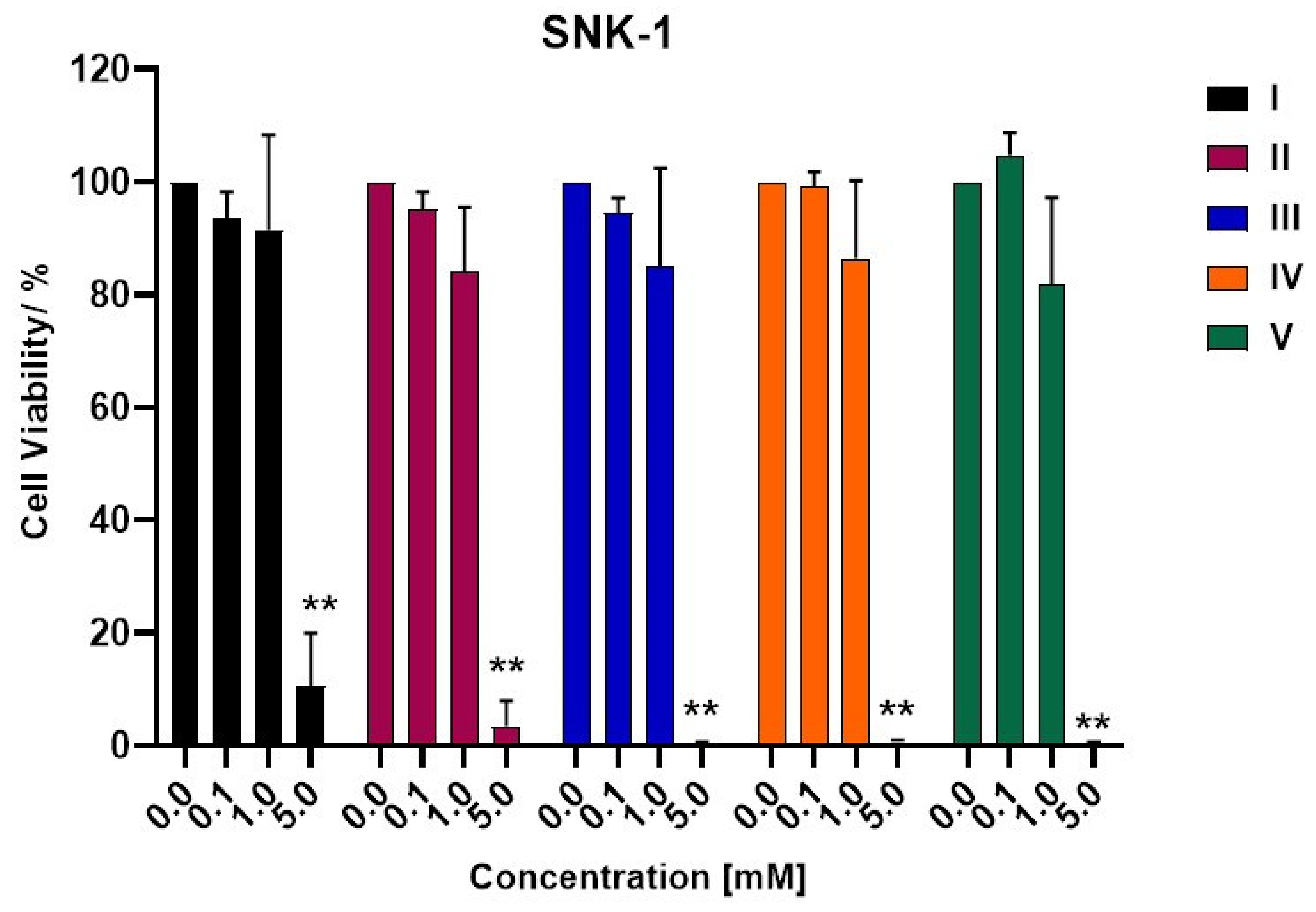
2.4.2. Cell Viability Assay
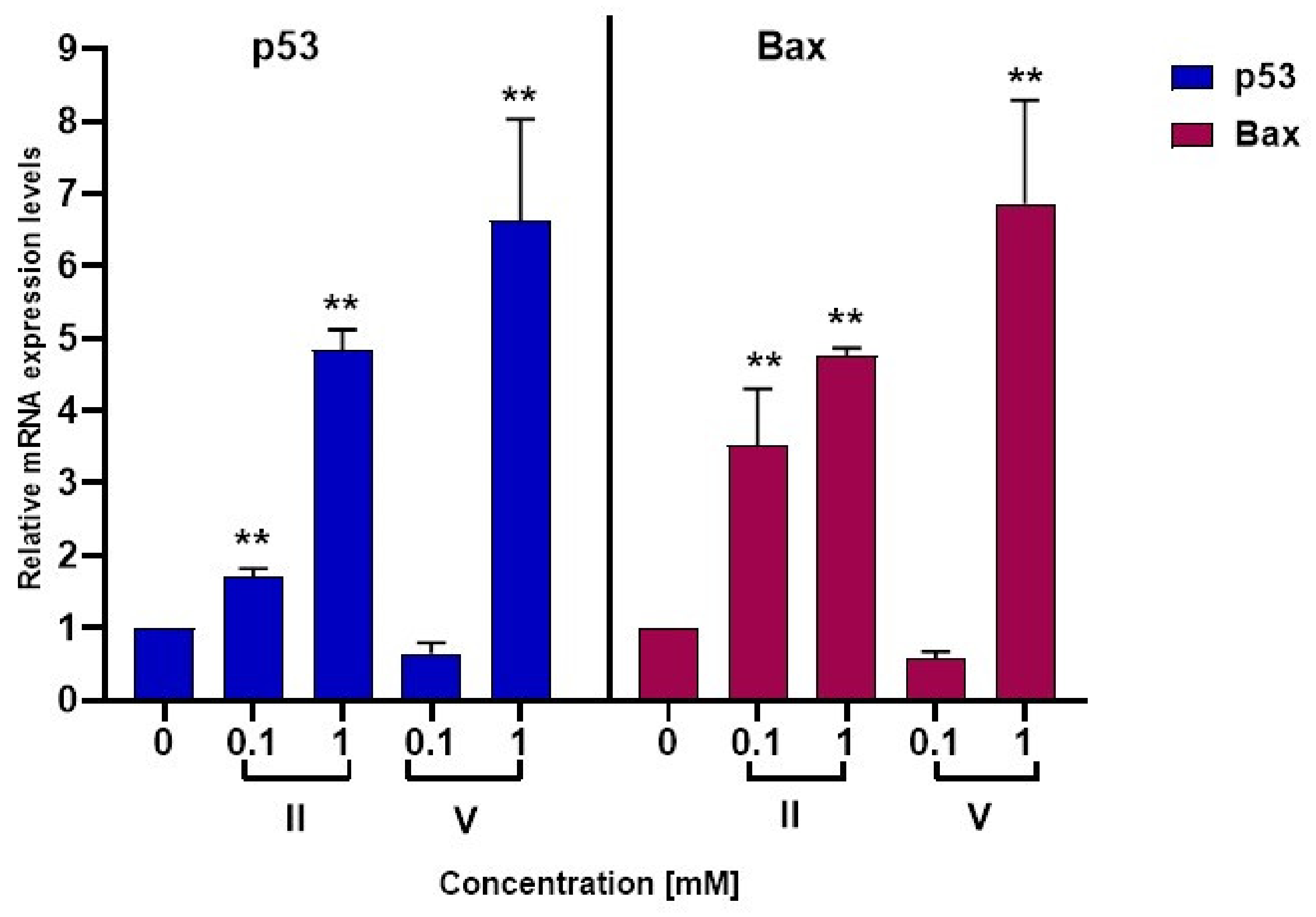
2.4.3. Quantitative RT-PCR Analyses
2.4.4. Statistical Analyses
2.5. Computational Study
2.5.1. Molecular Docking
2.5.2. ADMET Property Prediction
3. Results and Discussion
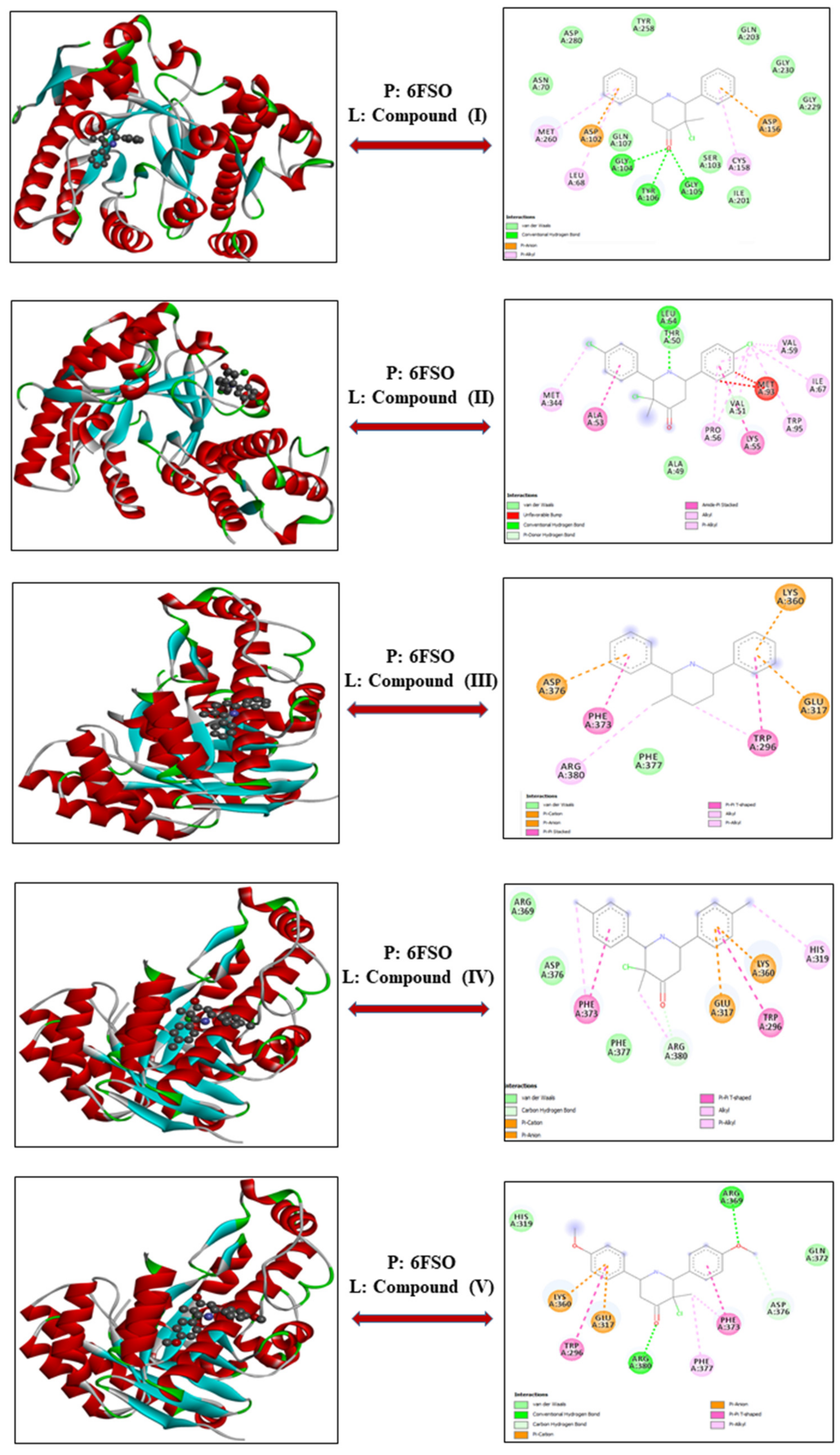
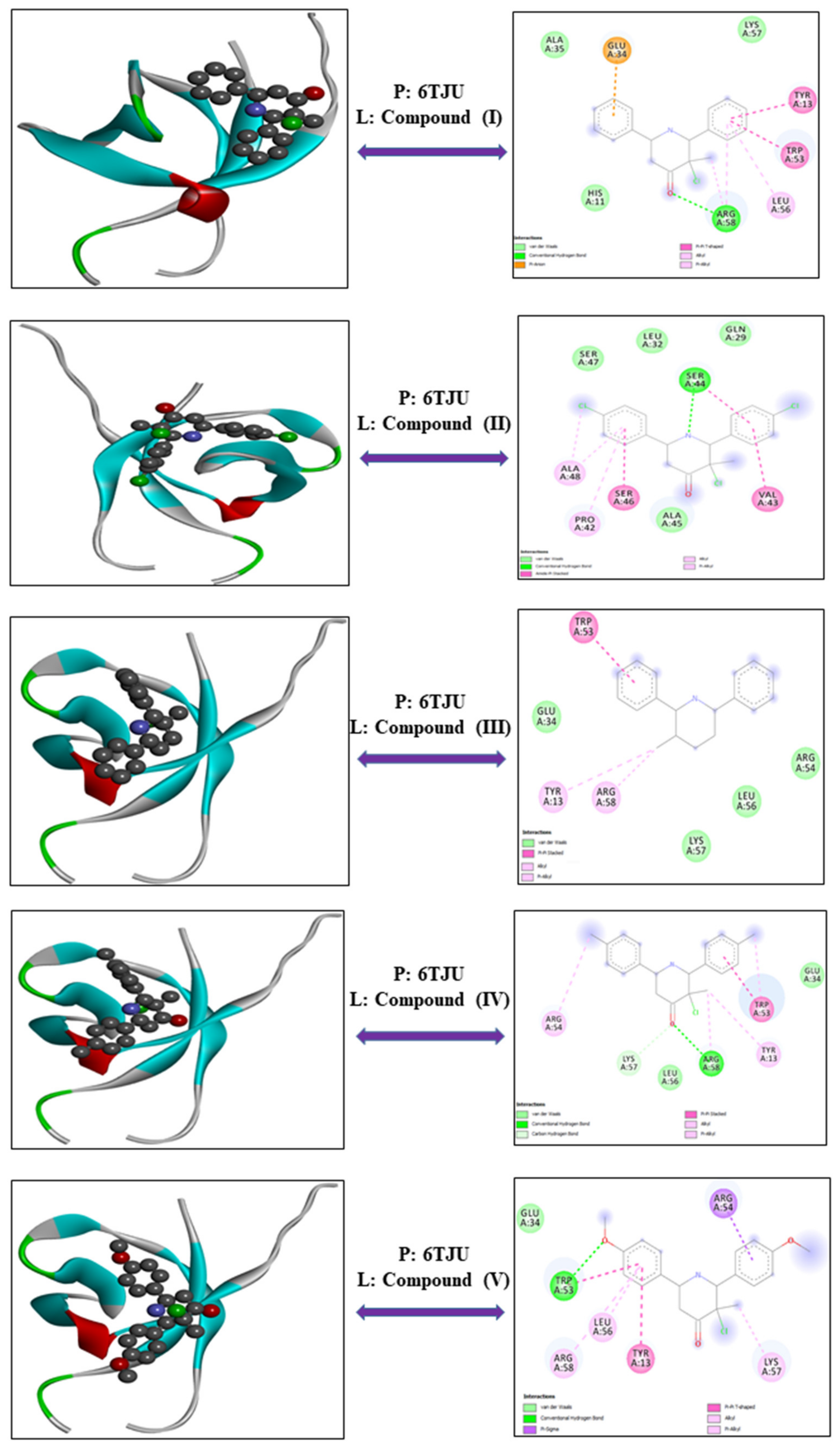
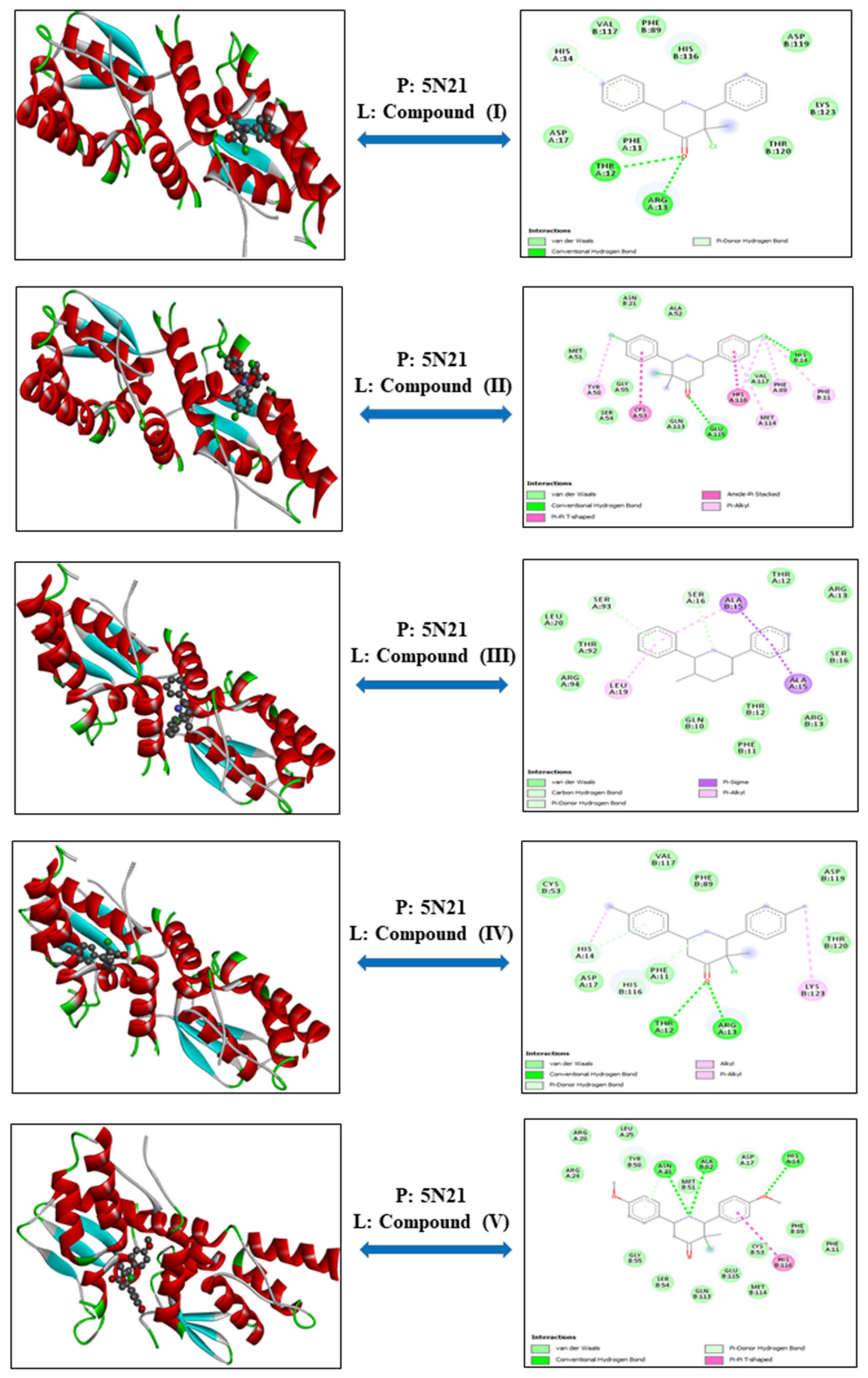
3.1. Molecular Docking Study
3.1.1. Myeloma—Proteins: 6FS1 and 6FSO
3.1.2. Leukemia—Protein: 6TJU
3.1.3. NKT Lymphoma—Proteins: 1OLL and 5N21
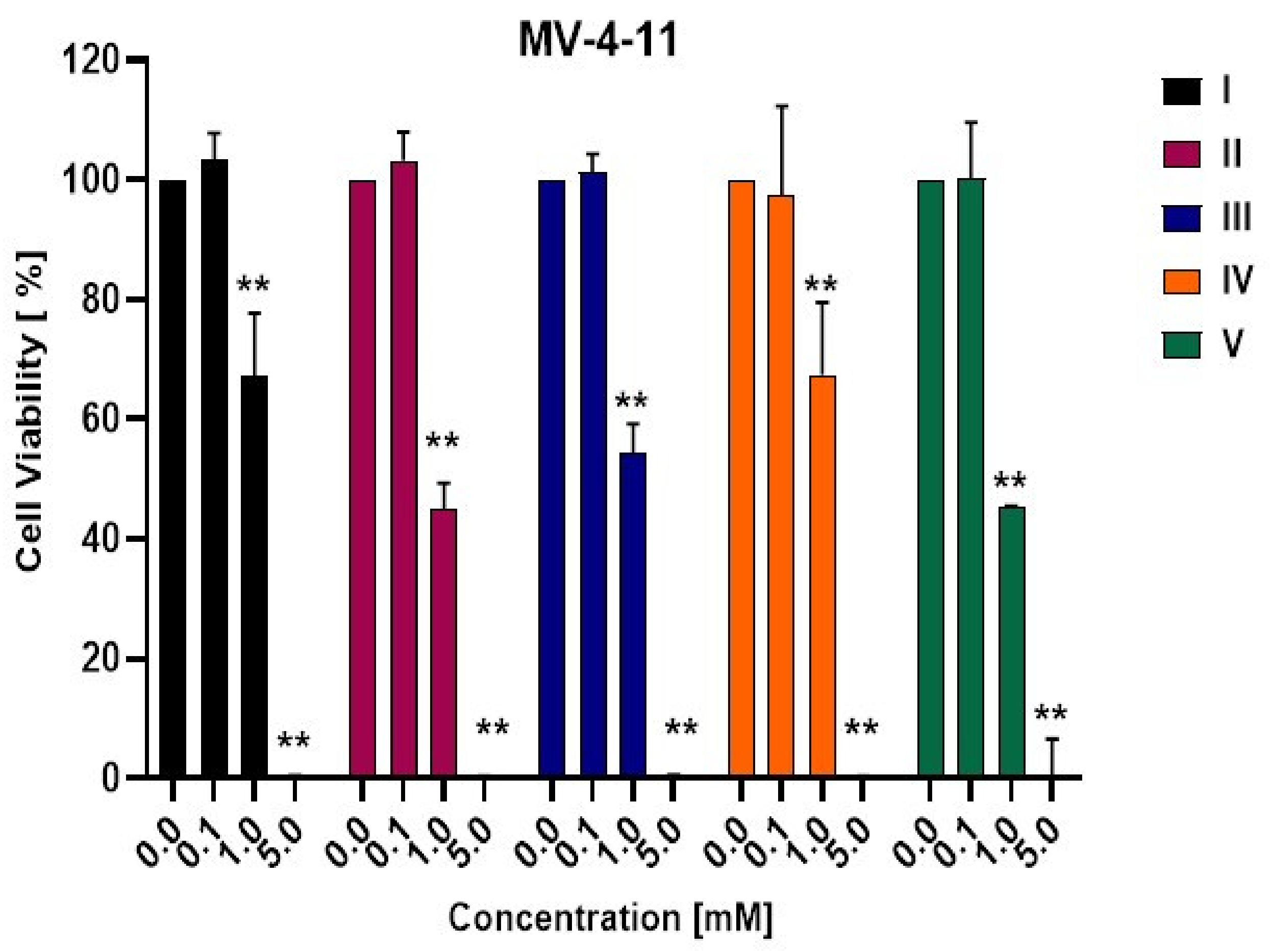
3.2. Biological Activity
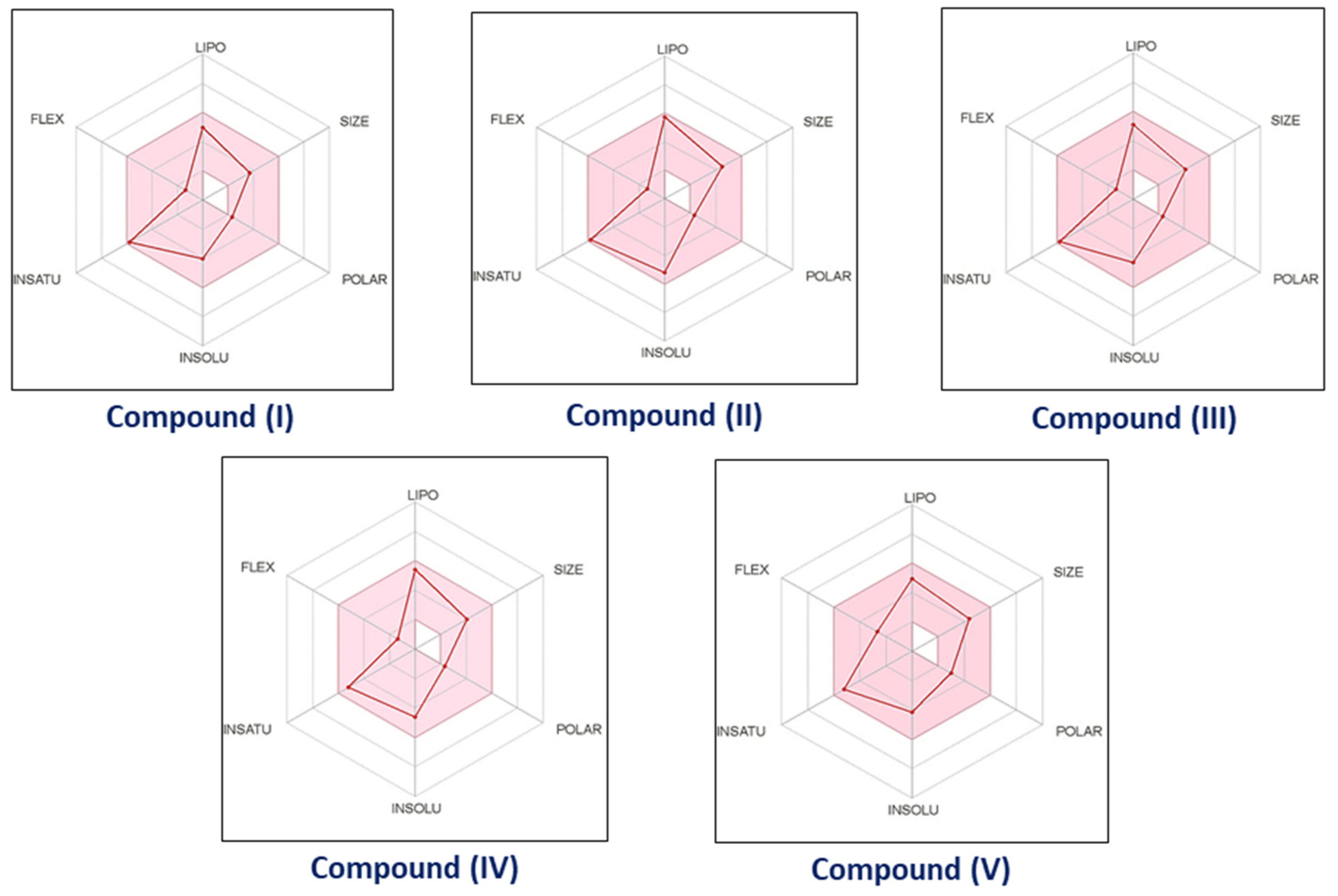
3.3. In Silico ADMET Study
3.3.1. Pharmacokinetic and Drug-like Properties
3.3.2. Gastrointestinal Absorption and Brain Penetration prediction [BOILED-Egg]
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Watson, P.S.; Jiang, B.; Scott, B. A diastereoselective synthesis of 2,4-disubstituted piperidines: Scaffolds for drug discovery. Org. Lett. 2000, 2, 3679–3681. [Google Scholar] [CrossRef]
- Aeluri, R.; Alla, M.; Bommena, V.R.; Murthy, R.; Jain, N. Synthesis and antiproliferative activity of polysubstituted tetrahydropyridineand piperidin-4-one-3-carboxylate derivatives. Asian J. Org. Chem. 2012, 1, 71–79. [Google Scholar] [CrossRef]
- Arulraj, R.; Sivakumar, S.; Mouna, M.; Omar, A.D.; Noureddine, I.; Marek, J.W. Study of a new piperidone as an anti-Alzheimer agent: Molecular docking, electronic and intermolecular interaction investigations by DFT method. J. King Saud Univ.–Sci. 2021, 33, 101632. [Google Scholar] [CrossRef]
- Anitha, K.; Sivakumar, S.; Arulraj, R.; Rajkumar, K.; Dhandapani, A.; Oyeneyin, O.E. Synthesis, molecular docking of 3-(2-chloroethyl)-2,6- diphenylpiperidin-4-one: Hirshfeld surface, spectroscopic and DFT based analyses. J. Mol. Struct. 2022, 1262, 132993. [Google Scholar] [CrossRef]
- Goel, P.; Alam, O.; Naim, M.J.; Nawaz, F.; Iqbal, M.; Alam, M.I. Recent advancement of piperidine moiety in treatment of cancer- A review. Eur. J. Med. Chem. 2018, 157, 480–502. [Google Scholar] [CrossRef]
- Arulraj, R.; Sivakumar, S.; Suresh, S.; Anitha, K. Synthesis, Vibrational spectra, DFT calculations, Hirshfeld surface analysis and Molecular docking study of 3-chloro-3-methyl-2,6-diphenylpiperidin-4-one. Spectrochim. Acta A 2020, 232, 118166. [Google Scholar] [CrossRef]
- Das, S.; Da Silva, C.J.; De Silva, M.M.; De Dantas, M.D.A.; Fátima, A.D.; Gois Ruiz, A.L.T.; Da Silva, C.M.; De Carvalho, J.E.; Santos, J.C.C.; Figueiredo, I.M.; et al. Highly functionalized piperidines: Free radical scavenging, anticancer activity, DNA interaction and correlation with biological activity. J. Adv. Res. 2018, 9, 51–61. [Google Scholar] [CrossRef]
- Kim, H.Y.; Park, E.J.; Joe, E.H.; Jou, I. Curcumin Suppresses Janus Kinase-STAT Inflammatory Signaling through Activation of Src Homology 2 Domain-Containing Tyrosine Phosphatase 2 in Brain Microglia. J. Immunol. 2003, 171, 6072–6079. [Google Scholar] [CrossRef] [Green Version]
- Lacronique, V.; Boureux, A.; Valle, V.D.; Poirel, H.; Quang, C.T.; Mauchauffé, M.; Berthou, C.; Lessard, M.; Berger, R.; Ghysdael, J.; et al. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science 1997, 278, 1309–1312. [Google Scholar] [CrossRef] [Green Version]
- Coppo, P.; Gouilleux-Gruart, V.; Huang, Y.; Bouhlal, H.; Bouamar, H.; Bouchet, S.; Perrot, C.; Vieillard, V.; Dartigues, P.; Gaulard, P.; et al. STAT3 transcription factor is constitutively activated and is oncogenic in nasal-type NK/T-cell. Leukemia 2009, 23, 1667–1678. [Google Scholar] [CrossRef] [Green Version]
- Cirmena, G.; Aliano, S.; Fugazza, G. A BCR-JAK2 fusion gene as the result of a t (9;22) (p24;q11) in a patient with acute myeloid leukemia. Cancer Genet Cytogenet 183:105-108, 2008 Elsevier Inc.: United States. Cancer Genet. Cytogenet. 2008, 183, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Bommert, K.; Bargou, R.C.; Stuhmer, T. Signalling and survival pathways in multiple myeloma. Eur. J. Cancer 2006, 42, 1574–1580. [Google Scholar] [CrossRef] [PubMed]
- Anthwal, A.; Singh, K.; Rawat, M.S.M.; Tyagi, A.K.; Haque, A.; Ali, I.; Rawat, D.S. Synthesis of 4-piperidone Based Curcuminoids with Anti-inflammatory and Anti-Proliferation Potential in Human Cancer Cell Lines. Anticancer Agents Med. Chem. 2016, 16, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.M.; Chen, C.C. GEMDOCK: A generic evolutionary method for molecular docking. Proteins Struct. Funct. Bioinform. 2004, 55, 288–304. [Google Scholar] [CrossRef] [PubMed]
- Discovery Studio Visualizer 2; Accelrys Software Inc.: San Diego, CA, USA, 2005.
- Protein Data Bank (PDB). Available online: http://www.rcsb.org/ (accessed on 10 February 2021).
- Arulraj, R.; Sivakumar, S.; Kaur, M.; Thiruvalluvar, A.; Jasinski, J.P. Crystal structures of three 3-chloro-3-methyl-2,6-di aryl piperidin-4-ones. Acta Cryst. E 2017, 73, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Arulraj, R.; Sivakumar, S.; Thiruvalluvar, A.; Kaur, M.; Jasinski, J.P. 3-Chloro-r-2,c-6-bis (4-fluoro phen yl)-3-methyl piperidin-4-one. IUCrData 2016, 1, x161580. [Google Scholar] [CrossRef] [Green Version]
- Arulraj, R.; Sivakumar, S.; Rajkumar, K.; Jasinski, J.P.; Kaur, M.; Thiruvalluvar, A. Synthesis, Crystal Structure, DFT Calculations and Hirshfeld Surface Analysis of 3-Chloro-3-methyl-r(2),c(6)-bis(p-methoxyphenyl)piperidin-4-one. J. Chem. Crystallogr. 2020, 50, 41–51. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, druglikeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery, A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple selection criteria for drug-like chemical matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. 2000, 44, 235–249. [Google Scholar] [CrossRef]
- GBD 2017 Risk Factor Collaborators. Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks for 195 countries and territories, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1923–1994. [Google Scholar] [CrossRef] [Green Version]
- Noureddine, O.; Issaoui, N.; Gatfaoui, S.; Al-Dossary, O.; Marouani, H. Quantum chemical calculations, spectroscopic properties and molecular docking studies of a novel piperazine derivative. J. King Saud Univ.-Sci. 2021, 33, 101283. [Google Scholar] [CrossRef]
- Noureddine, O.; Gatfaoui, S.; Brandán, S.A.; Marouani, H.; Issaoui, N. Structural, docking and spectroscopic studies of a new piperazine derivative, 1-Phenylpiperazine-1, 4-diium bis (hydrogen sulfate). J. Mol. Struct. 2020, 1202, 127351. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Kita, K.; Miwa, H.; Nishii, K.; Oka, K.; Ohno, T.; Shirakawa, S.; Fukumoto, M. Frequent expression of P-glycoprotein/MDR1 by nasal T-cell lymphoma cells. Cancer 1995, 76, 2351–2356. [Google Scholar] [CrossRef]
- Drénou, B.; Lamy, T.; Amiot, L.; Fardel, O.; Caulet-Maugendre, S.; Sasportes, M.; Diebold, J.; Le Prisé, P.Y.; Fauchet, R. CD3- CD56+ non-Hodgkin’s lymphomas with an aggressive behavior related to multidrug resistance. Blood 1997, 89, 2966–2974. [Google Scholar] [CrossRef] [Green Version]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef] [Green Version]
- Tait, S.; Green, D. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef]
- Muller, M.; Wilder, S.; Bannasch, D.; Israeli, D.; Lehlbach, K.; Li-Weber, M.; Friedman, S.L.; Galle, P.R.; Stremmel, W.; Oren, M.; et al. p53 Activates the CD95 (APO-1/Fas) Gene in Response to DNA Damage by Anticancer Drugs. J. Exp. Med. 1998, 188, 2033–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303, 1010–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anchoori, R.K.; Karanam, B.; Peng, S.; Wang, J.W.; Jiang, R.; Tanno, T.; Orlowski, R.Z.; Matsui, W.; Zhao, M.; Rudek, M.A.; et al. A bis-benzylidine piperidone targeting proteasome ubiquitin receptor RPN13/ADRM1 as a therapy for cancer. Cancer Cell. 2013, 24, 791–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiyama, Y. Druggability: Selecting optimized drug candidates. Drug Discov. Today 2005, 10, 1577–1579. [Google Scholar] [CrossRef]
- Borchardt, R.T. Pharmaceutical Profiling in Drug Discovery for Lead Selection; American Association of Pharmaceutical Scientists: Arlington, VA, USA, 2004. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliver. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Leeson, P.D.; Davis, A.M. Time-related differences in the physical property profiles of oral drugs. J. Med. Chem. 2004, 47, 6338–6348. [Google Scholar] [CrossRef]
- Congreve, M.; Carr, R.; Murray, C.; Jhoti, H. A rule of three for fragment-based lead discovery? Drug Discov. Today 2003, 8, 876–877. [Google Scholar] [CrossRef]
- Wenlock, M.C.; Austin, R.P.; Barton, P.; Davis, A.M.; Leeson, P.D. A comparison of physiochemical property profiles of development and marketed oral drugs. J. Med. Chem. 2003, 46, 1250–1256. [Google Scholar] [CrossRef]
- Manallack, D.T.; Prankerd, R.J.; Yuriev, E.; Oprea, T.I.; Chalmers, D.K. The significance of acid/base properties in drug discovery. Chem. Soc. Rev. 2013, 42, 485–496. [Google Scholar] [CrossRef] [Green Version]
- Han, V.W.; Smith, D.A.; Beaumont, K.; Walker, D.K. Property-based design: Optimization of drug absorption and pharmacokinetics. J. Med. Chem. 2001, 44, 1313–1333. [Google Scholar] [CrossRef]
- Young, R.J.; Campbell, M.; Borthwick, A.D.; Brown, D.; Burns-Kurtis, C.L.; Chan, C.; Convery, M.A.; Crowe, M.C.; Dayal, S.; Diallo, H.; et al. Structure- and property-based design of factor Xa inhibitors: Pyrrolidin-2-ones with acyclic alanyl amides as P4 motifs. Bioorg. Med. Chem. Lett. 2006, 16, 5953–5957. [Google Scholar] [CrossRef] [PubMed]
- Young, R.J.; Borthwick, A.D.; Brown, D.; Burns-Kurtis, C.L.; Campbell, M.; Chan, C.; Charbaut, M.; Chung, C.W.; Convery, M.A.; Kelly, H.A.; et al. Structure and property based design of factor Xa inhibitors: Pyrrolidin-2-ones with biaryl P4 motifs. Bioorg. Med. Chem. Lett. 2008, 18, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Eljack, F.T.; Eden, M.R.; Kazantzi, V.; Qin, X.Y.; El-Halwagi, M.A. Simultaneous process and molecular design—A property based approach. AIChe. J. 2007, 53, 1232–1239. [Google Scholar] [CrossRef]
- Jiang, H.; Zuo, Y.; Zhang, L.; Li, J.D.; Zhang, A.M.; Li, Y.B.; Yang, X.C. Property-based design: Optimization and characterization of polyvinyl alcohol (PVA) hydrogel and PVA-matrix composite for artificial cornea. J. Mater. Sci. Mater. Med. 2014, 25, 941–952. [Google Scholar] [CrossRef]
- Dowling, J.E.; Alimzhanov, M.; Bao, L.; Block, M.H.; Chuaqui, C.; Cooke, E.L.; Denz, C.R.; Hird, A.; Huang, S.; Larsen, N.A.; et al. Structure and property based design of pyrazolo 1,5-a pyrimidine Inhibitors of CK2 kinase with activity in vivo. ACS Med. Chem. Lett. 2013, 4, 800–805. [Google Scholar] [CrossRef] [Green Version]
- Larsen, S.D.; Wilson, M.W.; Abe, A.; Shu, L.M.; George, C.H.; Kirchhoff, P.; Showalter, H.D.H.; Xiang, J.M.; Keep, R.F.; Shayman, J.A. Property-based design of a glucosylceramide synthase inhibitor that reduces glucosylceramide in the brain. J. Lipid Res. 2012, 53, 282–291. [Google Scholar] [CrossRef] [Green Version]
- Fraley, M.E.; Hoffman, W.F.; Arrington, K.L.; Hungate, R.W.; Hartman, G.D.; McFall, R.C.; Coll, K.E.; Rickert, K.; Thomas, K.A.; McGaughey, G.B. Property-based design of KDR kinase inhibitors. Curr. Med. Chem. 2004, 11, 709–719. [Google Scholar] [CrossRef]
- Balakin, K.V.; Tkachenko, S.E.; Lang, S.A.; Okun, I.; Ivashchenko, A.A.; Savchuk, N.P. Property-based design of GPCR-targeted library. J. Chem. Inf. Comp. Sci. 2002, 42, 1332–1342. [Google Scholar] [CrossRef]
- Abraham, M.H.; Ibrahim, A.; Zissimos, A.M.; Zhao, Y.H.; Comer, J.; Reynolds, D.P. Application of hydrogen bonding calculations in property based drug design. Drug Discov. Today 2002, 7, 1056–1063. [Google Scholar] [CrossRef]
- Daina, A.; Zoete, V. A BOILED-Egg to Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [Green Version]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. Prediction of Hydrophobic (Lipophilic) Properties of Small Organic Molecules Using Fragmental Methods: An Analysis of ALOGP and CLOGP Methods. J. Phys. Chem. A 1998, 102, 3762–3772. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Proteins | Compounds | Energy | VDW | H-Bond | Electronic |
|---|---|---|---|---|---|---|
| Myeloma | 6FS1 | Compound—I | −92.28 | −92.17 | −0.12 | 0 |
| Compound—II | −97.40 | −97.31 | −0.09 | 0 | ||
| Compound—III | −93.16 | −93.03 | −0.12 | 0 | ||
| Compound—IV | −89.41 | −89.29 | −0.12 | 0 | ||
| Compound—V | −89.33 | −89.22 | −0.12 | 0 | ||
| 6FSO | Compound—I | −84.44 | −84.34 | −0.09 | 0 | |
| Compound—II | −92.75 | −92.65 | −0.10 | 0 | ||
| Compound—III | −89.45 | −89.37 | −0.08 | 0 | ||
| Compound—IV | −91.33 | −91.23 | −0.10 | 0 | ||
| Compound—V | −95.79 | −95.67 | −0.12 | 0 | ||
| Leukemia | 6TJU | Compound—I | −76.66 | −70.23 | −6.43 | 0 |
| Compound—II | −75.93 | −72.43 | −3.5 | 0 | ||
| Compound—III | −65.70 | −65.70 | 0 | 0 | ||
| Compound—IV | −75.99 | −72.49 | −3.5 | 0 | ||
| Compound—V | −87.11 | −74.93 | −12.18 | 0 | ||
| NK Lymphoma | 5N21 | Compound—I | −95.51 | −95.42 | −0.08 | 0 |
| Compound—II | −93.91 | −93.80 | −0.11 | 0 | ||
| Compound—III | −99.01 | −98.92 | −0.09 | 0 | ||
| Compound—IV | −97.50 | −97.41 | −0.09 | 0 | ||
| Compound—V | −109.10 | −108.97 | −0.13 | 0 | ||
| 1OLL | Compound—I | −77.67 | −77.58 | −0.09 | 0 | |
| Compound—II | −90.05 | −89.94 | −0.11 | 0 | ||
| Compound—III | −78.81 | −78.71 | −0.10 | 0 | ||
| Compound—IV | −81.41 | −81.32 | −0.09 | 0 | ||
| Compound—V | −91.34 | −91.21 | −0.14 | 0 |
| Parameters | Compound (I) | Compound (II) | Compound (III) | Compound (IV) | Compound (V) |
|---|---|---|---|---|---|
| Physicochemical Properties | |||||
| Formula | C18H18ClNO | C18H16Cl3NO | C18H16ClF2NO | C20H22ClNO | C20H22ClNO3 |
| Molecular weight | 299.79 g/mol | 368.68 g/mol | 335.78 g/mol | 327.85 g/mol | 359.85 g/mol |
| SMILES | CC1(Cl)C(NC(CC1=O)C1=CC=CC=C1)C1=CC=CC=C1 | CC1(Cl)C(NC(CC1=O)C1=CC=C(Cl)C=C1)C1=CC=C(Cl)C=C1 | CC1(Cl)C(NC(CC1=O)C1=CC=C(F)C=C1)C1=CC=C(F)C=C1 | CC1=CC=C(C=C1)C1CC(=O)C(C)(Cl)C(N1)C1=CC=C(C)C=C1 | COC1=CC=C(C=C1)C1CC(=O)C(C)(Cl)C(N1)C1=CC=C(OC)C=C1 |
| Num. heavy atoms | 21 | 23 | 23 | 23 | 25 |
| Num. rotatable bonds | 2 | 2 | 2 | 2 | 4 |
| Num. H-bond acceptors | 2 | 2 | 4 | 2 | 4 |
| Num. H-bond donors | 1 | 1 | 1 | 1 | 1 |
| Molar Refractivity | 89.57 | 99.59 | 89.48 | 99.50 | 102.55 |
| Lipophilicity | |||||
| Log Po/w (iLOGP) | 2.70 | 3.20 | 2.90 | 3.20 | 3.25 |
| Log Po/w (XLOGP3) | 3.22 | 4.48 | 3.42 | 3.95 | 3.16 |
| Log Po/w (SILICOS-IT) | 4.21 | 5.46 | 5.02 | 5.22 | 4.28 |
| Consensus Log Po/w | 3.26 | 4.32 | 3.88 | 3.92 | 3.22 |
| Water Solubility | |||||
| Log S (ESOL) | −4.02 | −5.20 | −4.33 | −4.62 | −4.15 |
| Solubility | 2.88 × 10−02 mg/mL; 9.59 × 10−05 mol/L | 2.31 × 10−03 mg/mL; 6.28 × 10−06 mol/L | 1.57 × 10−02 mg/mL; 4.67 × 10−05 mol/L | 7.95 × 10−03 mg/mL; 2.43 × 10−05 mol/L | 2.53 × 10−02 mg/mL; 7.03 × 10−05 mol/L |
| Pharmacokinetics | |||||
| GI absorption | High | High | High | High | High |
| BBB permeant | Yes | Yes | Yes | Yes | Yes |
| P-gp substrate | Yes | Yes | Yes | Yes | Yes |
| CYP1A2 inhibitor | No | No | No | No | No |
| CYP2C19 inhibitor | Yes | Yes | Yes | Yes | Yes |
| CYP2C9 inhibitor | No | No | No | No | No |
| CYP2D6 inhibitor | Yes | Yes | Yes | Yes | Yes |
| CYP3A4 inhibitor | No | Yes | Yes | No | Yes |
| Log Kp (skin permeation) | −5.84 cm/s | −5.37 cm/s | −5.92 cm/s | −5.50 cm/s | −6.25 cm/s |
| Druglikeness | |||||
| Lipinski | Yes; 0 violation | Yes; 0 violation | Yes; 0 violation | Yes; 0 violation | Yes; 0 violation |
| Ghose | Yes | Yes | Yes | Yes | Yes |
| Veber | Yes | Yes | Yes | Yes | Yes |
| Egan | Yes | Yes | Yes | Yes | Yes |
| Muegge | Yes | Yes | Yes | Yes | Yes |
| Bioavailability Score | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 |
| Medicinal Chemistry | |||||
| Leadlikeness | Yes | No; 2 violations: MW > 350, XLOGP3 > 3.5 | Yes | No; 1 violation: XLOGP3 > 3.5 | No; 1 violation: MW > 350 |
| Synthetic accessibility | 3.08 | 3.16 | 3.15 | 3.30 | 3.36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramalingam, A.; Mustafa, N.; Chng, W.J.; Medimagh, M.; Sambandam, S.; Issaoui, N. 3-Chloro-3-methyl-2,6-diarylpiperidin-4-ones as Anti-Cancer Agents: Synthesis, Biological Evaluation, Molecular Docking, and In Silico ADMET Prediction. Biomolecules 2022, 12, 1093. https://doi.org/10.3390/biom12081093
Ramalingam A, Mustafa N, Chng WJ, Medimagh M, Sambandam S, Issaoui N. 3-Chloro-3-methyl-2,6-diarylpiperidin-4-ones as Anti-Cancer Agents: Synthesis, Biological Evaluation, Molecular Docking, and In Silico ADMET Prediction. Biomolecules. 2022; 12(8):1093. https://doi.org/10.3390/biom12081093
Chicago/Turabian StyleRamalingam, Arulraj, Nurulhuda Mustafa, Wee Joo Chng, Mouna Medimagh, Sivakumar Sambandam, and Noureddine Issaoui. 2022. "3-Chloro-3-methyl-2,6-diarylpiperidin-4-ones as Anti-Cancer Agents: Synthesis, Biological Evaluation, Molecular Docking, and In Silico ADMET Prediction" Biomolecules 12, no. 8: 1093. https://doi.org/10.3390/biom12081093