
The Blocking of Drug Resistance Channels by Selected Hydrophobic Statins in Chemoresistance Human Melanoma
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Cultures and Preparation of Biological Material
2.3. Performing the MTT Test
2.4. Determination of Protein Concentration
2.5. Pgp Protein ATPase Activity Test
2.6. Microscopic Test of the Annexin V-Propidium Iodide Staining of Apoptotic Cells
2.7. Evaluation of the Genotoxicity of the Drugs Used in Melanoma Cells
2.7.1. Description of Experimental Groups
2.7.2. Comet System 3.0. Quantitative DNA Damage Analysis [28,29] (Description in the Supplementary Materials)
2.8. Analysis of the Absorption of Pharmaceuticals into Human Melanoma Cells
2.9. In Vivo Studies in SCID Mice
2.10. Statistical Methods
3. Results
3.1. Effect of Docetaxel and the Combination of Docetaxel with Statins on Melanoma Cell Viability (MTT Test)
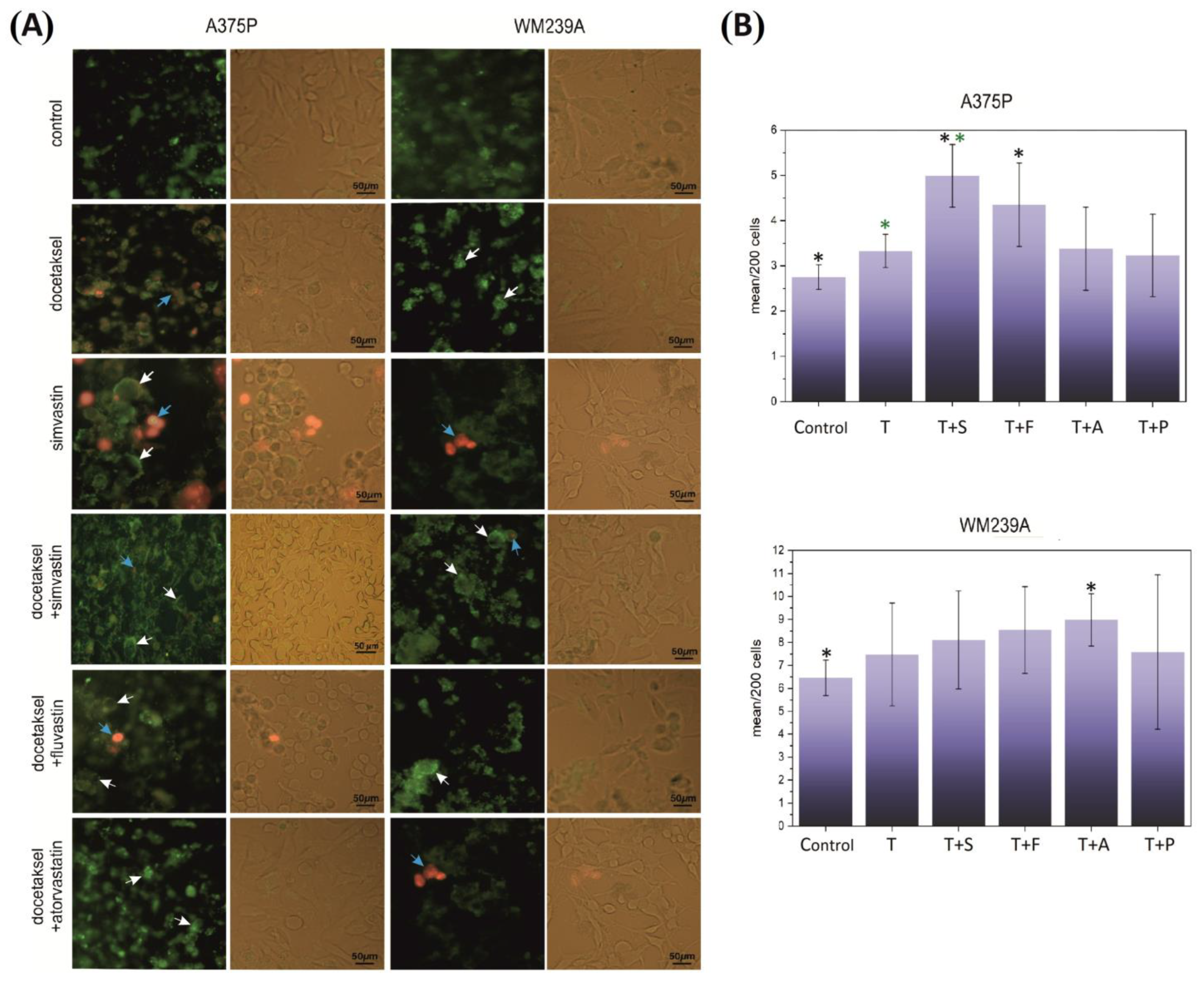
3.2. Effect of Docetaxel and the Combination of Docetaxel with Statins on the Necroptosis of Metastatic Melanoma Cells

3.3. Effect of Docetaxel and the Combination of Docetaxel with Simvastatin on the Level of DNA Damage Measured by the Comet Assay
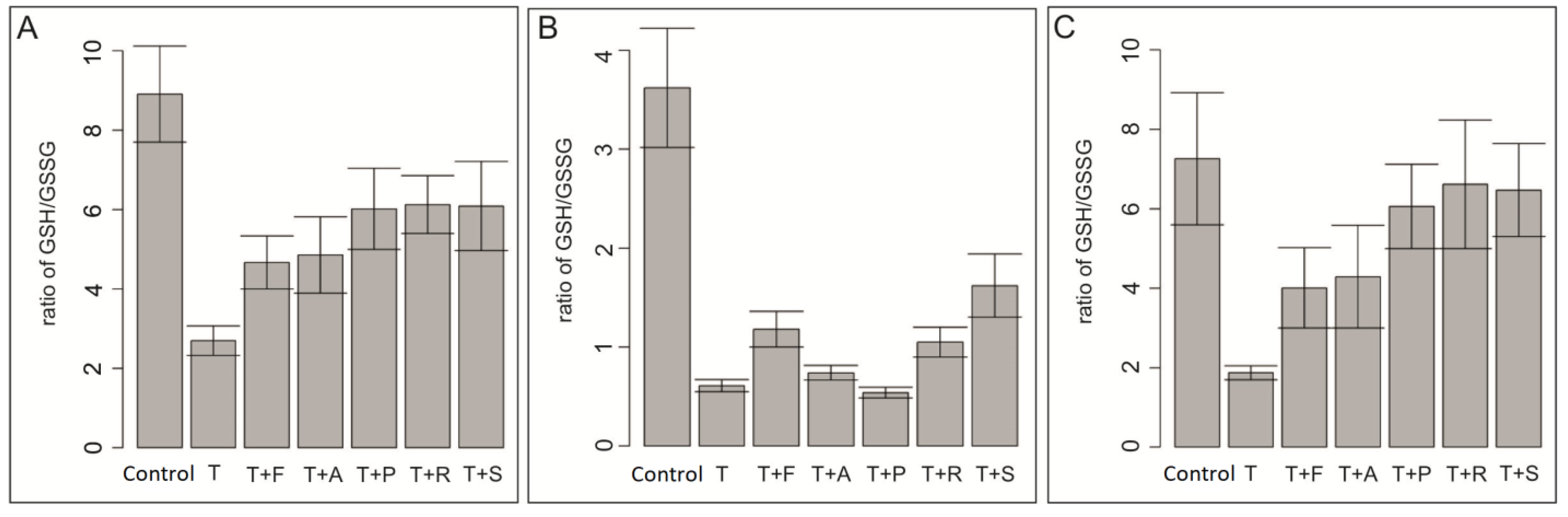
3.4. Effect of Docetaxel and the Combination of Docetaxel with Simvastatin on the Level of Total Glutathione in the Cells
3.5. Effect of Docetaxel and the Combination of Docetaxel with Simvastatin on Tumor Growth in Mice Induced from the A375P Cell Line
3.6. Effect of Docetaxel and the Combination of Docetaxel with Simvastatin on the Action Potential of the ABCB1 Drug Channel
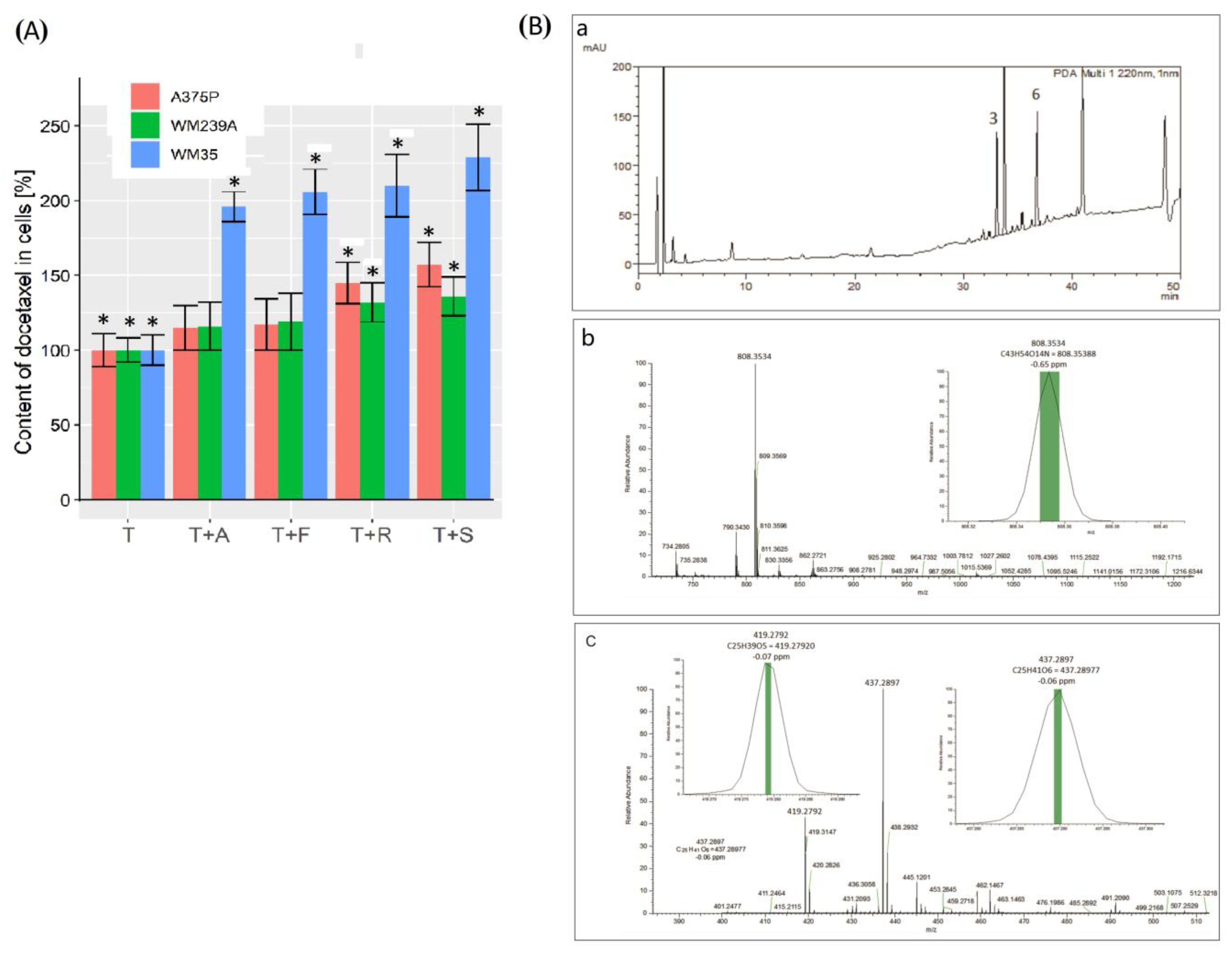
3.7. Recovery of Pharmaceuticals from Human Melanoma Cells
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pathania, S.; Bhatia, R.; Baldi, A.; Singh, R.; Rawal, R.K. Drug Metabolizing Enzymes and Their Inhibitors’ Role in Cancer Resistance. Biomed. Pharmacother. 2018, 105, 53–65. [Google Scholar] [CrossRef]
- Ye, Z.; Lu, Y.; Wu, T. The Impact of ATP-Binding Cassette Transporters on Metabolic Diseases. Nutr. Metab. 2020, 17, 61. [Google Scholar] [CrossRef] [PubMed]
- Locher, K.P. Mechanistic Diversity in ATP-Binding Cassette (ABC) Transporters. Nat. Struct. Mol. Biol. 2016, 23, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Locher, K.P. Structure and Mechanism of ATP-Binding Cassette Transporters. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 239–245. [Google Scholar] [CrossRef] [PubMed]
- ABCB1 ATP Binding Cassette Subfamily B Member 1 [Homo Sapiens (Human)]—Gene—NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/5243 (accessed on 23 January 2023).
- Chen, K.G.; Gottesman, M.M. How Melanoma Cells Evade Chemotherapy Role of Transporter-Dependent and -Independent Resistance Mechanisms. In From Melanocytes to Melanoma: The Progression to Malignancy; Humana Press Inc.: Totowa, NJ, USA, 2006; pp. 591–603. [Google Scholar] [CrossRef]
- Zong, Y.; Zhou, S.; Sorrentino, B.P. Loss of P-Glycoprotein Expression in Hematopoietic Stem Cells Does Not Improve Responses to Imatinib in a Murine Model of Chronic Myelogenous Leukemia. Leukemia 2005, 19, 1590–1596. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Kim, Y.K. Cancer Stem Cells as a Potential Target to Overcome Multidrug Resistance. Front. Oncol. 2020, 10, 764. [Google Scholar] [CrossRef]
- Vasan, N.; Baselga, J.; Hyman, D.M. A View on Drug Resistance in Cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef]
- Seelig, A. P-Glycoprotein: One Mechanism, Many Tasks and the Consequences for Pharmacotherapy of Cancers. Front. Oncol. 2020, 10, 576559. [Google Scholar] [CrossRef]
- Microtubules—The Cell—NCBI Bookshelf. Available online: https://www.ncbi.nlm.nih.gov/books/NBK9932/ (accessed on 23 January 2023).
- Sazonova, E.V.; Petrichuk, S.V.; Kopeina, G.S.; Zhivotovsky, B. A Link between Mitotic Defects and Mitotic Catastrophe: Detection and Cell Fate. Biol. Direct 2021, 16, 25. [Google Scholar] [CrossRef]
- Attard, G.; Greystoke, A.; Kaye, S.; De Bono, J. Update on Tubulin-Binding Agents. Pathol. Biol. 2006, 54, 72–84. [Google Scholar] [CrossRef]
- Khrapunovich-Baine, M.; Menon, V.; Yang, C.P.H.; Northcote, P.T.; Miller, J.H.; Angeletti, R.H.; Fiser, A.; Horwitz, S.B.; Xiao, H. Hallmarks of Molecular Action of Microtubule Stabilizing Agents: Effects of Epothilone B, Ixabepilone, Peloruside A, and Laulimalide on Microtubule Conformation. J. Biol. Chem. 2011, 286, 11765–11778. [Google Scholar] [CrossRef] [PubMed]
- Abraham, J.; Agrawal, M.; Bakke, S.; Rutt, A.; Edgerly, M.; Balis, F.M.; Widemann, B.; Davis, L.; Damle, B.; Sonnichsen, D.; et al. Phase I Trial and Pharmacokinetic Study of BMS-247550, an Epothilone B Analog, Administered Intravenously on a Daily Schedule for Five Days. J. Clin. Oncol. 2003, 21, 1866–1873. [Google Scholar] [CrossRef]
- Sessa, C.; Perotti, A.; Lladò, A.; Cresta, S.; Capri, G.; Voi, M.; Marsoni, S.; Corradino, I.; Gianni, L. Phase I Clinical Study of the Novel Epothilone B Analogue BMS-310705 given on a Weekly Schedule. Ann. Oncol. 2007, 18, 1548–1553. [Google Scholar] [CrossRef] [PubMed]
- Roché, H.; Yelle, L.; Cognetti, F.; Mauriac, L.; Bunnell, C.; Sparano, J.; Kerbrat, P.; Delord, J.P.; Vahdat, L.; Peck, R.; et al. Phase II Clinical Trial of Ixabepilone (BMS-247550), an Epothilone B Analog, as First-Line Therapy in Patients with Metastatic Breast Cancer Previously Treated with Anthracycline Chemotherapy. J. Clin. Oncol. 2007, 25, 3415–3420. [Google Scholar] [CrossRef]
- Hamed, A.R.; Abdel-Azim, N.S.; Shams, K.A.; Hammouda, F.M. Targeting Multidrug Resistance in Cancer by Natural Chemosensitizers. Bull. Natl. Res. Cent. 2019, 43, 8. [Google Scholar] [CrossRef]
- Krishna, R.; Mayer, L.D. Modulation of P-Glycoprotein (PGP) Mediated Multidrug Resistance (MDR) Using Chemosensitizers: Recent Advances in the Design of Selective MDR Modulators. Curr. Med. Chem. Anticancer Agents 2001, 1, 163–174. [Google Scholar] [CrossRef] [PubMed]
- An, C.S.; Kim, S.Y.; Park, C.M. A Role of Cyclosporine A That Suppresses Multi-Drug Resistance (MDR) in the Secondary Chemotherapy Drug Resistant Cell Line of Ovarian Cancer. Korean J. Gynecol. Oncol. 2006, 17, 286. [Google Scholar] [CrossRef]
- Hofsli, E.; Nissen-Meyer, J. Effect of Erythromycin and Tumour Necrosis Factor on the Drug Resistance of Multidrug-Resistant Cells: Reversal of Drug Resistance by Erythromycin. Int. J. Cancer 1989, 43, 520–525. [Google Scholar] [CrossRef]
- Dönmez, Y.; Akhmetova, L.; İşeri, Ö.D.; Kars, M.D.; Gündüz, U. Effect of MDR Modulators Verapamil and Promethazine on Gene Expression Levels of MDR1 and MRP1 in Doxorubicin-Resistant MCF-7 Cells. Cancer Chemother. Pharmacol. 2011, 67, 823–828. [Google Scholar] [CrossRef]
- Nanayakkara, A.K.; Follit, C.A.; Chen, G.; Williams, N.S.; Vogel, P.D.; Wise, J.G. Targeted Inhibitors of P-Glycoprotein Increase Chemotherapeutic-Induced Mortality of Multidrug Resistant Tumor Cells. Sci. Rep. 2018, 8, 967. [Google Scholar] [CrossRef]
- Liscovitch, M.; Lavie, Y. Cancer Multidrug Resistance: A Review of Recent Drug Discovery Research. IDrugs 2002, 5, 349–355. [Google Scholar]
- Wong, J.; Quinn, C.M.; Gelissen, I.C.; Jessup, W.; Brown, A.J. The Effect of Statins on ABCA1 and ABCG1 Expression in Human Macrophages Is Influenced by Cellular Cholesterol Levels and Extent of Differentiation. Atherosclerosis 2008, 196, 180–189. [Google Scholar] [CrossRef]
- Holtzman, C.W.; Wiggins, B.S.; Spinler, S.A. Role of P-Glycoprotein in Statin Drug Interactions. Pharmacotherapy 2006, 26, 1601–1607. [Google Scholar] [CrossRef] [PubMed]
- Sieczkowski, E.; Lehner, C.; Ambros, P.F.; Hohenegger, M. Double Impact on P-Glycoprotein by Statins Enhances Doxorubicin Cytotoxicity in Human Neuroblastoma Cells. Int. J. Cancer 2010, 126, 2025–2035. [Google Scholar] [CrossRef] [PubMed]
- Kopjar, N.; Milas, I.; Garaj-Vrhovac, V.; Gamulin, M. Alkaline Comet Assay Study with Breast Cancer Patients: Evaluation of Baseline and Chemotherapy-Induced DNA Damage in Non-Target Cells. Clin. Exp. Med. 2006, 6, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Miszczyk, J.; Panek, A.; Rawojć, K.; Swakoń, J.; Prasanna, P.G.S.; Rydygier, M.; Gałaś, A. Effects of 60 MeV Protons and 250 KV X-Rays on Cell Viability. Acta Phys. Pol. A 2016, 129, 222–225. [Google Scholar] [CrossRef]
- Bronowicka-Adamska, P.; Zagajewski, J.; Wróbel, M. An Application of RP-HPLC for Determination of the Activity of Cystathionine β-Synthase and γ-Cystathionase in Tissue Homogenates. Nitric Oxide 2015, 46, 186–191. [Google Scholar] [CrossRef]
- Placha, W.; Zagajewski, J.; Suder, P.; Piwowar, M. A Method of Isolating and Analysing Drugs from Cancer Cells for Preclinical Research. J. Chromatogr. A 2022, 1682, 463500. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2016; ISBN 3-900051-07-0. [Google Scholar]
- Zarros, A.; Ye, Y.; John, D.; Lane, R.; Salomone, S.; Falzone, L.; Libra, M. Evolution of Cancer Pharmacological Treatments at the Turn of the Third Millennium. Front. Pharmacol. 2018, 9, 1300. [Google Scholar] [CrossRef]
- Arslan Isik, N.; Emir, I. The Effect of Preoperative Anxiety on Postoperative Symptoms in Patients Without a History of Anxiety Scheduled for Coronary Artery Bypass Grafting. Galician Med. J. 2022, 29, E202237. [Google Scholar] [CrossRef]
- Saaki, A.; Yokomatsu, Y.; Kojima, K.A.; Yoshikawa, T.; Sannomiya, Y.; Tatsumi, J.; Hashimoto, K.; Park, K.; Kawarabayashi, K.; Yoshikawa, C. [Combination Chemotherapy with Cyclophosphamide, Adriamycin, Vincristine and Prednisolone (VEPA) for Non-Hodgkin’s Lymphoma]. Gan Kagaku Ryoho. 1986, 13, 65–69. [Google Scholar]
- Sacks, D.; Baxter, B.; Campbell, B.C.V.; Carpenter, J.S.; Cognard, C.; Dippel, D.; Eesa, M.; Fischer, U.; Hausegger, K.; Hirsch, J.A.; et al. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int. J. Stroke Off. J. Int. Stroke Soc. 2018, 13, 612–632. [Google Scholar] [CrossRef]
- Dallavalle, S.; Dobričić, V.; Lazzarato, L.; Gazzano, E.; Machuqueiro, M.; Pajeva, I.; Tsakovska, I.; Zidar, N.; Fruttero, R. Improvement of Conventional Anti-Cancer Drugs as New Tools against Multidrug Resistant Tumors. Drug Resist. Updat. Rev. Comment. Antimicrob. Anticancer Chemother. 2020, 50, 100682. [Google Scholar] [CrossRef] [PubMed]
- Bethesda (MD) LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. In National Institute of Diabetes and Digestive and Kidney Diseases; Bathesda: Montgomer, MD, USA, 2022.
- Nussinov, R.; Tsai, C.-J.; Jang, H. Anticancer Drug Resistance: An Update and Perspective. Drug Resist. Updat. Rev. Comment. Antimicrob. Anticancer Chemother. 2021, 59, 100796. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Tomás, R. Multidrug Resistance: Retrospect and Prospects in Anti-Cancer Drug Treatment. Curr. Med. Chem. 2006, 13, 1859–1876. [Google Scholar] [CrossRef]
- Istvan, E. Statin Inhibition of HMG-CoA Reductase: A 3-Dimensional View. Atheroscler. Suppl. 2003, 4, 3–8. [Google Scholar] [CrossRef]
- Toyoda, S.; Haruyama, A.; Inami, S.; Arikawa, T.; Saito, F.; Watanabe, R.; Sakuma, M.; Abe, S.; Nakajima, T.; Tanaka, A.; et al. Effects of Carvedilol vs Bisoprolol on Inflammation and Oxidative Stress in Patients with Chronic Heart Failure. J. Cardiol. 2020, 75, 140–147. [Google Scholar] [CrossRef]
- Libby, P.; Aikawa, M. Mechanisms of Plaque Stabilization with Statins. Am. J. Cardiol. 2003, 91, 4B–8B. [Google Scholar] [CrossRef]
- Nilsson, J. Atherosclerotic Plaque Vulnerability in the Statin Era. Eur. Heart J. 2017, 38, 1638–1644. [Google Scholar] [CrossRef]
- Undas, A. Statins in Prevention of Thromboembolic Events: From Seminal Studies to Recent Advances. Pol. Arch. Intern. Med. 2022, 132, 16208. [Google Scholar] [CrossRef]
- Rao, A.K.; Del Carpio-Cano, F.; Janapati, S.; Zhao, H.; Voelker, H.; Lu, X.; Criner, G. Effects of Simvastatin on Tissue Factor Pathway of Blood Coagulation in STATCOPE (Simvastatin in the Prevention of COPD Exacerbations) Trial. J. Thromb. Haemost. 2021, 19, 1709–1717. [Google Scholar] [CrossRef] [PubMed]
- Wiesbauer, F.; Kaun, C.; Zorn, G.; Maurer, G.; Huber, K.; Wojta, J. HMG CoA Reductase Inhibitors Affect the Fibrinolytic System of Human Vascular Cells in Vitro: A Comparative Study Using Different Statins. Br. J. Pharmacol. 2002, 135, 284–292. [Google Scholar] [CrossRef]
- Opelz, G.; Wujciak, T.; Ritz, E. Association of Chronic Kidney Graft Failure with Recipient Blood Pressure. Collaborative Transplant Study. Kidney Int. 1998, 53, 217–222. [Google Scholar] [CrossRef]
- Zeiser, R. Immune Modulatory Effects of Statins. Immunology 2018, 154, 69–75. [Google Scholar] [CrossRef]
- Amarenco, P.; Tonkin, A.M. Statins for Stroke Prevention: Disappointment and Hope. Circulation 2004, 109, III44-9. [Google Scholar] [CrossRef] [PubMed]
- Ezad, S.; Cheema, H.; Collins, N. Statin-Induced Rhabdomyolysis: A Complication of a Commonly Overlooked Drug Interaction. Oxf. Med. Case Rep. 2018, 2018, omx104. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Kimura, J. Mechanism of Statin-Induced Rhabdomyolysis. J. Pharmacol. Sci. 2013, 123, 289–294. [Google Scholar] [CrossRef]
- Tilija Pun, N.; Jeong, C.-H. Statin as a Potential Chemotherapeutic Agent: Current Updates as a Monotherapy, Combination Therapy, and Treatment for Anti-Cancer Drug Resistance. Pharmaceuticals 2021, 14, 470. [Google Scholar] [CrossRef]
- Zhang, Y.; Tacheva-Grigorova, S.K.; Sutton, J.; Melton, Z.; Mak, Y.S.L.; Lay, C.; Smith, B.A.; Sai, T.; Van Blarcom, T.; Sasu, B.J.; et al. Allogeneic CAR T Cells Targeting DLL3 Are Efficacious and Safe in Preclinical Models of Small Cell Lung Cancer. Clin. Cancer Res. 2023, 29, 971–985. [Google Scholar] [CrossRef]
- Sopik, V.; Narod, S.A. The Relationship between Tumour Size, Nodal Status and Distant Metastases: On the Origins of Breast Cancer. Breast Cancer Res. Treat. 2018, 170, 647–656. [Google Scholar] [CrossRef]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular Principles of Metastasis: A Hallmark of Cancer Revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef]
- Szczurek, E.; Krüger, T.; Klink, B.; Beerenwinkel, N. A Mathematical Model of the Metastatic Bottleneck Predicts Patient Outcome and Response to Cancer Treatment. PLoS Comput. Biol. 2020, 16, e1008056. [Google Scholar] [CrossRef]
- Norton, L.; Simon, R.; Brereton, H.D.; Bogden, A.E. Predicting the Course of Gompertzian Growth. Nature 1976, 264, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Laird, A.K. Dynamics of Tumor Growth. Br. J. Cancer 1964, 18, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.T.; Karlseder, J. DNA Damage Associated with Mitosis and Cytokinesis Failure. Oncogene 2013, 32, 4593–4601. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu, H.; Olsson, M.; Zhivotovsky, B. Death through a Tragedy: Mitotic Catastrophe. Cell Death Differ. 2008, 15, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, L.-B.; Zhang, J.; Tang, D.-P.; Wei, J.-J.; Zhuang, Z.-H. Simvastatin, but Not Pravastatin, Inhibits the Proliferation of Esophageal Adenocarcinoma and Squamous Cell Carcinoma Cells: A Cell-Molecular Study. Lipids Health Dis. 2018, 17, 290. [Google Scholar] [CrossRef]
- Dreyer, C.; Placha, W.; Zgajewski, J.; Dygut, J.; Stangel-Wójcikiewicz, K.; Piwowar, M. The Similarity of Selected Statins—A Comparative Analysis. Bio-Algorithms Med-Syst. 2018, 14, 1–5. [Google Scholar] [CrossRef]
- Bonovas, S.; Sitaras, N.M. Does Pravastatin Promote Cancer in Elderly Patients? A Meta-Analysis. Can. Med. Assoc. J. 2007, 176, 649–654. [Google Scholar] [CrossRef]
- Aharoni-Simon, M.; Hann-Obercyger, M.; Pen, S.; Madar, Z.; Tirosh, O. Fatty Liver Is Associated with Impaired Activity of PPARγ-Coactivator 1α (PGC1α) and Mitochondrial Biogenesis in Mice. Lab. Investig. 2011, 91, 1018–1028. [Google Scholar] [CrossRef]
- Czerwinska, P.; Jaworska, A.M.; Wlodarczyk, N.A.; Mackiewicz, A.A. Melanoma Stem Cell-Like Phenotype and Significant Suppression of Immune Response within a Tumor Are Regulated by TRIM28 Protein. Cancers 2020, 12, 2998. [Google Scholar] [CrossRef] [PubMed]
- Kalirai, H.; Damato, B.E.; Coupland, S.E. Uveal Melanoma Cell Lines Contain Stem-like Cells That Self-Renew, Produce Differentiated Progeny, and Survive Chemotherapy. Investig. Opthalmol. Vis. Sci. 2011, 52, 8458–8466. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Placha, W.; Suder, P.; Panek, A.; Bronowicka-Adamska, P.; Zarzycka, M.; Szczygieł, M.; Zagajewski, J.; Piwowar, M.W. The Blocking of Drug Resistance Channels by Selected Hydrophobic Statins in Chemoresistance Human Melanoma. Biomolecules 2023, 13, 1682. https://doi.org/10.3390/biom13121682
Placha W, Suder P, Panek A, Bronowicka-Adamska P, Zarzycka M, Szczygieł M, Zagajewski J, Piwowar MW. The Blocking of Drug Resistance Channels by Selected Hydrophobic Statins in Chemoresistance Human Melanoma. Biomolecules. 2023; 13(12):1682. https://doi.org/10.3390/biom13121682
Chicago/Turabian StylePlacha, Wojciech, Piotr Suder, Agnieszka Panek, Patrycja Bronowicka-Adamska, Marta Zarzycka, Małgorzata Szczygieł, Jacek Zagajewski, and Monika Weronika Piwowar. 2023. "The Blocking of Drug Resistance Channels by Selected Hydrophobic Statins in Chemoresistance Human Melanoma" Biomolecules 13, no. 12: 1682. https://doi.org/10.3390/biom13121682