

The Induction Mechanism of Ferroptosis, Necroptosis, and Pyroptosis in Inflammatory Bowel Disease, Colorectal Cancer, and Intestinal Injury

Abstract
:1. Introduction
2. Mechanisms of Ferroptosis, Necroptosis, and Pyroptosis
2.1. Mechanism of Ferroptosis
2.1.1. GSH/Gpx4–Lipid Peroxidation Pathway
2.1.2. Iron Metabolism Imbalance Pathway
2.1.3. Other Mechanisms of Ferroptosis
2.2. Mechanism of Necroptosis
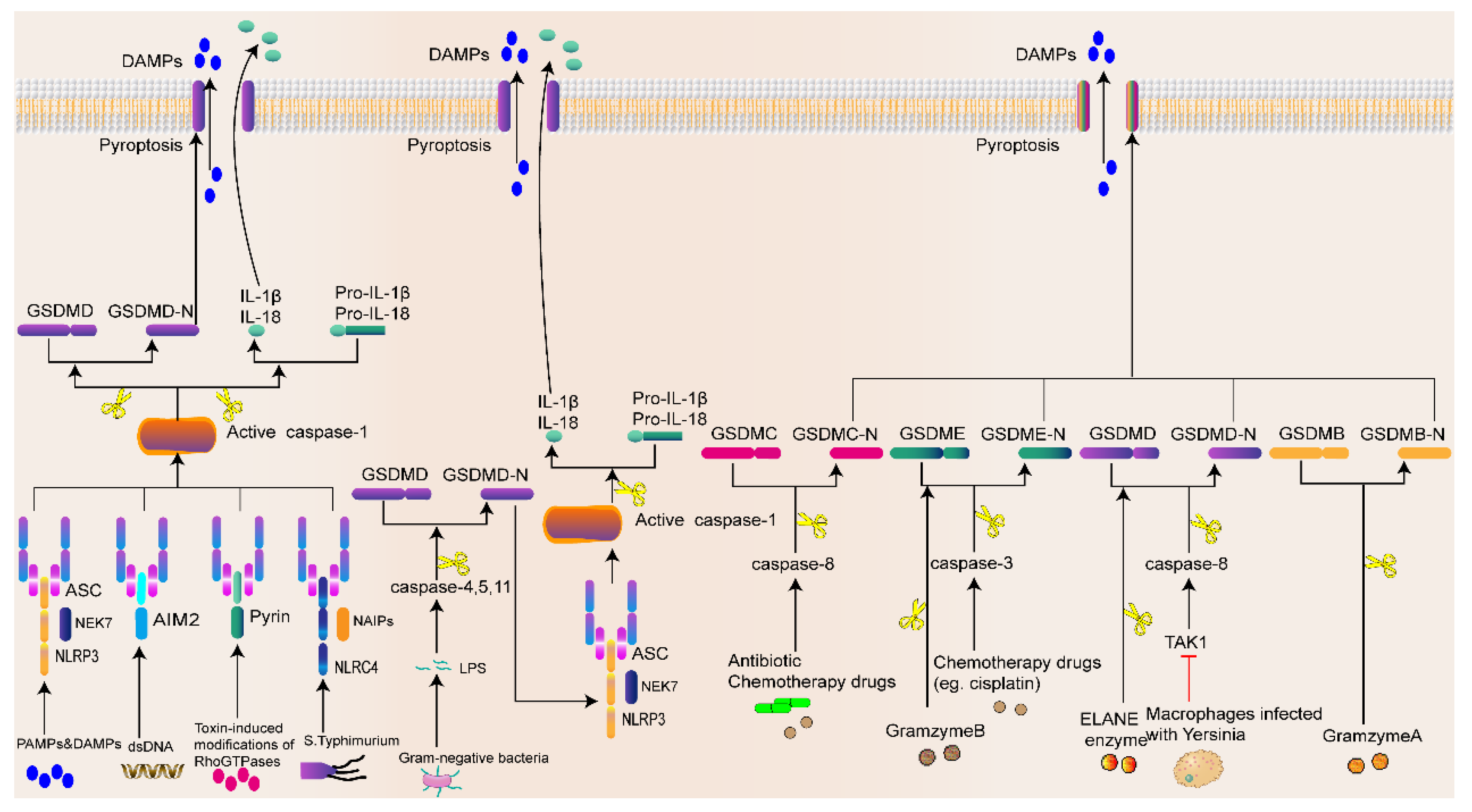
2.3. Mechanisms of Pyroptosis
2.3.1. Inflammasome-Activated Pyroptosis Pathway
2.3.2. Noninflammasome-Activated Pyroptosis Pathway
3. Role of Ferroptosis, Necroptosis, and Pyroptosis in Intestinal Diseases
3.1. Role of Ferroptosis in Intestinal Diseases
3.1.1. Ferroptosis and IBD
3.1.2. Ferroptosis and CRC
3.1.3. Ferroptosis and Intestinal Injury

{kind=link}
{kind=link}
{kind=link}
| Compound/Target | Model | Effect | Mechanism | Ref. |
|---|---|---|---|---|
| Fer-1 | TNBS-induced colitis mice | Inhibition | Ferrostatin-1 alleviates TNBS-induced colitis via the inhibition of ferroptosis. | [76] |
| Fer-1/Lip-1/DFP | DSS-induced colitis mice | Inhibition | Ferroptosis-mediated DSS-induced ulcerative colitis is associated with the Nrf2–HO-1 signaling pathway. | [79] |
| Furin | DSS-induced colitis mice; NCM460 cells | Inhibition | Furin inhibits epithelial cell injury and alleviates experimental colitis by activating the Nrf2–Gpx4 signaling pathway. | [81] |
| APS | DSS-induced colitis mice; Caco-2 cells | Inhibition | Astragalus polysaccharide prevents ferroptosis in a murine model of experimental colitis and human Caco-2 cells by inhibiting the NRf2–HO-1 pathway. | [84] |
| Curculigoside | DSS-induced colitis mice; IEC6-cells | Inhibition | Curculigoside inhibits ferroptosis in ulcerative colitis through the induction of GPX4. | [85] |
| RSL3 | HCT116/LoVo/HT29 CRC cells | Induction | RSL3 drives ferroptosis through GPX4 inactivation and ROS production in colorectal cancer. | [6] |
| SLC7A11 | HT29-cells | Induction | Targeting SLC7A11 specifically suppresses the progression of colorectal cancer stem cells by inducing ferroptosis. | [13] |
| Resibufogenin | HT29/SW480/NCM460 cells | Induction | Resibufogenin inhibits colorectal cancer cell growth and tumorigenesis by triggering ferroptosis and ROS production mediated by GPX4 inactivation. | [86] |
| MiR-19a | HT29-cells | Inhibition | MiR-19a suppresses the ferroptosis of colorectal cancer cells by targeting IREB2. | [90] |
| TIGAR | SW620/HCT116 cells | Induction | TIGAR drives colorectal cancer ferroptosis resistance through the ROS–AMPK–SCD1 pathway. | [92] |
| β-Elemonic acid | SW480/HCT116/HT29 cells; BALB/c nude mice | Induction | EA at high concentrations induces ferroptosis by downregulating FTL and upregulating TF, CP, and ACSL4. | [94] |
| ACSL4 | I/R mice; Caco-2 cells | Induction | Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated intestinal injury in intestinal ischemia–reperfusion. | [14] |
| Ionizing radiation | Balb/c mice | Induction | Ferroptosis can promote ionizing radiation-induced intestinal damage. | [100] |
| P. aeruginosa | Female C57BL/6 mice; Caco-2 cells | Induction | The colonization of the gut with P. aeruginosa induces ferroptosis in host cells and significantly reduces survival in irradiated mice. | [101] |
3.2. Role of Necroptosis in Intestinal Diseases
3.2.1. Necroptosis and IBD
3.2.2. Necroptosis and CRC
3.2.3. Necroptosis and Intestinal Injury
| Compound/Target | Model | Effect | Mechanism | Ref. |
|---|---|---|---|---|
| Wu-Mei-Wan | TNBS-induced colitis mice | Inhibition | Wu-Mei-Wan alleviates TNBS-induced colitis in mice by inhibiting necroptosis through increasing RIPK3 O-GlcNAcylation. | [108] |
| ABIN3 | DSS-induced colitis mice; AOM/DSS mice | Inhibition | ABIN3 regulates the intestinal inflammatory response by interacting with A20 and regulating the K63 deubiquitination modification of necroptosis in IBD. | [109] |
| Resibufogenin | SW480/MC38 cells;The splenic capsule of mice | Induction | Resibufogenin inhabits CRC growth through RIP3-mediated necroptosis. | [112] |
| FMRP | WT and Fmr1 KO mice were injected with the AOM; CRC samples from patients | Induction | FMRP regulates RIPK1 and CRC resistance to necroptosis. | [113] |
| LPS | Weaned pigs | Induction | LPS activates the necroptosis signaling pathway in the gut and leads to impaired gut morphology and function. | [124] |
| Flaxseed Oil | Weaned pigs | Inhibition | Flaxseed oil attenuates intestinal damage and inflammation by regulating necroptosis and TLR4/NOD signaling pathways. | [125] |
| SpvB | C57BL/6J female mice; Caco-2 cells | Inhibition | SpvB accumulates a large amount of RIPK3 by inhibiting the degradation of RIPK3, which eventually leads to the aggravation of intestinal damage. | [127] |
| Cadmium | IPEC-J2 cells; weaned swine | Induction | Cadmium induces necroptosis in pigs by increasing ROS accumulation and Th1/Th2 imbalance, thereby aggravating small intestinal injuries in pigs. | [128] |
| Nrf2 | C57BL/6 Nrf2 KO mice; human intestinal epithelial crypt (HIEC) cells | Inhibition | Nrf2 attenuates radiation-induced rectal injury by negatively regulating necroptosis. | [129] |
3.3. Role of Pyroptosis in Intestinal Diseases
3.3.1. Pyroptosis and IBD
3.3.2. Pyroptosis and CRC
3.3.3. Pyroptosis and Intestinal Injury
| Compound/Target | Model | Effect | Mechanism | Ref. |
|---|---|---|---|---|
| NR4A1 | Marrow-derived macrophages (BMDMs)from WT mice and NR4A1−/− mice | Inhibition | NR4A1 inhabits pyroptosis by transcriptionally inhibiting NLRP3 and IL-1β and colocalizing with NLRP3 in trans-Golgi to alleviate pathogenic bacteria-induced colitis. | [131] |
| NEK7 | MODE-K cells; DSS-induced colitis mice | Induction | NEK7 interacts with NLRP3 to regulate pyroptosis in IBD via NF-κB signaling | [132] |
| JQ1 | LPS-induced endotoxemia mice | Inhibition | JQ1 blocks inflammatory pyroptosis-related acute colon injury induced by LPS. | [133] |
| hucMSC-derived exosomes | DSS-induced colitis mice | Inhibition | hucMSC-derived exosomes attenuate colitis by modulating macrophage pyroptosis via the miR–378a–5p–NLRP3 axis. | [134] |
| SDG | HCT116 cells | Induction | SDG induces pyroptosis by activating caspase-1 to cleave GSDMD in CRC cells. | [136] |
| Nanostructured toxins | CRC samples from patients | Induction | Nanostructured toxins for the selective destruction of drug-resistant human CXCR4(+) colorectal cancer stem cells. | [137] |
| GW4064 | HT-29, SW480/HCT1 16/CACO-2/RKO cells | Induction | GW4064 enhances the chemosensitivity of colorectal cancer to oxaliplatin by inducing pyroptosis. | [142] |
| A438079 | HCT-116/SW620 cells; xenografted mice | Induction | A438079 affects CRC cell proliferation, migration, apoptosis, and pyroptosis by inhibiting the P2X7 receptor. | [143] |
| Metformin | I/R mice; Caco-2 cells | Inhibition | Metformin treatment protects gut barrier function from intestinal I/R injury and reduces oxidative stress and inflammatory responses by modulating pyroptosis. | [145] |
| CB2 receptor | LPS induced-sepsis mice | Inhibition | CB2 receptor activation plays a protective role in sepsis-induced intestinal injury by inhibiting pyroptosis. | [146] |
| Berberine | DSS-induced colitis mouse; Caco-2 cells | Inhibition | Berberine negatively regulates the Wnt/β-catenin pathway, thereby attenuating colitis-stimulated pyroptosis and intestinal mucosal barrier defects. | [148] |
| GSDME | CT26 cells; GSDME-KO CT26 cells | Induction | GSDME enhances radiation-induced pyroptosis in CRC cells and normal epithelial cells via a caspase-3-dependent pathway. | [151] |
| Dopamine-derived nanoparticles | X-ray-induced mice | Inhibition | PDA-NPs inhibit pyroptosis and alleviate ionizing radiation-induced intestinal damage. | [152] |
| p-Coumaric acid | X-ray-induced mice | Inhibition | p-Coumaric acid (CA) ameliorates ionizing radiation-induced intestinal damage by modulating oxidative stress, inflammation, and pyroptosis. | [153] |
4. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kist, M.; Vucic, D. Cell death pathways: Intricate connections and disease implications. EMBO J. 2021, 40, e106700. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, Y.; Yasui, H.; Higashikawa, K.; Miyamoto, N.; Kuge, Y. Erastin, a ferroptosis-inducing agent, sensitized cancer cells to X-ray irradiation via glutathione starvation in vitro and in vivo. PLoS ONE 2019, 14, e0225931. [Google Scholar] [CrossRef] [Green Version]
- Sui, X.; Zhang, R.; Liu, S.; Duan, T.; Zhai, L.; Zhang, M.; Han, X.; Xiang, Y.; Huang, X.; Lin, H.; et al. RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front. Pharmacol. 2018, 9, 1371. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Zhang, Z.; Tsai, H.I.; Liu, Y.; Gao, J.; Wang, M.; Song, L.; Cao, X.; Xu, Z.; Chen, H.; et al. Branched-chain amino acid aminotransferase 2 regulates ferroptotic cell death in cancer cells. Cell Death Differ. 2021, 28, 1222–1236. [Google Scholar] [CrossRef]
- Ooko, E.; Saeed, M.E.; Kadioglu, O.; Sarvi, S.; Colak, M.; Elmasaoudi, K.; Janah, R.; Greten, H.J.; Efferth, T. Artemisinin derivatives induce iron-dependent cell death (ferroptosis) in tumor cells. Phytomedicine 2015, 22, 1045–1054. [Google Scholar] [CrossRef]
- Lane, D.J.R.; Metselaar, B.; Greenough, M.; Bush, A.I.; Ayton, S.J. Ferroptosis and NRF2: An emerging battlefield in the neurodegeneration of Alzheimer’s disease. Essays Biochem. 2021, 65, 925–940. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Zhang, X.; Wei, C.; Zheng, D.; Lu, X.; Yang, Y.; Luo, A.; Zhang, K.; Duan, X.; Wang, Y. Targeting SLC7A11 specifically suppresses the progression of colorectal cancer stem cells via inducing ferroptosis. Eur. J. Pharm. Sci. 2020, 152, 105450. [Google Scholar] [CrossRef]
- Li, Y.; Feng, D.; Wang, Z.; Zhao, Y.; Sun, R.; Tian, D.; Liu, D.; Zhang, F.; Ning, S.; Yao, J.; et al. Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ. 2019, 26, 2284–2299. [Google Scholar] [CrossRef] [Green Version]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Reis, C.R.; Chen, P.H.; Bendris, N.; Schmid, S.L. TRAIL-death receptor endocytosis and apoptosis are selectively regulated by dynamin-1 activation. Proc. Natl. Acad. Sci. USA 2017, 114, 504–509. [Google Scholar] [CrossRef] [Green Version]
- Petrie, E.J.; Birkinshaw, R.W.; Koide, A.; Denbaum, E.; Hildebrand, J.M.; Garnish, S.E.; Davies, K.A.; Sandow, J.J.; Samson, A.L.; Gavin, X.; et al. Identification of MLKL membrane translocation as a checkpoint in necroptotic cell death using Monobodies. Proc. Natl. Acad. Sci. USA 2020, 117, 8468–8475. [Google Scholar] [CrossRef]
- Garnish, S.E.; Meng, Y.; Koide, A.; Sandow, J.J.; Denbaum, E.; Jacobsen, A.V.; Yeung, W.; Samson, A.L.; Horne, C.R.; Fitzgibbon, C.; et al. Conformational interconversion of MLKL and disengagement from RIPK3 precede cell death by necroptosis. Nat. Commun. 2021, 12, 2211. [Google Scholar] [CrossRef]
- D’Souza, C.A.; Heitman, J. Dismantling the Cryptococcus coat. Trends Microbiol. 2001, 9, 112–113. [Google Scholar] [CrossRef]
- Kovacs, S.B.; Miao, E.A. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol. 2017, 27, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; He, Y.; Lin, L.; Chen, P.; Chen, M.; Zhang, S. The emerging role of ferroptosis in intestinal disease. Cell Death Dis. 2021, 12, 289. [Google Scholar] [CrossRef]
- Subramanian, S.; Geng, H.; Tan, X.D. Cell death of intestinal epithelial cells in intestinal diseases. Sheng Li Xue Bao 2020, 72, 308–324. [Google Scholar] [PubMed]
- Li, M.; Wang, Z.W.; Fang, L.J.; Cheng, S.Q.; Wang, X.; Liu, N.F. Programmed cell death in atherosclerosis and vascular calcification. Cell Death Dis. 2022, 13, 467. [Google Scholar] [CrossRef] [PubMed]
- Christgen, S.; Tweedell, R.E.; Kanneganti, T.D. Programming inflammatory cell death for therapy. Pharmacol. Ther. 2022, 232, 108010. [Google Scholar] [CrossRef]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell. Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef]
- Oestreicher, J.; Morgan, B. Glutathione: Subcellular distribution and membrane transport. Biochem. Cell Biol. 2019, 97, 270–289. [Google Scholar] [CrossRef] [Green Version]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.J.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.J.; Stockwell, B.R. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef] [Green Version]
- McBean, G.J. The transsulfuration pathway: A source of cysteine for glutathione in astrocytes. Amino Acids 2012, 42, 199–205. [Google Scholar] [CrossRef] [Green Version]
- Bridges, R.J.; Natale, N.R.; Patel, S.A. System xc− cystine/glutamate antiporter: An update on molecular pharmacology and roles within the CNS. Br. J. Pharmacol. 2012, 165, 20–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lachaier, E.; Louandre, C.; Godin, C.; Saidak, Z.; Baert, M.; Diouf, M.; Chauffert, B.; Galmiche, A. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res. 2014, 34, 6417–6422. [Google Scholar]
- Gout, P.W.; Buckley, A.R.; Simms, C.R.; Bruchovsky, N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: A new action for an old drug. Leukemia 2001, 15, 1633–1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Zhu, S.; Song, X.; Sun, X.; Fan, Y.; Liu, J.; Zhong, M.; Yuan, H.; Zhang, L.; Billiar, T.R.; et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 2017, 20, 1692–1704. [Google Scholar] [CrossRef] [Green Version]
- Kong, R.; Wang, N.; Han, W.; Bao, W.; Lu, J. IFNγ-mediated repression of system xc(-) drives vulnerability to induced ferroptosis in hepatocellular carcinoma cells. J. Leukoc. Biol. 2021, 110, 301–314. [Google Scholar] [CrossRef]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent. Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Stoyanovsky, D.A.; Tyurina, Y.Y.; Shrivastava, I.; Bahar, I.; Tyurin, V.A.; Protchenko, O.; Jadhav, S.; Bolevich, S.B.; Kozlov, A.V.; Vladimirov, Y.A.; et al. Iron catalysis of lipid peroxidation in ferroptosis: Regulated enzymatic or random free radical reaction? Free Radic. Biol. Med. 2019, 133, 153–161. [Google Scholar] [CrossRef]
- Coffey, R.; Ganz, T. Iron homeostasis: An anthropocentric perspective. J. Biol. Chem. 2017, 292, 12727–12734. [Google Scholar] [CrossRef] [Green Version]
- Qiao, B.; Sugianto, P.; Fung, E.; Del-Castillo-Rueda, A.; Moran-Jimenez, M.J.; Ganz, T.; Nemeth, E. Hepcidin-induced endocytosis of ferroportin is dependent on ferroportin ubiquitination. Cell Metab. 2012, 15, 918–924. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Zhou, Y.; Li, Y.; Xia, J.; Chen, Y.; Chen, S.; Wang, X.; Sun, W.; Wang, T.; Ren, X.; et al. Identification of Frataxin as a regulator of ferroptosis. Redox Biol. 2020, 32, 101483. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; He, H.; Wang, J.; Guo, X.; Lin, H.; Chen, H.; Jiang, C.; Chen, L.; Yao, P.; Tang, Y. Oxidative stress-dependent frataxin inhibition mediated alcoholic hepatocytotoxicity through ferroptosis. Toxicology 2020, 445, 152584. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ou, Z.; Xie, M.; Kang, R.; Fan, Y.; Niu, X.; Wang, H.; Cao, L.; Tang, D. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene 2015, 34, 5617–5625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem. Biophys. Res. Commun. 2016, 478, 838–844. [Google Scholar] [CrossRef]
- Jiang, L.; Hickman, J.H.; Wang, S.J.; Gu, W. Dynamic roles of p53-mediated metabolic activities in ROS-induced stress responses. Cell Cycle 2015, 14, 2881–2885. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, Y.; Zhang, R.; Wang, F.; Wang, T.; Jiao, Y. The Role of Erastin in Ferroptosis and Its Prospects in Cancer Therapy. OncoTargets Ther. 2020, 13, 5429–5441. [Google Scholar] [CrossRef]
- Tyurina, Y.Y.; Poloyac, S.M.; Tyurin, V.A.; Kapralov, A.A.; Jiang, J.; Anthonymuthu, T.S.; Kapralova, V.I.; Vikulina, A.S.; Jung, M.Y.; Epperly, M.W.; et al. A mitochondrial pathway for biosynthesis of lipid mediators. Nat. Chem. 2014, 6, 542–552. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.Q.; Chang, S.Y.; Wu, Q.; Gou, Y.J.; Jia, L.; Cui, Y.M.; Yu, P.; Shi, Z.H.; Wu, W.S.; Gao, G.; et al. The Protective Role of Mitochondrial Ferritin on Erastin-Induced Ferroptosis. Front. Aging Neurosci. 2016, 8, 308. [Google Scholar] [CrossRef] [Green Version]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Stockwell, B.R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018, 16, e2006203. [Google Scholar] [CrossRef]
- Yuan, J.; Amin, P.; Ofengeim, D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat. Rev. Neurosci. 2019, 20, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Fan, Z.; Luo, G.; Yang, C.; Huang, Q.; Fan, K.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. The role of necroptosis in cancer biology and therapy. Mol. Cancer 2019, 18, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mompeán, M.; Li, W.; Li, J.; Laage, S.; Siemer, A.B.; Bozkurt, G.; Wu, H.; McDermott, A.E. The Structure of the Necrosome RIPK1-RIPK3 Core, a Human Hetero-Amyloid Signaling Complex. Cell 2018, 173, 1244–1253.e1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.F.; Wang, F.S.; Wang, X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef] [Green Version]
- Conos, S.A.; Chen, K.W.; De Nardo, D.; Hara, H.; Whitehead, L.; Núñez, G.; Masters, S.L.; Murphy, J.M.; Schroder, K.; Vaux, D.L.; et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc. Natl. Acad. Sci. USA 2017, 114, E961–E969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, K.D.; Davis, M.A.; Daniels, B.P.; Olsen, T.M.; Ralli-Jain, P.; Tait, S.W.; Gale, M., Jr.; Oberst, A. MLKL Activation Triggers NLRP3-Mediated Processing and Release of IL-1β Independently of Gasdermin-D. J. Immunol. 2017, 198, 2156–2164. [Google Scholar] [CrossRef] [Green Version]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef]
- Liston, A.; Masters, S.L. Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nat. Rev. Immunol. 2017, 17, 208–214. [Google Scholar] [CrossRef]
- Levy, M.; Thaiss, C.A.; Zeevi, D.; Dohnalová, L.; Zilberman-Schapira, G.; Mahdi, J.A.; David, E.; Savidor, A.; Korem, T.; Herzig, Y.; et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 2015, 163, 1428–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, I.; Zhang, Y.; Krantz, B.A.; Miao, E.A. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J. Exp. Med. 2016, 213, 2113–2128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sollberger, G.; Strittmatter, G.E.; Garstkiewicz, M.; Sand, J.; Beer, H.D. Caspase-1: The inflammasome and beyond. Innate Immun. 2014, 20, 115–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afonina, I.S.; Zhong, Z.; Karin, M.; Beyaert, R. Limiting inflammation-the negative regulation of NF-κB and the NLRP3 inflammasome. Nat. Immunol. 2017, 18, 861–869. [Google Scholar] [CrossRef] [PubMed]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Aglietti, R.A.; Estevez, A.; Gupta, A.; Ramirez, M.G.; Liu, P.S.; Kayagaki, N.; Ciferri, C.; Dixit, V.M.; Dueber, E.C. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc. Natl. Acad. Sci. USA 2016, 113, 7858–7863. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef]
- Orning, P.; Weng, D.; Starheim, K.; Ratner, D.; Best, Z.; Lee, B.; Brooks, A.; Xia, S.; Wu, H.; Kelliher, M.A.; et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 2018, 362, 1064–1069. [Google Scholar] [CrossRef] [Green Version]
- Sarhan, J.; Liu, B.C.; Muendlein, H.I.; Li, P.; Nilson, R.; Tang, A.Y.; Rongvaux, A.; Bunnell, S.C.; Shao, F.; Green, D.R.; et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl. Acad. Sci. USA 2018, 115, E10888–E10897. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Zhao, R.; Xia, W.; Chang, C.W.; You, Y.; Hsu, J.M.; Nie, L.; Chen, Y.; Wang, Y.C.; Liu, C.; et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat. Cell Biol. 2020, 22, 1264–1275. [Google Scholar] [CrossRef] [PubMed]
- Kambara, H.; Liu, F.; Zhang, X.; Liu, P.; Bajrami, B.; Teng, Y.; Zhao, L.; Zhou, S.; Yu, H.; Zhou, W.; et al. Gasdermin D Exerts Anti-inflammatory Effects by Promoting Neutrophil Death. Cell Rep. 2018, 22, 2924–2936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; He, H.; Wang, K.; Shi, X.; Wang, Y.; Su, Y.; Wang, Y.; Li, D.; Liu, W.; Zhang, Y.; et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 2020, 368, eaaz7548. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, Y.; Xia, S.; Kong, Q.; Li, S.; Liu, X.; Junqueira, C.; Meza-Sosa, K.F.; Mok, T.M.Y.; Ansara, J.; et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 2020, 579, 415–420. [Google Scholar] [CrossRef]
- Xu, M.; Tao, J.; Yang, Y.; Tan, S.; Liu, H.; Jiang, J.; Zheng, F.; Wu, B. Ferroptosis involves in intestinal epithelial cell death in ulcerative colitis. Cell Death Dis. 2020, 11, 86. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Liu, S.; Cui, Z.; Wang, X.; Ning, T.; Wang, T.; Zhang, N.; Xie, S.; Min, L.; Zhang, S.; et al. Ferrostatin-1 alleviated TNBS induced colitis via the inhibition of ferroptosis. Biochem. Biophys. Res. Commun. 2021, 573, 48–54. [Google Scholar] [CrossRef]
- Li, N.; Wang, W.; Zhou, H.; Wu, Q.; Duan, M.; Liu, C.; Wu, H.; Deng, W.; Shen, D.; Tang, Q. Ferritinophagy-mediated ferroptosis is involved in sepsis-induced cardiac injury. Free Radic. Biol. Med. 2020, 160, 303–318. [Google Scholar] [CrossRef]
- Triantafillidis, J.; Vagianos, C.; Agrogiannis, G.; Gikas, A.; Douvi, G.; Syrmos, N.; Patsouris, E.; Papalois, A. Effect of Infliximab and Adalimumab on Experimental Colitis Following Orally Supplemented Iron. J. Investig. Surg. 2017, 30, 6–12. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, P.; Chen, W.; Chen, G. Ferroptosis mediated DSS-induced ulcerative colitis associated with Nrf2/HO-1 signaling pathway. Immunol. Lett. 2020, 225, 9–15. [Google Scholar] [CrossRef]
- Dong, S.; Lu, Y.; Peng, G.; Li, J.; Li, W.; Li, M.; Wang, H.; Liu, L.; Zhao, Q. Furin inhibits epithelial cell injury and alleviates experimental colitis by activating the Nrf2-Gpx4 signaling pathway. Dig. Liver Dis. 2021, 53, 1276–1285. [Google Scholar] [CrossRef]
- Mayr, L.; Grabherr, F.; Schwärzler, J.; Reitmeier, I.; Sommer, F.; Gehmacher, T.; Niederreiter, L.; He, G.W.; Ruder, B.; Kunz, K.T.R.; et al. Dietary lipids fuel GPX4-restricted enteritis resembling Crohn’s disease. Nat. Commun. 2020, 11, 1775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, D.J.; Chen, C.; Yuan, W.Q.; Yang, Y.H.; Han, L. Integrative analysis of ferroptosis-related genes in ulcerative colitis. J. Int. Med. Res. 2021, 49, 3000605211042975. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Zhu, J.; Fang, S.; Wang, Y.; Vinothkumar, R.; Li, M.; Weng, Q.; Zheng, L.; Yang, Y.; Qiu, R.; et al. Pharmacological inhibition of MELK restricts ferroptosis and the inflammatory response in colitis and colitis-propelled carcinogenesis. Free Radic. Biol. Med. 2021, 172, 312–329. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, J.; Li, J.; Zhu, J.; Wang, R.; Xi, Q.; Wu, H.; Shi, T.; Chen, W. Astragalus polysaccharide prevents ferroptosis in a murine model of experimental colitis and human Caco-2 cells via inhibiting NRF2/HO-1 pathway. Eur. J. Pharmacol. 2021, 911, 174518. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, W.; Wang, J.; Bai, X. Curculigoside inhibits ferroptosis in ulcerative colitis through the induction of GPX4. Life Sci. 2020, 259, 118356. [Google Scholar] [CrossRef]
- Shen, L.D.; Qi, W.H.; Bai, J.J.; Zuo, C.Y.; Bai, D.L.; Gao, W.D.; Zong, X.L.; Hao, T.T.; Ma, Y.; Cao, G.C. Resibufogenin inhibited colorectal cancer cell growth and tumorigenesis through triggering ferroptosis and ROS production mediated by GPX4 inactivation. Anat. Rec. 2021, 304, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Lee, D.H.; Jeong, S.Y.; Park, S.H.; Oh, S.C.; Park, Y.S.; Yu, J.; Choudry, H.A.; Bartlett, D.L.; Lee, Y.J. Ferroptosis-inducing agents enhance TRAIL-induced apoptosis through upregulation of death receptor 5. J. Cell Biochem. 2019, 120, 928–939. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.; Ai, R.; Mu, S.; Niu, X.; Guo, Z.; Liu, L. MiR-19a suppresses ferroptosis of colorectal cancer cells by targeting IREB2. Bioengineered 2022, 13, 12021–12029. [Google Scholar] [CrossRef]
- Liu, L.; Yao, H.; Zhou, X.; Chen, J.; Chen, G.; Shi, X.; Wu, G.; Zhou, G.; He, S. MiR-15a-3p regulates ferroptosis via targeting glutathione peroxidase GPX4 in colorectal cancer. Mol. Carcinog. 2022, 61, 301–310. [Google Scholar] [CrossRef]
- Sharma, P.; Shimura, T.; Banwait, J.K.; Goel, A. Andrographis-mediated chemosensitization through activation of ferroptosis and suppression of β-catenin/Wnt-signaling pathways in colorectal cancer. Carcinogenesis 2020, 41, 1385–1394. [Google Scholar] [CrossRef]
- He, Z.; Yang, J.; Sui, C.; Zhang, P.; Wang, T.; Mou, T.; Sun, K.; Wang, Y.; Xu, Z.; Li, G.; et al. FAM98A promotes resistance to 5-fluorouracil in colorectal cancer by suppressing ferroptosis. Arch. Biochem. Biophys. 2022, 722, 109216. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.Y.; Li, H.M.; Wang, X.Y.; Xia, R.; Li, X.; Ma, Y.J.; Wang, M.; Zhang, H.S. TIGAR drives colorectal cancer ferroptosis resistance through ROS/AMPK/SCD1 pathway. Free Radic. Biol. Med. 2022, 182, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Peng, J.; Kang, F.; Zhang, W.; Peng, E.; He, Q. Ferroptosis-Related Gene MT1G as a Novel Biomarker Correlated With Prognosis and Immune Infiltration in Colorectal Cancer. Front. Cell Dev. Biol. 2022, 10, 881447. [Google Scholar] [CrossRef]
- Xia, Y.; Yang, J.; Li, C.; Hao, X.; Fan, H.; Zhao, Y.; Tang, J.; Wan, X.; Lian, S.; Yang, J. TMT-Based Quantitative Proteomics Analysis Reveals the Panoramic Pharmacological Molecular Mechanism of β-Elemonic Acid Inhibition of Colorectal Cancer. Front. Pharmacol. 2022, 13, 830328. [Google Scholar] [CrossRef] [PubMed]
- Malfa, G.A.; Tomasello, B.; Acquaviva, R.; Genovese, C.; La Mantia, A.; Cammarata, F.P.; Ragusa, M.; Renis, M.; Di Giacomo, C. Betula etnensis Raf. (Betulaceae) Extract Induced HO-1 Expression and Ferroptosis Cell Death in Human Colon Cancer Cells. Int. J. Mol. Sci. 2019, 20, 2723. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.X.; Chen, L.H.; Zhuang, H.B.; Shi, Z.S.; Chen, Z.C.; Pan, J.P.; Hong, Z.S. Auriculasin enhances ROS generation to regulate colorectal cancer cell apoptosis, ferroptosis, oxeiptosis, invasion and colony formation. Biochem. Biophys. Res. Commun. 2022, 587, 99–106. [Google Scholar] [CrossRef]
- Hu, Y.; Mao, Z.; Xu, L.; Yin, L.; Tao, X.; Tang, Z.; Qi, Y.; Sun, P.; Peng, J. Protective effect of dioscin against intestinal ischemia/reperfusion injury via adjusting miR-351-5p-mediated oxidative stress. Pharmacol. Res. 2018, 137, 56–63. [Google Scholar] [CrossRef]
- Balogh, N.; Krausz, F.; Lévai, P.; Ribiczeyné, P.S.; Vajdovich, P.; Gaál, T. Effect of deferoxamine and L-arginine treatment on lipid peroxidation in an intestinal ischaemia-reperfusion model in rats. Acta Vet. Hung. 2002, 50, 343–356. [Google Scholar] [CrossRef]
- Deng, S.; Wu, D.; Li, L.; Li, J.; Xu, Y. TBHQ attenuates ferroptosis against 5-fluorouracil-induced intestinal epithelial cell injury and intestinal mucositis via activation of Nrf2. Cell. Mol. Biol. Lett. 2021, 26, 48. [Google Scholar] [CrossRef]
- Wang, L.; Wang, A.; Fu, Q.; Shi, Z.; Chen, X.; Wang, Y.; Xu, W.; Wang, T.; Zhang, S.; Hu, S. Ferroptosis plays an important role in promoting ionizing radiation-induced intestinal injuries. Biochem. Biophys. Res. Commun. 2022, 595, 7–13. [Google Scholar] [CrossRef]
- Dar, H.H.; Epperly, M.W.; Tyurin, V.A.; Amoscato, A.A.; Anthonymuthu, T.S.; Souryavong, A.B.; Kapralov, A.A.; Shurin, G.V.; Samovich, S.N.; St Croix, C.M.; et al. P. aeruginosa augments irradiation injury via 15-lipoxygenase-catalyzed generation of 15-HpETE-PE and induction of theft-ferroptosis. JCI Insight 2022, 7, e156013. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.W.; Cai, S.; Zhao, T.S.; Li, M.; Tian, Y. Green tea derivative (−)-epigallocatechin-3-gallate (EGCG) confers protection against ionizing radiation-induced intestinal epithelial cell death both in vitro and in vivo. Free Radic. Biol. Med. 2020, 161, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Yue, M.; Shao, L.; Cheng, J.; Fan, Y.; Cai, X.; Li, H.; Li, M.; Zhang, X.; Fu, A.; Huang, Y.; et al. Prostaglandin E2 accelerated recovery of chemotherapy-induced intestinal damage by increasing expression of cyclin D. Exp. Cell Res. 2020, 388, 111819. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wei, Y.; Hua, H.; Jing, X.; Zhu, H.; Xiao, K.; Zhao, J.; Liu, Y. Polyphenols Sourced from Ilex latifolia Thunb. Relieve Intestinal Injury via Modulating Ferroptosis in Weanling Piglets under Oxidative Stress. Antioxidants 2022, 11, 966. [Google Scholar] [CrossRef]
- Yu, Q.; Gao, J.; Shao, X.; Lu, W.; Chen, L.; Jin, L. The Effects of Alda-1 Treatment on Renal and Intestinal Injuries After Cardiopulmonary Resuscitation in Pigs. Front. Med. 2022, 9, 892472. [Google Scholar] [CrossRef]
- Alagbaoso, C.A.; Mizuno, M. Polysaccharides from Shiitake Culinary-Medicinal Mushroom Lentinus edodes (Agaricomycetes) Suppress pMLKL-Mediated Necroptotic Cell Death and Colitis in Mice. Int. J. Med. Mushrooms 2021, 23, 13–26. [Google Scholar] [CrossRef]
- Negroni, A.; Colantoni, E.; Pierdomenico, M.; Palone, F.; Costanzo, M.; Oliva, S.; Tiberti, A.; Cucchiara, S.; Stronati, L. RIP3 AND pMLKL promote necroptosis-induced inflammation and alter membrane permeability in intestinal epithelial cells. Dig. Liver Dis. 2017, 49, 1201–1210. [Google Scholar] [CrossRef]
- Wu, F.; Shao, Q.; Cheng, Z.; Xiong, X.; Fang, K.; Zhao, Y.; Dong, R.; Xu, L.; Lu, F.; Chen, G. Traditional herbal formula Wu-Mei-Wan alleviates TNBS-induced colitis in mice by inhibiting necroptosis through increasing RIPK3 O-GlcNAcylation. Chin. Med. 2021, 16, 78. [Google Scholar] [CrossRef]
- Zhou, M.; He, J.; Shi, Y.; Liu, X.; Luo, S.; Cheng, C.; Ge, W.; Qu, C.; Du, P.; Chen, Y. ABIN3 Negatively Regulates Necroptosis-induced Intestinal Inflammation Through Recruiting A20 and Restricting the Ubiquitination of RIPK3 in Inflammatory Bowel Disease. J. Crohns Colitis 2021, 15, 99–114. [Google Scholar] [CrossRef]
- Lee, C.; An, M.; Joung, J.G.; Park, W.Y.; Chang, D.K.; Kim, Y.H.; Hong, S.N. TNFα Induces LGR5+ Stem Cell Dysfunction In Patients with Crohn’s Disease. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 789–808. [Google Scholar] [CrossRef]
- Patankar, J.V.; Müller, T.M.; Kantham, S.; Acera, M.G.; Mascia, F.; Scheibe, K.; Mahapatro, M.; Heichler, C.; Yu, Y.; Li, W.; et al. E-type prostanoid receptor 4 drives resolution of intestinal inflammation by blocking epithelial necroptosis. Nat. Cell Biol. 2021, 23, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Ma, Y.; Wang, H.; Dai, Y.; Chen, C.; Liu, Y.; Jing, L.; Sun, X. Resibufogenin suppresses colorectal cancer growth and metastasis through RIP3-mediated necroptosis. J. Transl. Med. 2018, 16, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Grazia, A.; Marafini, I.; Pedini, G.; Di Fusco, D.; Laudisi, F.; Dinallo, V.; Rosina, E.; Stolfi, C.; Franzè, E.; Sileri, P.; et al. The Fragile X Mental Retardation Protein Regulates RIPK1 and Colorectal Cancer Resistance to Necroptosis. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 639–658. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.Q.; Thonn, V.; Patankar, J.V.; Thoma, O.M.; Waldner, M.; Zielinska, M.; Bao, L.L.; Gonzalez-Acera, M.; Wallmüller, S.; Engel, F.B.; et al. SMYD2 targets RIPK1 and restricts TNF-induced apoptosis and necroptosis to support colon tumor growth. Cell Death Dis. 2022, 13, 52. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Hu, D.; Sakya, J.; Sun, T.; Wang, D.; Wang, L.; Mao, X.; Su, Z. ABIN-1 is a key regulator in RIPK1-dependent apoptosis (RDA) and necroptosis, and ABIN-1 deficiency potentiates necroptosis-based cancer therapy in colorectal cancer. Cell Death Dis. 2021, 12, 140. [Google Scholar] [CrossRef]
- Wu, X.; Lu, Y.; Qin, X. Combination of Compound Kushen Injection and cisplatin shows synergistic antitumor activity in p53-R273H/P309S mutant colorectal cancer cells through inducing apoptosis. J. Ethnopharmacol. 2022, 283, 114690. [Google Scholar] [CrossRef]
- Zhang, Z.; Ju, F.; Chen, F.; Wu, H.; Chen, J.; Zhong, J.; Shao, L.; Zheng, S.; Wang, L.; Xue, M. GDC-0326 Enhances the Effects of 5-Fu in Colorectal Cancer Cells by Inducing Necroptotic Death. OncoTargets Ther. 2021, 14, 2519–2530. [Google Scholar] [CrossRef]
- Zhao, Q.; Yu, X.; Li, M.; Liu, Y.; Han, Y.; Zhang, X.; Li, X.M.; Wu, X.; Qin, J.; Fang, J.; et al. MLKL attenuates colon inflammation and colitis-tumorigenesis via suppression of inflammatory responses. Cancer Lett. 2019, 459, 100–111. [Google Scholar] [CrossRef]
- Zhao, Q.; Cheng, X.; Guo, J.; Bi, Y.; Kuang, L.; Ren, J.; Zhong, J.; Pan, L.; Zhang, X.; Guo, Y.; et al. MLKL inhibits intestinal tumorigenesis by suppressing STAT3 signaling pathway. Int. J. Biol. Sci. 2021, 17, 869–881. [Google Scholar] [CrossRef]
- Wen, S.; Ling, Y.; Yang, W.; Shen, J.; Li, C.; Deng, W.; Liu, W.; Liu, K. Necroptosis is a key mediator of enterocytes loss in intestinal ischaemia/reperfusion injury. J. Cell Mol. Med. 2017, 21, 432–443. [Google Scholar] [CrossRef]
- Hu, Q.; Ren, H.; Ren, J.; Liu, Q.; Wu, J.; Wu, X.; Li, G.; Wang, G.; Gu, G.; Guo, K.; et al. Released Mitochondrial DNA Following Intestinal Ischemia Reperfusion Induces the Inflammatory Response and Gut Barrier Dysfunction. Sci. Rep. 2018, 8, 7350. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, J.; Liu, Q.; Li, X.; Li, S.; Chen, J.; Hong, Z.; Wu, X.; Zhao, Y.; Ren, J. mtDNA-STING pathway promotes necroptosis-dependent enterocyte injury in intestinal ischemia reperfusion. Cell Death Dis. 2020, 11, 1050. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ling, Y.; Cao, Z.; Shen, J.; Chen, S.; Liu, W.; Yuan, B.; Wen, S. Targeting intestinal epithelial cell-programmed necrosis alleviates tissue injury after intestinal ischemia/reperfusion in rats. J. Surg. Res. 2018, 225, 108–117. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Xu, Q.; Wang, Y.; Liang, T.; Li, X.; Wang, D.; Wang, X.; Zhu, H.; Xiao, K. Necroptosis is active and contributes to intestinal injury in a piglet model with lipopolysaccharide challenge. Cell Death Dis. 2021, 12, 62. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, H.; Wang, S.; Tu, Z.; Zhang, L.; Wang, X.; Hou, Y.; Wang, C.; Chen, J.; Liu, Y. Flaxseed Oil Attenuates Intestinal Damage and Inflammation by Regulating Necroptosis and TLR4/NOD Signaling Pathways Following Lipopolysaccharide Challenge in a Piglet Model. Mol. Nutr. Food Res. 2018, 62, e1700814. [Google Scholar] [CrossRef] [PubMed]
- Martens, E.C.; Neumann, M.; Desai, M.S. Interactions of commensal and pathogenic microorganisms with the intestinal mucosal barrier. Nat. Rev. Microbiol. 2018, 16, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Dong, K.; Zhu, Y.; Deng, Q.; Sun, L.; Yang, S.; Huang, K.; Cao, Y.; Li, Y.; Wu, S.; Huang, R. Salmonella pSLT-encoded effector SpvB promotes RIPK3-dependent necroptosis in intestinal epithelial cells. Cell Death Discov. 2022, 8, 44. [Google Scholar] [CrossRef]
- Chen, X.; Bi, M.; Yang, J.; Cai, J.; Zhang, H.; Zhu, Y.; Zheng, Y.; Liu, Q.; Shi, G.; Zhang, Z. Cadmium exposure triggers oxidative stress, necroptosis, Th1/Th2 imbalance and promotes inflammation through the TNF-α/NF-κB pathway in swine small intestine. J. Hazard. Mater. 2022, 421, 126704. [Google Scholar] [CrossRef]
- Xu, Y.; Tu, W.; Sun, D.; Chen, X.; Ge, Y.; Yao, S.; Li, B.; Zhenbo, Z.; Liu, Y. Nrf2 alleviates radiation-induced rectal injury by inhibiting of necroptosis. Biochem. Biophys. Res. Commun. 2021, 554, 49–55. [Google Scholar] [CrossRef]
- Demon, D.; Kuchmiy, A.; Fossoul, A.; Zhu, Q.; Kanneganti, T.D.; Lamkanfi, M. Caspase-11 is expressed in the colonic mucosa and protects against dextran sodium sulfate-induced colitis. Mucosal Immunol. 2014, 7, 1480–1491. [Google Scholar] [CrossRef] [Green Version]
- Deng, Z.; Yang, Z.; Cui, C.; Wei, H.; Wang, L.; Tian, D.; Xiao, F.; Peng, J. NR4A1 suppresses pyroptosis by transcriptionally inhibiting NLRP3 and IL-1β and co-localizing with NLRP3 in trans-Golgi to alleviate pathogenic bacteria-induced colitis. Clin. Transl. Med. 2021, 11, e639. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, G.; Yuan, Y.; Wu, G.; Wang, S.; Yuan, L. NEK7 interacts with NLRP3 to modulate the pyroptosis in inflammatory bowel disease via NF-κB signaling. Cell Death Dis. 2019, 10, 906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Zhong, X.; Cao, W.; Mao, M.; Li, W.; Yang, H.; Li, M.; Shi, M.; Zhang, Y.; Deng, Y.; et al. JQ1 as a BRD4 Inhibitor Blocks Inflammatory Pyroptosis-Related Acute Colon Injury Induced by LPS. Front. Immunol. 2021, 12, 609319. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Zhang, Z.Y.; Yuan, J.T.; Ocansey, D.K.W.; Tu, Q.; Zhang, X.; Qian, H.; Xu, W.R.; Qiu, W.; Mao, F. hucMSC-derived exosomes attenuate colitis by regulating macrophage pyroptosis via the miR-378a-5p/NLRP3 axis. Stem Cell. Res. Ther. 2021, 12, 416. [Google Scholar] [CrossRef]
- Tan, G.; Huang, C.; Chen, J.; Chen, B.; Zhi, F. Gasdermin-E-mediated pyroptosis participates in the pathogenesis of Crohn’s disease by promoting intestinal inflammation. Cell Rep. 2021, 35, 109265. [Google Scholar] [CrossRef]
- Chen, T.; Wang, Z.; Zhong, J.; Zhang, L.; Zhang, H.; Zhang, D.; Xu, X.; Zhong, X.; Wang, J.; Li, H. Secoisolariciresinol diglucoside induces pyroptosis by activating caspase-1 to cleave GSDMD in colorectal cancer cells. Drug Dev. Res. 2022, 83, 1152–1166. [Google Scholar] [CrossRef]
- Sala, R.; Rioja-Blanco, E.; Serna, N.; Sánchez-García, L.; Álamo, P.; Alba-Castellón, L.; Casanova, I.; López-Pousa, A.; Unzueta, U.; Céspedes, M.V.; et al. GSDMD-dependent pyroptotic induction by a multivalent CXCR4-targeted nanotoxin blocks colorectal cancer metastases. Drug Deliv. 2022, 29, 1384–1397. [Google Scholar] [CrossRef]
- Wu, L.S.; Liu, Y.; Wang, X.W.; Xu, B.; Lin, Y.L.; Song, Y.; Dong, Y.; Liu, J.L.; Wang, X.J.; Liu, S.; et al. LPS Enhances the Chemosensitivity of Oxaliplatin in HT29 Cells via GSDMD-Mediated Pyroptosis. Cancer Manag. Res. 2020, 12, 10397–10409. [Google Scholar] [CrossRef]
- Mu, M.; Yu, Q.; Zhang, Q.; Guo, J.; Wang, X.; Sun, X.; Yu, J. A pan-cancer analysis of molecular characteristics and oncogenic role of gasdermins. Cancer Cell Int. 2022, 22, 80. [Google Scholar] [CrossRef]
- Tan, G.; Huang, C.; Chen, J.; Zhi, F. HMGB1 released from GSDME-mediated pyroptotic epithelial cells participates in the tumorigenesis of colitis-associated colorectal cancer through the ERK1/2 pathway. J. Hematol. Oncol. 2020, 13, 149. [Google Scholar] [CrossRef]
- Liu, Z.; Li, Y.; Zhu, Y.; Li, N.; Li, W.; Shang, C.; Song, G.; Li, S.; Cong, J.; Li, T.; et al. Apoptin induces pyroptosis of colorectal cancer cells via the GSDME-dependent pathway. Int. J. Biol. Sci. 2022, 18, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zheng, J.; Mu, M.; Chen, Z.; Xu, Z.; Zhao, C.; Yang, K.; Qin, X.; Sun, X.; Yu, J. GW4064 enhances the chemosensitivity of colorectal cancer to oxaliplatin by inducing pyroptosis. Biochem. Biophys. Res. Commun. 2021, 548, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, F.; Wang, L.; Lou, Y. A438079 affects colorectal cancer cell proliferation, migration, apoptosis, and pyroptosis by inhibiting the P2X7 receptor. Biochem. Biophys. Res. Commun. 2021, 558, 147–153. [Google Scholar] [CrossRef]
- Tan, Y.; Zuo, W.; Huang, L.; Zhou, B.; Liang, H.; Zheng, S.; Jia, W.; Chen, S.; Liu, J.; Yang, X.; et al. Nervilifordin F alleviates intestinal ischemia/reperfusion-induced acute lung injury via inhibiting inflammasome and mTOR pathway. Int. Immunopharmacol. 2020, 89, 107014. [Google Scholar] [CrossRef]
- Jia, Y.; Cui, R.; Wang, C.; Feng, Y.; Li, Z.; Tong, Y.; Qu, K.; Liu, C.; Zhang, J. Metformin protects against intestinal ischemia-reperfusion injury and cell pyroptosis via TXNIP-NLRP3-GSDMD pathway. Redox Biol. 2020, 32, 101534. [Google Scholar] [CrossRef]
- Zhang, B.; Zheng, F.; Liu, A.; Li, Z.; Zheng, F.; Liu, Q.; Yang, L.; Chen, K.; Wang, Y.; Zhang, Z.; et al. Activation of CB2 receptor inhibits pyroptosis and subsequently ameliorates cecal ligation and puncture-induced sepsis. Int. Immunopharmacol. 2021, 99, 108038. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Yuan, L.; Liu, J.; Muhammad, I.; Cao, C.; Shi, C.; Zhang, Y.; Li, R.; Li, C.; Liu, F. Dihydromyricetin attenuates Escherichia coli lipopolysaccharide-induced ileum injury in chickens by inhibiting NLRP3 inflammasome and TLR4/NF-κB signalling pathway. Vet. Res. 2020, 51, 72. [Google Scholar] [CrossRef]
- Zhao, X.; Cui, D.; Yuan, W.; Chen, C.; Liu, Q. Berberine represses Wnt/β-catenin pathway activation via modulating the microRNA-103a-3p/Bromodomain-containing protein 4 axis, thereby refraining pyroptosis and reducing the intestinal mucosal barrier defect induced via colitis. Bioengineered 2022, 13, 7392–7409. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, W.; Shen, C.; Wang, X.; Pu, Z.; Yin, Q. Network pharmacology for systematic understanding of Schisandrin B reduces the epithelial cells injury of colitis through regulating pyroptosis by AMPK/Nrf2/NLRP3 inflammasome. Aging 2021, 13, 23193–23209. [Google Scholar] [CrossRef]
- Lu, L.; Li, W.; Chen, L.; Su, Q.; Wang, Y.; Guo, Z.; Lu, Y.; Liu, B.; Qin, S. Radiation-induced intestinal damage: Latest molecular and clinical developments. Future Oncol. 2019, 15, 4105–4118. [Google Scholar] [CrossRef]
- Tan, G.; Lin, C.; Huang, C.; Chen, B.; Chen, J.; Shi, Y.; Zhi, F. Radiosensitivity of colorectal cancer and radiation-induced gut damages are regulated by gasdermin E. Cancer Lett. 2022, 529, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.; Dong, S.; Liu, H.; Yu, H.; Chen, Z.; Wang, S.; Li, W.; Peng, R.; Li, F.; Jiang, Q.; et al. Dopamine-derived nanoparticles for the protection of irradiation-induced intestinal injury by maintaining intestinal homeostasis. Biomater. Sci. 2022, 10, 3309–3322. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; He, Q.; Chen, Y.Z.; Du, Y.F.; Guo, Y.X.; Xu, J.Y.; Qin, L.Q. p-Coumaric acid ameliorates ionizing radiation-induced intestinal injury through modulation of oxidative stress and pyroptosis. Life Sci. 2021, 278, 119546. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, P.; Zhang, S.; Wang, M.; Zhou, J. The Induction Mechanism of Ferroptosis, Necroptosis, and Pyroptosis in Inflammatory Bowel Disease, Colorectal Cancer, and Intestinal Injury. Biomolecules 2023, 13, 820. https://doi.org/10.3390/biom13050820
Zhou P, Zhang S, Wang M, Zhou J. The Induction Mechanism of Ferroptosis, Necroptosis, and Pyroptosis in Inflammatory Bowel Disease, Colorectal Cancer, and Intestinal Injury. Biomolecules. 2023; 13(5):820. https://doi.org/10.3390/biom13050820
Chicago/Turabian StyleZhou, Ping, Shun Zhang, Maohua Wang, and Jun Zhou. 2023. "The Induction Mechanism of Ferroptosis, Necroptosis, and Pyroptosis in Inflammatory Bowel Disease, Colorectal Cancer, and Intestinal Injury" Biomolecules 13, no. 5: 820. https://doi.org/10.3390/biom13050820