
In Silico Identification of Potential Inhibitors of the SARS-CoV-2 Main Protease among a PubChem Database of Avian Infectious Bronchitis Virus 3CLPro Inhibitors

Abstract
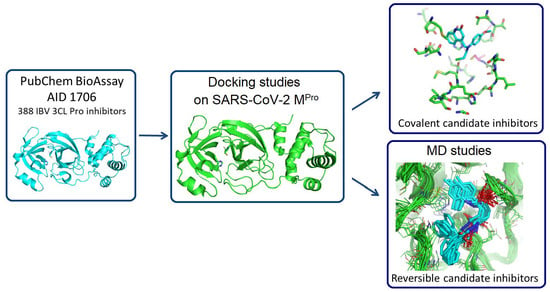
:
1. Introduction
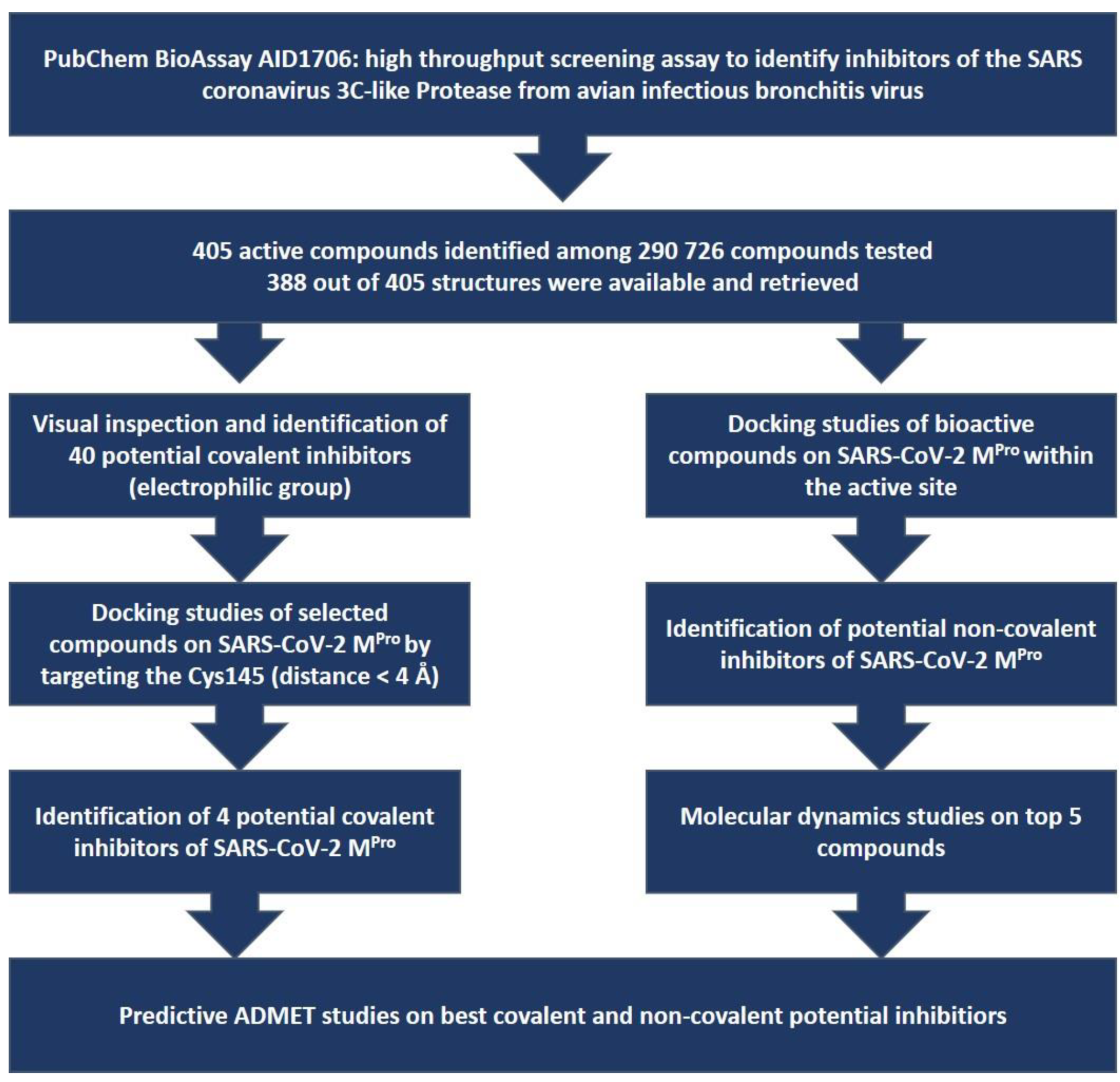
2. Materials and Methods
2.1. Homology Studies
2.2. Docking Studies
2.2.1. Docking Studies for Covalent Inhibition
2.2.2. Docking Studies for Non-Covalent Inhibition
2.2.3. Molecular Dynamics Studies
3. Results and Discussion
3.1. Homology Studies
3.2. Docking Studies of the 388 Active Compounds from the PubChem BioAssay AID 1706 within the SARS-CoV-2 Protease Active Site
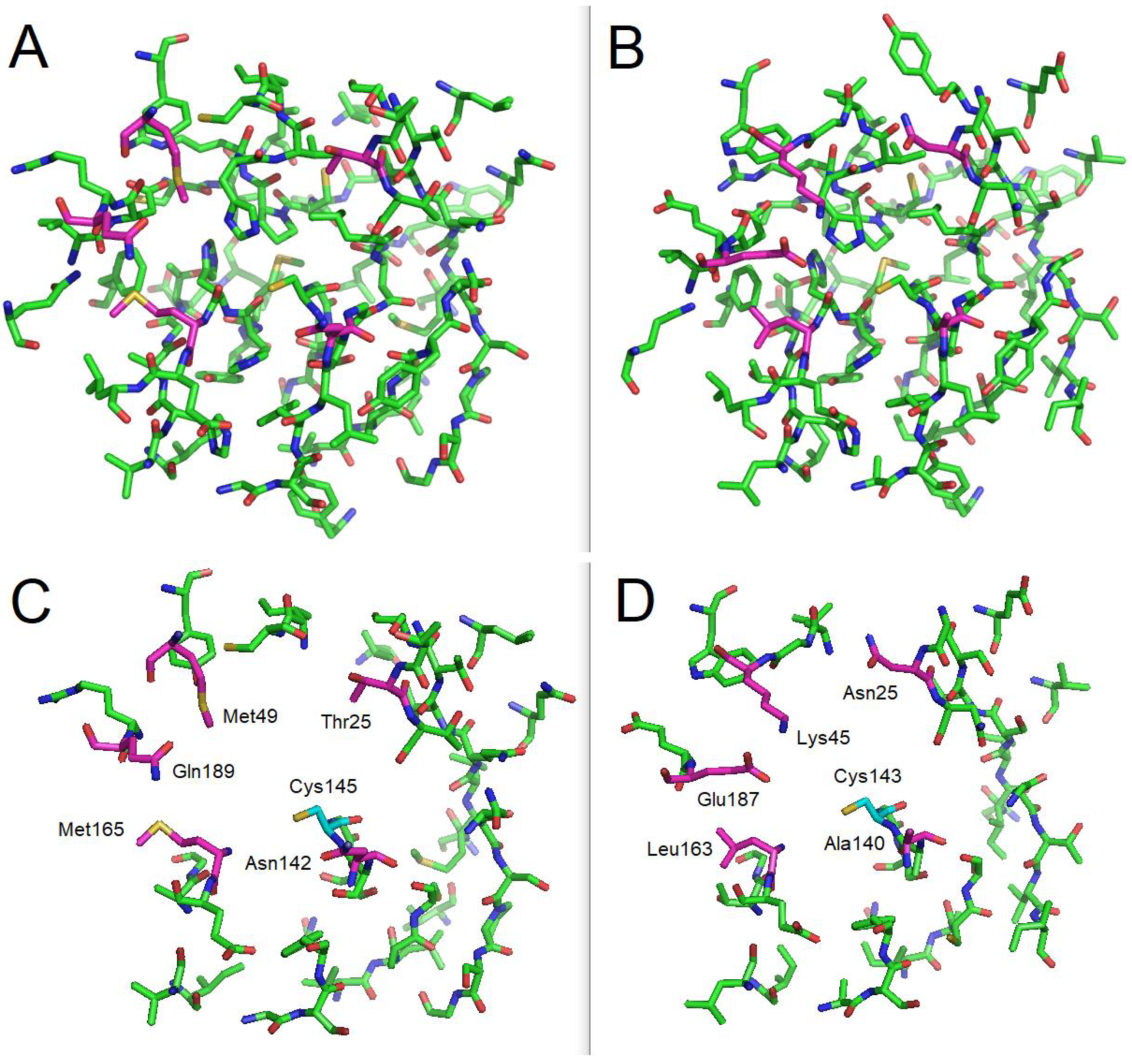
3.2.1. Docking Studies Looking for Covalent Inhibitors
3.2.2. Docking Studies Looking for Non-Covalent Inhibitors
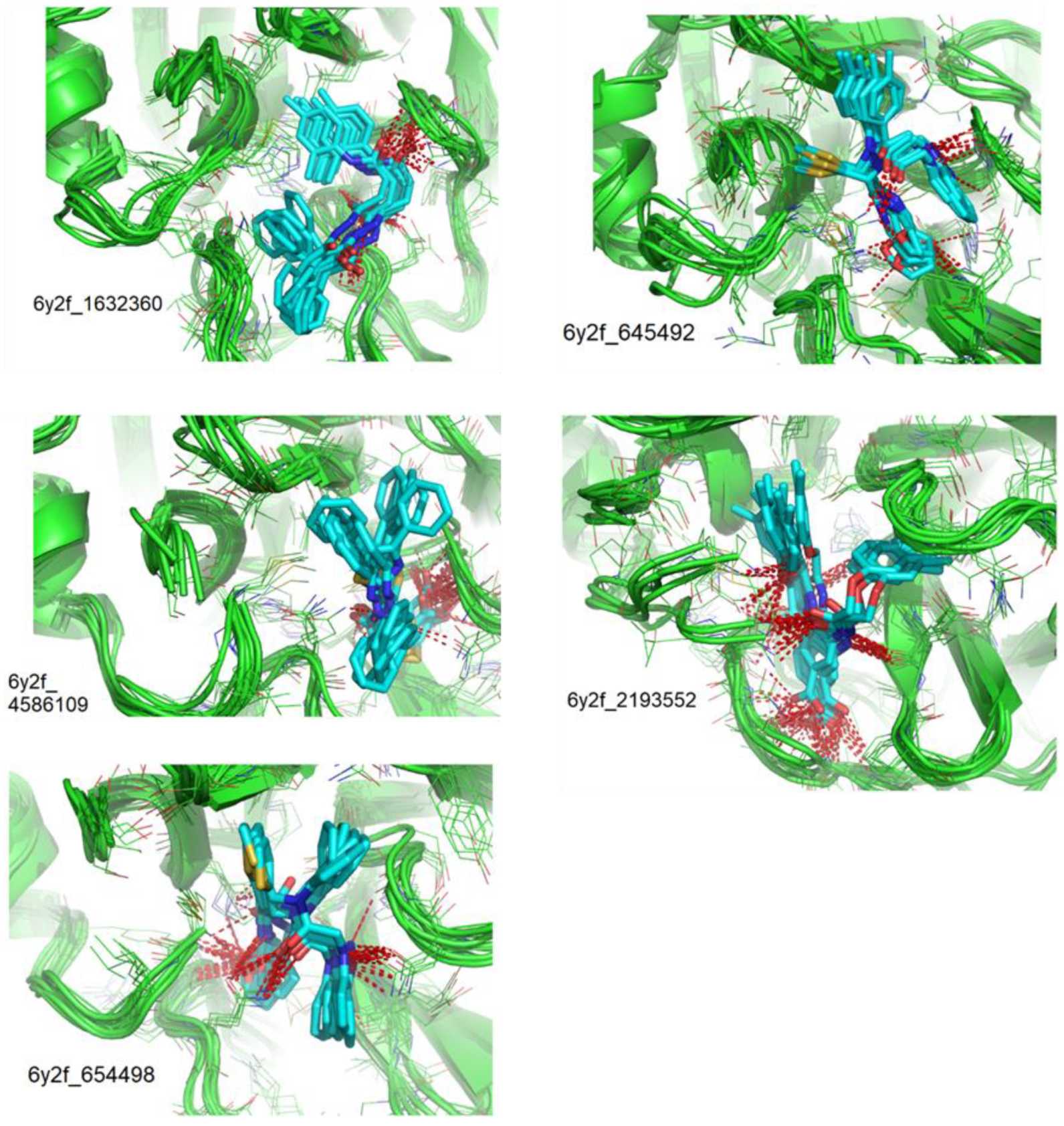
3.3. Molecular Dynamics Studies (Non-Covalent Inhibitors)
3.4. ADMET Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pillaiyar, T.; Meenakshisundaram, S.; Manickam, M. Recent discovery and development of inhibitors targeting coronaviruses. Drug Discov. Today 2020, 25, 668–688. [Google Scholar] [CrossRef]
- Menéndez, J.C. Approaches to the Potential Therapy of COVID-19: A General Overview from the Medicinal Chemistry Perspective. Molecules 2022, 27, 658. [Google Scholar] [CrossRef]
- Castillo-Garit, J.A.; Cañizares-Carmenate, Y.; Pham-The, H.; Pérez-Doñate, V.; Torrens, F.; Pérez-Giménez, F. A Review of Computational Approaches Targeting SARS-CoV-2 Main Protease to the Discovery of New Potential Antiviral Compounds. Curr. Top. Med. Chem. 2023, 23, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Suryavanshi, H.; Chaudhari, R.D.; Patil, V.; Majumdar, S.; Debnath, S.; Biswas, G. Design, synthesis and docking study of Vortioxetine derivatives as a SARS-CoV-2 main protease inhibitor. DARU J. Pharm. Sci. 2022, 30, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Mslati, H.; Gentile, F.; Perez, C.; Cherkasov, A. Comprehensive Consensus Analysis of SARS-CoV-2 Drug Repurposing Campaigns. J. Chem. Inf. Model. 2021, 61, 3771–3788. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef]
- Zhang, S.; Amahong, K.; Sun, X.; Lian, X.; Liu, J.; Sun, H.; Lou, Y.; Zhu, F.; Qiu, Y. The miRNA: A small but powerful RNA for COVID-19. Brief. Bioinform. 2021, 22, 1137–1149. [Google Scholar] [CrossRef]
- Zhang, S.; Amahong, K.; Zhang, C.; Li, F.; Gao, J.; Qiu, Y.; Zhu, F. RNA–RNA interactions between SARS-CoV-2 and host benefit viral development and evolution during COVID-19 infection. Brief. Bioinform. 2022, 23, bbab397. [Google Scholar] [CrossRef]
- Gimeno, A.; Mestres-Truyol, J.; Ojeda-Montes, M.J.; Macip, G.; Saldivar-Espinoza, B.; Cereto-Massagué, A.; Pujadas, G.; Garcia-Vallvé, S. Prediction of Novel Inhibitors of the Main Protease (M-pro) of SARS-CoV-2 through Consensus Docking and Drug Reposition. Int. J. Mol. Sci. 2020, 21, 3793. [Google Scholar] [CrossRef]
- Yamamoto, K.Z.; Yasuo, N.; Sekijima, M. Screening for Inhibitors of Main Protease in SARS-CoV-2: In Silico and In Vitro Approach Avoiding Peptidyl Secondary Amides. J. Chem. Inf. Model 2022, 62, 350–358. [Google Scholar] [CrossRef]
- Mengist, H.M.; Dilnessa, T.; Jin, T. Structural Basis of Potential Inhibitors Targeting SARS-CoV-2 Main Protease. Front. Chem. 2021, 9, 622898. [Google Scholar] [CrossRef] [PubMed]
- Citarella, A.; Scala, A.; Piperno, A.; Micale, N. SARS-CoV-2 Mpro: A Potential Target for Peptidomimetics and Small-Molecule Inhibitors. Biomolecules 2021, 11, 607. [Google Scholar] [CrossRef] [PubMed]
- Agost-Beltrán, L.; de la Hoz-Rodríguez, S.; Bou-Iserte, L.; Rodríguez, S.; Fernández-de-la-Pradilla, A.; González, F.V. Advances in the Development of SARS-CoV-2 Mpro Inhibitors. Molecules 2022, 27, 2523. [Google Scholar] [CrossRef] [PubMed]
- Antonopoulou, I.; Sapountzaki, E.; Rova, U.; Christakopoulos, P. Inhibition of the main protease of SARS-CoV-2 (M(pro)) by repurposing/designing drug-like substances and utilizing nature’s toolbox of bioactive compounds. Comput. Struct. Biotechnol. J. 2022, 20, 1306–1344. [Google Scholar] [CrossRef]
- Liu, S.; Zheng, Q.; Wang, Z. Potential covalent drugs targeting the main protease of the SARS-CoV-2 coronavirus. Bioinformatics 2020, 36, 3295–3298. [Google Scholar] [CrossRef]
- Soulère, L.; Barbier, T.; Queneau, Y. Docking-based virtual screening studies aiming at the covalent inhibition of SARS-CoV-2 M(Pro) by targeting the cysteine 145. Comput. Biol. Chem. 2021, 92, 107463. [Google Scholar] [CrossRef]
- Ton, A.T.; Gentile, F.; Hsing, M.; Ban, F.; Cherkasov, A. Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds. Mol. Inform. 2020, 39, e2000028. [Google Scholar] [CrossRef] [Green Version]
- Wang, J. Fast Identification of Possible Drug Treatment of Coronavirus Disease-19 (COVID-19) through Computational Drug Repurposing Study. J. Chem. Inf. Model. 2020, 60, 3277–3286. [Google Scholar] [CrossRef] [Green Version]
- Mishra, B.; Ballaney, P.; Saha, G.; Shinde, A.; Banerjee, S.; Thimmakondu, V.S.; Aduri, R. An in silico discovery of potential 3CL protease inhibitors of SARS-CoV-2 based upon inactivation of the cysteine 145-Histidine 41 catalytic dyad. J. Biomol. Struct. Dyn. 2022, 41, 3167–3186. [Google Scholar] [CrossRef]
- Awoonor-Williams, E.; Abu-Saleh, A.A.A. Covalent and non-covalent binding free energy calculations for peptidomimetic inhibitors of SARS-CoV-2 main protease. Phys. Chem. 2021, 23, 6746–6757. [Google Scholar] [CrossRef]
- Xie, X.Q. Exploiting PubChem for Virtual Screening. Expert Opin. Drug Discov. 2010, 5, 1205–1220. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xiao, J.; Suzek, T.O.; Zhang, J.; Wang, J.; Zhou, Z.; Han, L.; Karapetyan, K.; Dracheva, S.; Shoemaker, B.A.; et al. PubChem’s BioAssay Database. Nucleic Acids Res. 2012, 40, D400–D412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohapatra, S.; Nath, P.; Chatterjee, M.; Das, N.; Kalita, D.; Roy, P.; Satapathi, S. Repurposing therapeutics for COVID-19: Rapid prediction of commercially available drugs through machine learning and docking. PLoS ONE 2020, 15, e0241543. [Google Scholar] [CrossRef]
- Alves, V.M.; Bobrowski, T.; Melo-Filho, C.C.; Korn, D.; Auerbach, S.; Schmitt, C.; Muratov, E.N.; Tropsha, A. QSAR Modeling of SARS-CoV M(pro) Inhibitors Identifies Sufugolix, Cenicriviroc, Proglumetacin, and other Drugs as Candidates for Repurposing against SARS-CoV-2. Mol. Inform. 2021, 40, e2000113. [Google Scholar] [CrossRef] [PubMed]
- Delijewski, M.; Haneczok, J. AI drug discovery screening for COVID-19 reveals zafirlukast as a repurposing candidate. Med. Drug Discov. 2021, 9, 100077. [Google Scholar] [CrossRef] [PubMed]
- Nand, M.; Maiti, P.; Joshi, T.; Chandra, S.; Pande, V.; Kuniyal, J.C.; Ramakrishnan, M.A. Virtual screening of anti-HIV1 compounds against SARS-CoV-2: Machine learning modeling, chemoinformatics and molecular dynamics simulation based analysis. Sci. Rep. 2020, 10, 20397. [Google Scholar] [CrossRef] [PubMed]
- Santana, M.V.S.; Silva-Jr, F.P. De novo design and bioactivity prediction of SARS-CoV-2 main protease inhibitors using recurrent neural network-based transfer learning. BMC Chem. 2021, 15, 8. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Xue, X.; Yu, H.; Yang, H.; Xue, F.; Wu, Z.; Shen, W.; Li, J.; Zhou, Z.; Ding, Y.; Zhao, Q.; et al. Structures of two coronavirus main proteases: Implications for substrate binding and antiviral drug design. J. Virol. 2008, 82, 2515–2527. [Google Scholar] [CrossRef] [Green Version]
- Dalton, S.E.; Campos, S. Covalent Small Molecules as Enabling Platforms for Drug Discovery. Chembiochem 2020, 21, 1080–1100. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Samanta, I.; Mondal, A.; Liu, W.R. Covalent Inhibition in Drug Discovery. Chemmedchem 2019, 14, 889–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [Green Version]
- Thompson, M.A. ArgusLaB 4.0.1; WA planetaria Software LLC: Seattle, WA, USA, 2004. [Google Scholar]
- Oda, A.; Okayasu, M.; Kamiyama, Y.; Yoshida, T.; Takahashi, O.; Matsuzaki, H. Evaluation of docking accuracy and investigations of roles of parameters and each term in scoring functions for protein–ligand docking using ArgusLab software. Bull. Chem. Soc. Jpn. 2007, 80, 1920–1925. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Case, D.A.; Darden, T.A.; Cheatham, T.E.; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Crowley, M.; Walker, R.C.; Zhang, W. Amber 10; University of California: San Francisco, CA, USA, 2008. [Google Scholar]
- Gerber, P.R.; Müller, K. MAB, a generally applicable molecular force field for structure modelling in medicinal chemistry. J. Comput. Aided Mol. Des. 1995, 9, 251–268. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, R. An extended Hückel theory. I. hydrocarbons. J. Chem. Phys. 1963, 39, 1397–1412. [Google Scholar] [CrossRef]
- Sturgeon, J.B.; Laird, B.B. Symplectic algorithm for constant-pressure molecular dynamics using a Nosé–Poincaré thermostat. J. Chem. Phys. 2000, 112, 3474–3482. [Google Scholar] [CrossRef] [Green Version]
- Khelfaoui, H.; Harkati, D.; Saleh, B.A. Molecular docking, molecular dynamics simulations and reactivity, studies on approved drugs library targeting ACE2 and SARS-CoV-2 binding with ACE2. J. Biomol. Struct. Dyn. 2021, 39, 7246–7262. [Google Scholar] [CrossRef]
- Kurkcuoglu, Z.; Koukos, P.I.; Citro, N.; Trellet, M.E.; Rodrigues, J.; Moreira, I.S.; Roel-Touris, J.; Melquiond, A.S.J.; Geng, C.; Schaarschmidt, J.; et al. Performance of HADDOCK and a simple contact-based protein-ligand binding affinity predictor in the D3R Grand Challenge 2. J. Comput. Aided Mol. Des. 2018, 32, 175–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vangone, A.; Schaarschmidt, J.; Koukos, P.; Geng, C.; Citro, N.; Trellet, M.E.; Xue, L.C.; Bonvin, A. Large-scale prediction of binding affinity in protein-small ligand complexes: The PRODIGY-LIG web server. Bioinformatics 2019, 35, 1585–1587. [Google Scholar] [CrossRef] [Green Version]
- Douangamath, A.; Fearon, D.; Gehrtz, P.; Krojer, T.; Lukacik, P.; Owen, C.D.; Resnick, E.; Strain-Damerell, C.; Aimon, A.; Ábrányi-Balogh, P.; et al. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Nat. Commun. 2020, 11, 5047. [Google Scholar] [CrossRef]
- Grygorenko, O.O.; Volochnyuk, D.M.; Vashchenko, B.V. Emerging Building Blocks for Medicinal Chemistry: Recent Synthetic Advances. Eur. J. Org. Chem. 2021, 2021, 6478–6510. [Google Scholar] [CrossRef]
- Yoshino, R.; Yasuo, N.; Sekijima, M. Identification of key interactions between SARS-CoV-2 main protease and inhibitor drug candidates. Sci. Rep. 2020, 10, 12493. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions, and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions, or products referred to in the content. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound CID— H-Bonds and Interactions with Amino Acids | Binding Modes |
|---|---|
 |  |
 | 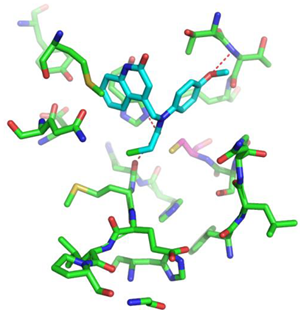 |
 |  |
 |  |
| Compound CID— H-Bonds and Interactions with Amino Acids | Binding Modes and Docking Score (kcal/mol) |
|---|---|
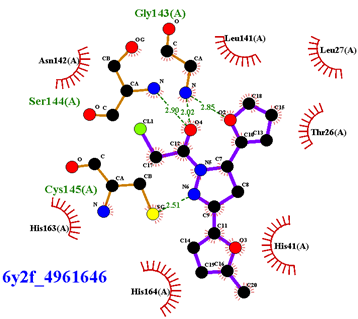
 |  |
 | 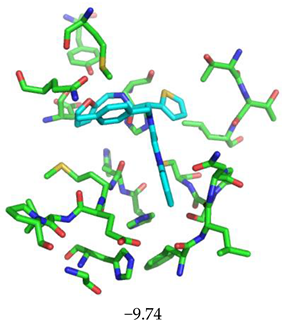 |
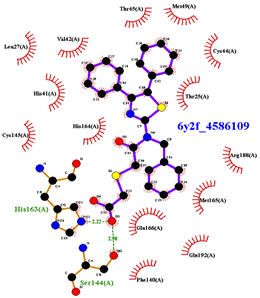 |  |
 | 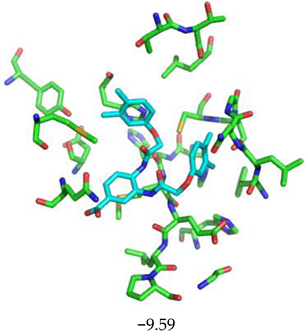 |
 |  |
| Compounds CID | Ligand RMSD (Å) | Residues Interacting via H-Bonds during the MD Simulations |
|---|---|---|
| 1632360 | 1.602 | Asn142, Gly143, Glu166 |
| 645492 | 1.087 | Met49, Ser144, Gln189 |
| 2193552 | 1.851 | His41, Met49, Asn51, Glu166, Asp187, Thr190, Gln192, Gln189 |
| 4586109 | 3.344 | Gly143, Ser144, His163, Glu166, Gln189, |
| 654498 | 1.717 | Met49, Glu166, Gln189, |
| Compounds CID | 1632360 | 645492 | 2193552 | 4586109 | 654498 |
|---|---|---|---|---|---|
| ΔGBinding t = 0 | −9.16 (Gly143) | −9.21 (Gln189, Met49, Cys145) | −10.23 (Glu166, Thr190, Gln192) | −9.42 (Gly143, Ser144, His163, Glu166) | −8.52 (Met49, Gln189) |
| ΔGBinding t = 1000 | −9.34 (Gly143, Glu166) | −9.24 (Gln189) | −9.95 (His41, Glu166, Asp187, Gln189, Thr190, Gln192) | −9.91 (Gly143, Ser144, His163, Gln189) | −9.08 (Met49, Glu166, Gln189) |
| ΔGBinding t = 2000 | −9.47 (Gly143) | −9.08 (Gln189) | −10.28 (His41, Glu166, Asp187, Gln189, Thr190, Gln192) | −9.89 (Gly143, Ser144, His163, Gln189) | −9.17 (Cys145) |
| ΔGBinding t = 3000 | −9.37 (Asn142) | −9.28 (Met49) | −10.33 (His41, Glu166, Gln189, Thr190, Gln192) | −10.20 (Gly143, Ser144, His163, Gln189) | −8.70 (Glu166, Gln189) |
| ΔGBinding t = 4000 | −9.16 (Cys145) | −9.09 (Cys145) | −10.63 (Met49 Glu166, Thr190, Gln192) | −9.79 (Gly143 Ser144, Cys145, His163) | −9.05 (Met49, Glu166, Gln189) |
| ΔGBinding t = 5000 | −9.12 (Glu166) | −9.22 (Ser144) | −10.52 (Met49, Asn51, Glu166, Thr190, Gln192) | −9.82 (Gly143 Ser144, His163, Gln189) | −9.36 (Glu166, Gln189) |
| Property | Unit | CID 843322 | CID 1154427 | CID 4868361 | CID 4961646 | CID 1632360 | CID 645492 | CID 2193552 | CID 4586109 | CID 654498 |
|---|---|---|---|---|---|---|---|---|---|---|
| Molecular weight | g/mol | 255.10 | 356.80 | 303.74 | 292.72 | 429.51 | 499.58 | 476.52 | 474.59 | 485.56 |
| LogP | 2.55 | 2.92 | 2.32 | 2.27 | 3.70 | 3.60 | 3.92 | 4.89 | 3.22 | |
| H-bond donors | 1 | 1 | 2 | 0 | 3 | 1 | 3 | 1 | 1 | |
| H-bond acceptors | 2 | 3 | 2 | 4 | 3 | 5 | 6 | 4 | 5 | |
| Rotatable bonds | 3 | 6 | 7 | 4 | 11 | 11 | 11 | 10 | 11 | |
| PSA | Å2 | 52.89 | 62.40 | 61.44 | 58.95 | 87.30 | 121.50 | 113.96 | 124.04 | 121.50 |
| Caco2 permeability | log Papp in 10–6 cm/s | 1.324 | 1.133 | 1.307 | 1.315 | 0.793 | 0.739 | 0.626 | 1.04 | 0.83 |
| Intestinal absorption (human) | % Absorbed | 91.937 | 96.64 | 91.279 | 96.937 | 93.663 | 94.393 | 70.344 | 93.421 | 93.919 |
| VDss (human) | log L/kg | 0.011 | −0.076 | −0.021 | −0.21 | −0.24 | 0.166 | −1.842 | −0.714 | 0.117 |
| Fraction unbound (human) | Fu | 0.319 | 0.038 | 0 | 0.34 | 0.024 | 0.086 | 0 | 0.278 | 0.072 |
| BBB permeability | log BB | 0.079 | −0.077 | 0.244 | 0.122 | −0.662 | −0.429 | −1.359 | −0.996 | −0.435 |
| CNS permeability | log PS | −2.781 | −2.35 | −2.088 | −2.877 | −2.191 | −2.239 | −2.896 | −2.257 | −2.308 |
| CYP2D6 inhibitor | Yes/No | No | No | No | No | No | No | No | No | No |
| CYP3A4 inhibitor | Yes/No | No | No | No | No | Yes | Yes | No | No | Yes |
| Total Clearance | log ml/min/kg | −0.054 | 0.086 | −0.069 | 0.189 | 0.241 | 0.035 | −0.045 | 0.624 | 0.091 |
| Hepatotoxicity | Yes/No (probability %) | No (61) | No (71) | No (54) | Yes (51) | No (59) | No (58) | No (75) | No (57) | No (58) |
| Carcinogenicity | Yes/No (probability %) | No (59) | Yes (52) | Yes (66) | Yes (66) | Yes (56) | Yes (52) | No (69) | No (53) | Yes (52) |
| Immunotoxicity | Yes/No (probability %) | No (99) | Yes (87) | No (99) | No (98) | No (99) | No (99) | No (99) | No (99) | No (99) |
| Mutagenicity | Yes/No (probability %) | Yes (55) | Yes (52) | No (55) | No (57) | No (52) | Yes (55) | No (79) | No (65) | Yes (56) |
| Cytotoxicity | Yes/No (probability %) | No (76) | Yes (57) | No (72) | No (68) | No (77) | No (61) | No (60) | No (66) | No (63) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soulère, L.; Barbier, T.; Queneau, Y. In Silico Identification of Potential Inhibitors of the SARS-CoV-2 Main Protease among a PubChem Database of Avian Infectious Bronchitis Virus 3CLPro Inhibitors. Biomolecules 2023, 13, 956. https://doi.org/10.3390/biom13060956
Soulère L, Barbier T, Queneau Y. In Silico Identification of Potential Inhibitors of the SARS-CoV-2 Main Protease among a PubChem Database of Avian Infectious Bronchitis Virus 3CLPro Inhibitors. Biomolecules. 2023; 13(6):956. https://doi.org/10.3390/biom13060956
Chicago/Turabian StyleSoulère, Laurent, Thibaut Barbier, and Yves Queneau. 2023. "In Silico Identification of Potential Inhibitors of the SARS-CoV-2 Main Protease among a PubChem Database of Avian Infectious Bronchitis Virus 3CLPro Inhibitors" Biomolecules 13, no. 6: 956. https://doi.org/10.3390/biom13060956