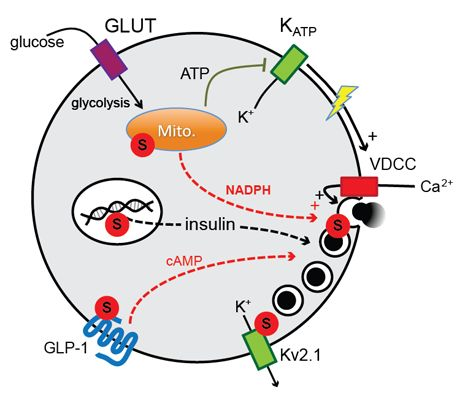
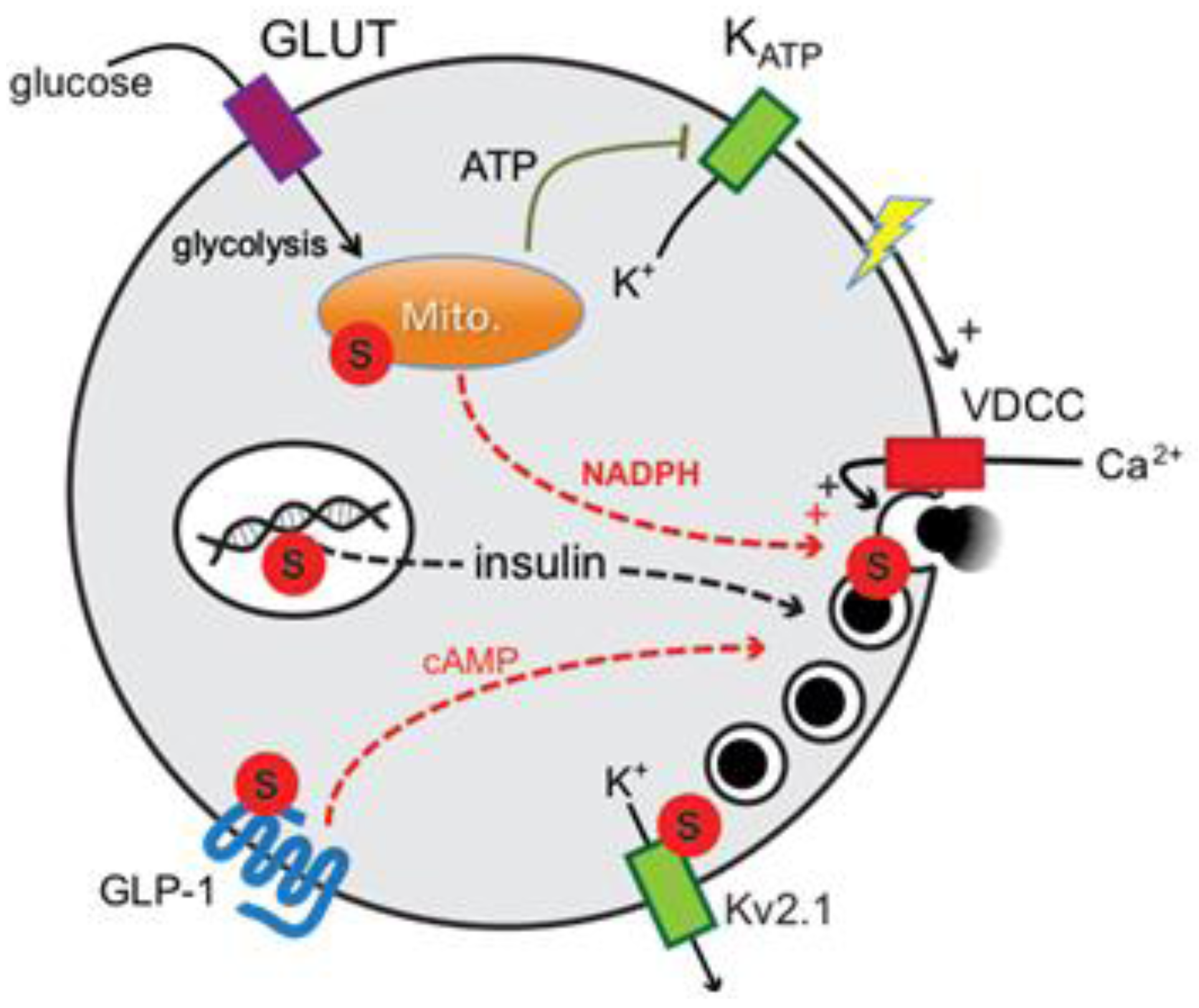
DeSUMOylation Controls Insulin Exocytosis in Response to Metabolic Signals

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

2. Results and Discussion
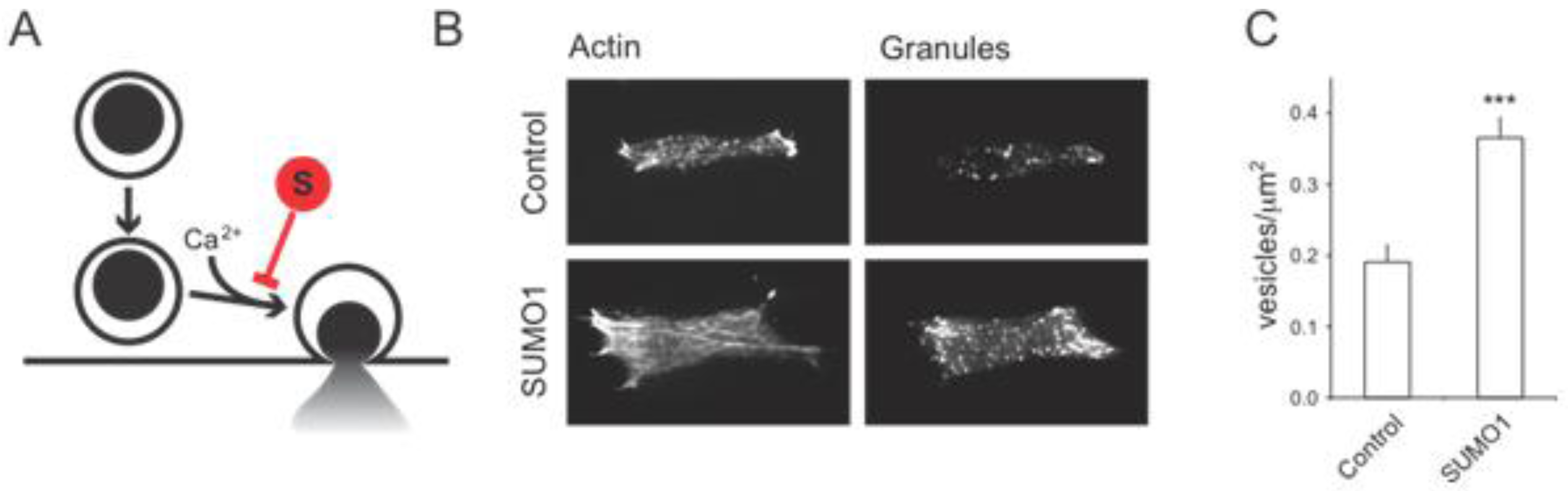
2.1. SUMO1 and Insulin Secretion
2.1.1. Acute and Reversible Inhibition of Insulin Exocytosis by SUMOylation

2.1.2. Possible SUMO Targets Inhibiting Insulin Secretion



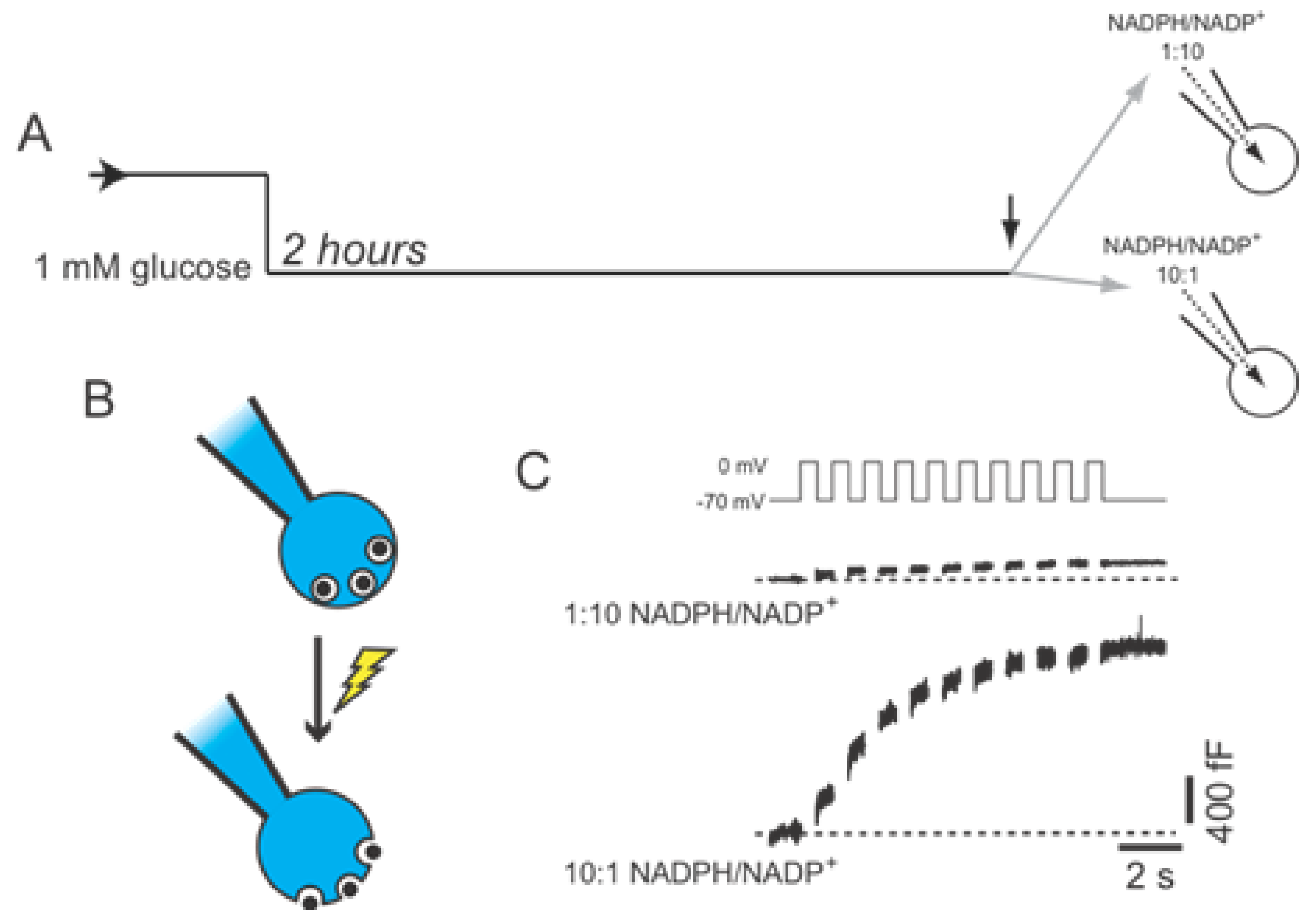
2.1.3. DeSUMOylation Is Required for Insulin Secretion
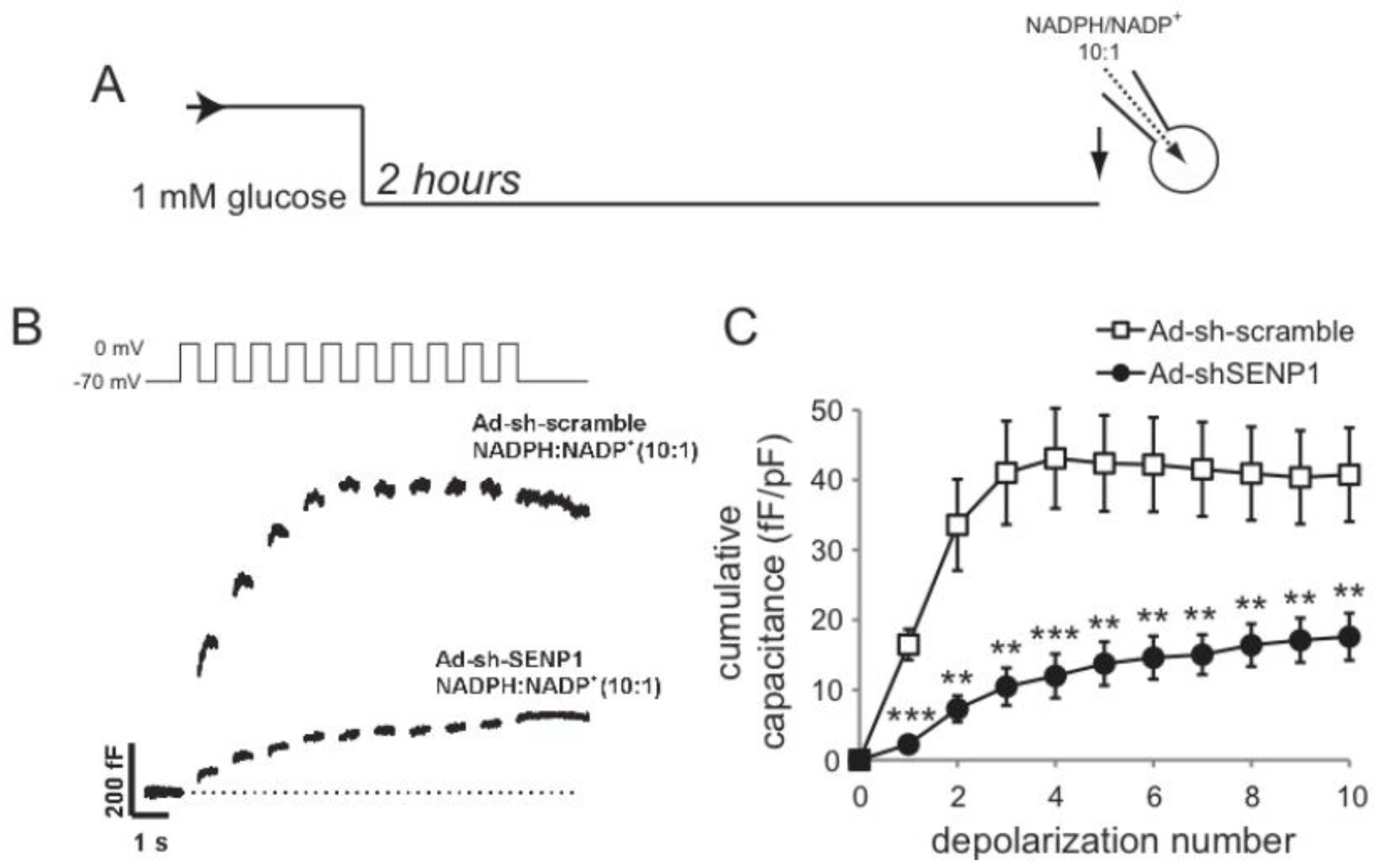
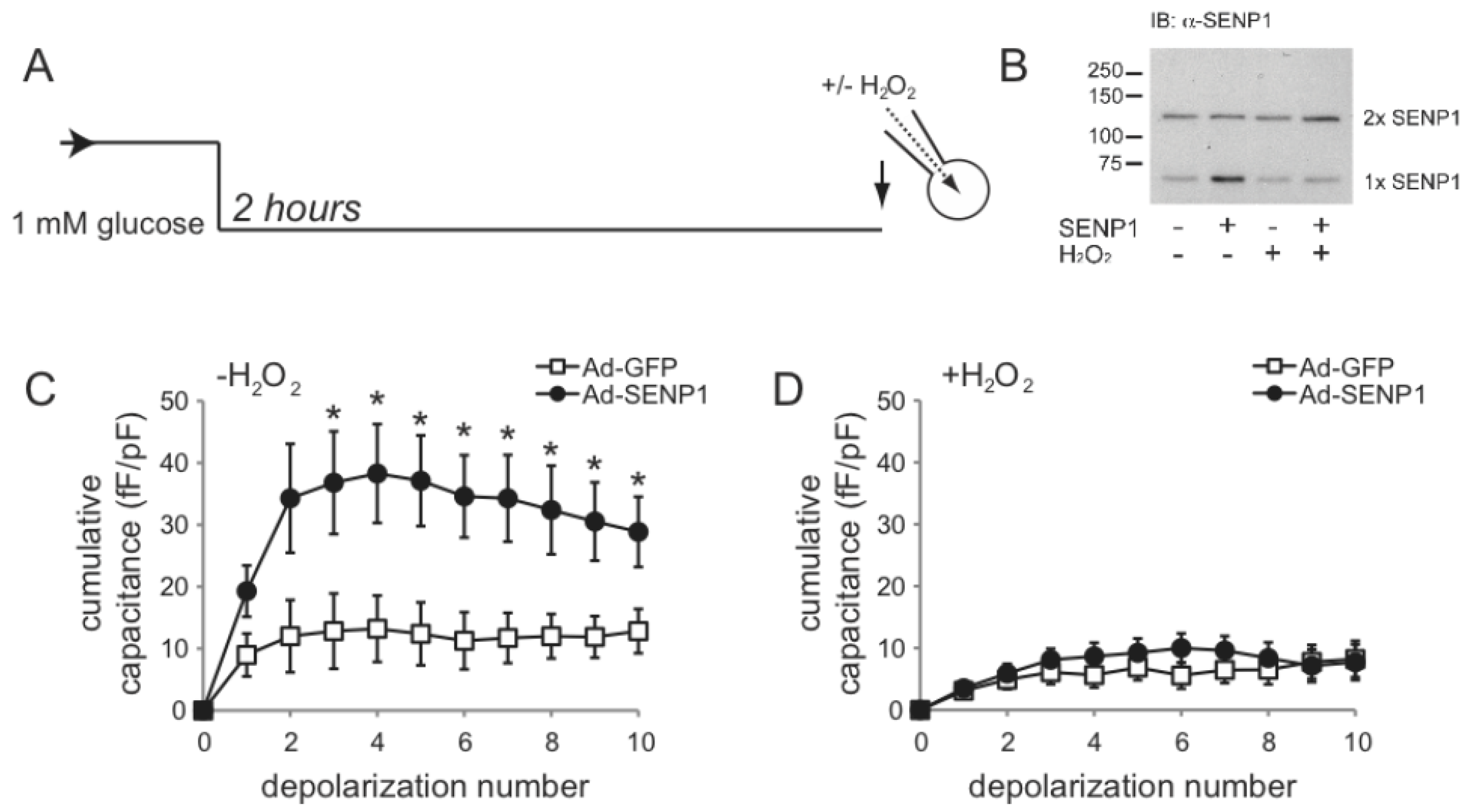
2.2. SENP1 Is Required for NADPH-Dependent Insulin Exocytosis
3. Experimental Section
3.1. Recombinant Adenoviruses, Islet Isolation and Cell Culture
3.2. Western Blotting
3.3. Electrophysiology
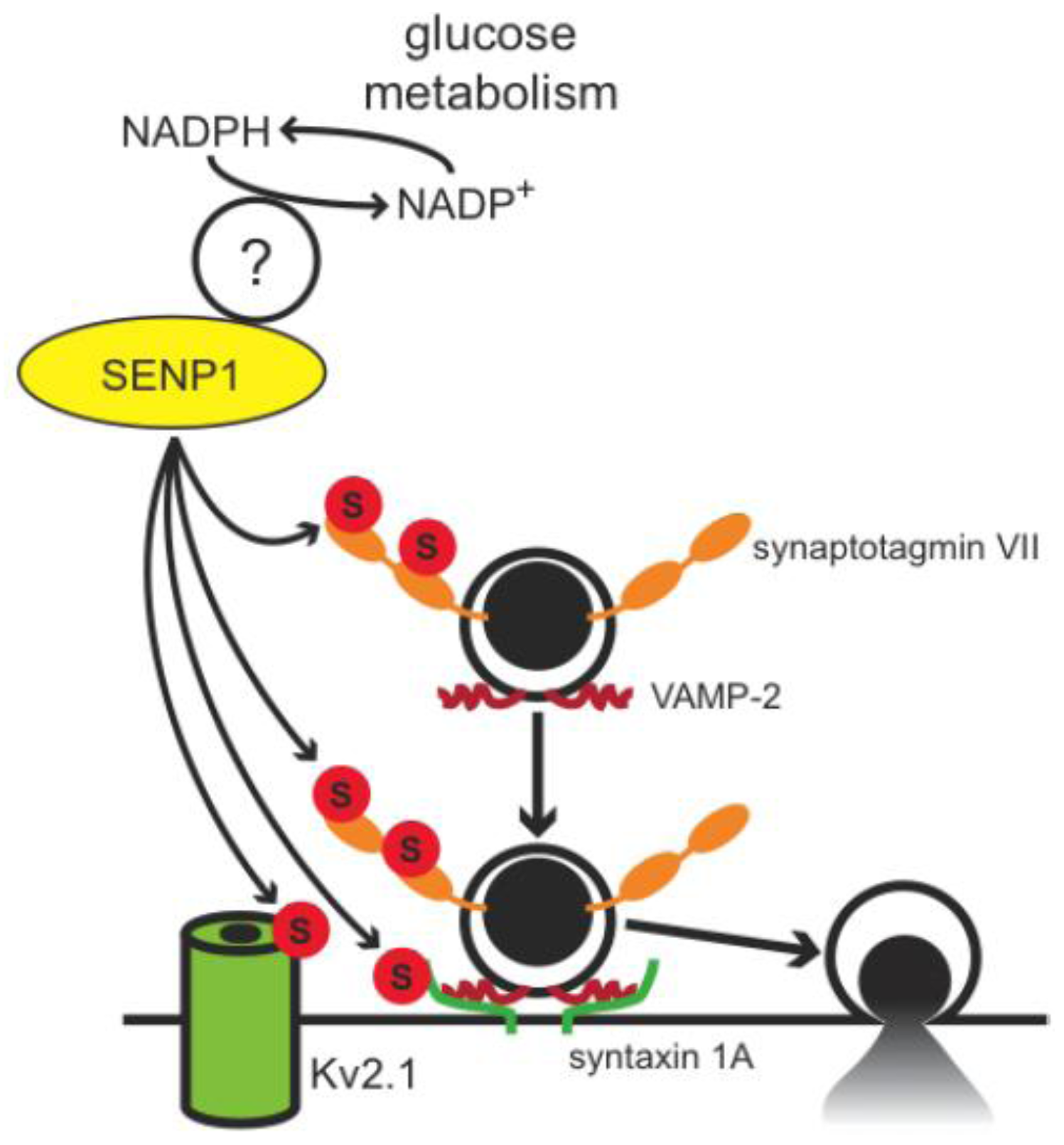
4. Conclusions

Acknowledgments
References
- Kahn, S.E. The importance of the beta-cell in the pathogenesis of type 2 diabetes mellitus. Am. J. Med. 2000, 108 Suppl. 6a, 2–8. [Google Scholar] [CrossRef]
- Henquin, J.C. The dual control of insulin secretion by glucose involves triggering and amplifying pathways in beta-cells. Diabetes Res. Clin. Pract. 2011, 93 Suppl. 1, S27–S31. [Google Scholar] [CrossRef]
- Henquin, J.C. Regulation of insulin secretion: A matter of phase control and amplitude modulation. Diabetologia 2009, 52, 739–751. [Google Scholar] [CrossRef]
- MacDonald, P.E.; Joseph, J.W.; Rorsman, P. Glucose-sensing mechanisms in pancreatic beta-cells. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005, 360, 2211–2225. [Google Scholar]
- Rorsman, P. The pancreatic beta-cell as a fuel sensor: An electrophysiologist's viewpoint. Diabetologia 1997, 40, 487–495. [Google Scholar] [CrossRef]
- MacDonald, P.E.; El-Kholy, W.; Riedel, M.J.; Salapatek, A.M.; Light, P.E.; Wheeler, M.B. The multiple actions of GLP-1 on the process of glucose-stimulated insulin secretion. Diabetes 2002, 51 Suppl. 3, S434–S442. [Google Scholar]
- Leech, C.A.; Dzhura, I.; Chepurny, O.G.; Kang, G.; Schwede, F.; Genieser, H.G.; Holz, G.G. Molecular physiology of glucagon-like peptide-1 insulin secretagogue action in pancreatic beta cells. Prog. Biophys. Mol. Biol. 2011, 107, 236–247. [Google Scholar] [CrossRef]
- Furman, B.; Ong, W.K.; Pyne, N.J. Cyclic AMP signaling in pancreatic islets. Adv. Exp. Med. Biol. 2010, 654, 281–304. [Google Scholar]
- MacDonald, M.J.; Fahien, L.A.; Brown, L.J.; Hasan, N.M.; Buss, J.D.; Kendrick, M.A. Perspective: Emerging evidence for signaling roles of mitochondrial anaplerotic products in insulin secretion. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E1–E15. [Google Scholar]
- Jensen, M.V.; Joseph, J.W.; Ronnebaum, S.M.; Burgess, S.C.; Sherry, A.D.; Newgard, C.B. Metabolic cycling in control of glucose-stimulated insulin secretion. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1287–E1297. [Google Scholar] [CrossRef]
- Ronnebaum, S.M.; Ilkayeva, O.; Burgess, S.C.; Joseph, J.W.; Lu, D.; Stevens, R.D.; Becker, T.C.; Sherry, A.D.; Newgard, C.B.; Jensen, M.V. A pyruvate cycling pathway involving cytosolic NADP-dependent isocitrate dehydrogenase regulates glucose-stimulated insulin secretion. J. Biol. Chem. 2006, 281, 30593–30602. [Google Scholar]
- Joseph, J.W.; Jensen, M.V.; Ilkayeva, O.; Palmieri, F.; Alarcon, C.; Rhodes, C.J.; Newgard, C.B. The mitochondrial citrate/isocitrate carrier plays a regulatory role in glucose-stimulated insulin secretion. J. Biol. Chem. 2006, 281, 35624–35632. [Google Scholar]
- Ivarsson, R.; Quintens, R.; Dejonghe, S.; Tsukamoto, K.; in 't Veld, P.; Renstrom, E.; Schuit, F.C. Redox control of exocytosis: Regulatory role of NADPH, thioredoxin, and glutaredoxin. Diabetes 2005, 54, 2132–2142. [Google Scholar]
- Reinbothe, T.M.; Ivarsson, R.; Li, D.Q.; Niazi, O.; Jing, X.; Zhang, E.; Stenson, L.; Bryborn, U.; Renstrom, E. Glutaredoxin-1 mediates NADPH-dependent stimulation of calcium-dependent insulin secretion. Mol. Endocrinol. 2009, 23, 893–900. [Google Scholar] [CrossRef]
- Lillig, C.H.; Berndt, C.; Holmgren, A. Glutaredoxin systems. Biochim. Biophys. Acta 1780, 1304–1317. [Google Scholar]
- Ulrich, H.D. The SUMO system: An overview. Methods Mol. Biol. 2009, 497, 3–16. [Google Scholar] [CrossRef]
- Gareau, J.R.; Lima, C.D. The SUMO pathway: Emerging mechanisms that shape specificity, conjugation and recognition. Nat. Rev. Mol. Cell. Biol. 2010, 11, 861–871. [Google Scholar]
- Kishi, A.; Nakamura, T.; Nishio, Y.; Maegawa, H.; Kashiwagi, A. Sumoylation of Pdx1 is associated with its nuclear localization and insulin gene activation. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E830–E840. [Google Scholar]
- Shao, C.; Cobb, M.H. Sumoylation regulates the transcriptional activity of MafA in pancreatic beta cells. J. Biol. Chem. 2009, 284, 3117–3124. [Google Scholar] [CrossRef]
- Martin, S.; Nishimune, A.; Mellor, J.R.; Henley, J.M. SUMOylation regulates kainate-receptor-mediated synaptic transmission. Nature 2007, 447, 321–325. [Google Scholar]
- Feligioni, M.; Nishimune, A.; Henley, J.M. Protein SUMOylation modulates calcium influx and glutamate release from presynaptic terminals. Eur. J. Neurosci. 2009, 29, 1348–1356. [Google Scholar] [CrossRef]
- Rajan, S.; Plant, L.D.; Rabin, M.L.; Butler, M.H.; Goldstein, S.A. Sumoylation silences the plasma membrane leak K+ channel K2P1. Cell 2005, 121, 37–47. [Google Scholar]
- Benson, M.D.; Li, Q.J.; Kieckhafer, K.; Dudek, D.; Whorton, M.R.; Sunahara, R.K.; Iniguez-Lluhi, J.A.; Martens, J.R. SUMO modification regulates inactivation of the voltage-gated potassium channel Kv1.5. Proc. Natl. Acad. Sci. USA 2007, 104, 1805–1810. [Google Scholar]
- Dai, X.Q.; Kolic, J.; Marchi, P.; Sipione, S.; Macdonald, P.E. SUMOylation regulates Kv2.1 and modulates pancreatic β-cell excitability. J. Cell Sci. 2009, 122, 775–779. [Google Scholar]
- Plant, L.D.; Dowdell, E.J.; Dementieva, I.S.; Marks, J.D.; Goldstein, S.A. SUMO modification of cell surface Kv2.1 potassium channels regulates the activity of rat hippocampal neurons. J. Gen. Physiol. 2011, 137, 441–454. [Google Scholar]
- Harder, Z.; Zunino, R.; McBride, H. Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Curr. Biol. 2004, 14, 340–345. [Google Scholar]
- Zunino, R.; Schauss, A.; Rippstein, P.; Andrade-Navarro, M.; McBride, H.M. The SUMO protease SENP5 is required to maintain mitochondrial morphology and function. J. Cell Sci. 2007, 120, 1178–1188. [Google Scholar]
- Braschi, E.; Zunino, R.; McBride, H.M. MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep. 2009, 10, 748–754. [Google Scholar] [CrossRef]
- Drag, M.; Salvesen, G.S. DeSUMOylating enzymes--SENPs. IUBMB Life 2008, 60, 734–742. [Google Scholar] [CrossRef]
- Kim, J.H.; Baek, S.H. Emerging roles of desumoylating enzymes. Biochim. Biophys. Acta 1792, 155–162. [Google Scholar]
- Xu, Z.; Lam, L.S.; Lam, L.H.; Chau, S.F.; Ng, T.B.; Au, S.W. Molecular basis of the redox regulation of SUMO proteases: A protective mechanism of intermolecular disulfide linkage against irreversible sulfhydryl oxidation. FASEB J. 2008, 22, 127–137. [Google Scholar]
- Xu, Z.; Chan, H.Y.; Lam, W.L.; Lam, K.H.; Lam, L.S.; Ng, T.B.; Au, S.W. SUMO proteases: Redox regulation and biological consequences. Antioxid. Redox. Signal. 2009, 11, 1453–1484. [Google Scholar]
- Hayden, M.R.; Sowers, J.R. Isletopathy in Type 2 diabetes mellitus: Implications of islet RAS, islet fibrosis, islet amyloid, remodeling, and oxidative stress. Antioxid. Redox. Signal. 2007, 9, 891–910. [Google Scholar]
- Ramirez, R.; Rasschaert, J.; Sener, A.; Malaisse, W.J. The coupling of metabolic to secretory events in pancreatic islets. Glucose-induced changes in mitochondrial redox state. Biochim. Biophys. Acta 1996, 1273, 263–267. [Google Scholar]
- Dai, X.Q.; Plummer, G.; Casimir, M.; Kang, Y.; Hajmrle, C.; Gaisano, H.; Manning-Fox, J.; MacDonald, P. E. SUMOylation regulates insulin exocytosis downstream of secretory granule docking in rodents and humans. Diabetes 2011, 60, 838–847. [Google Scholar]
- Rajan, S.; Torres, J.; Thompson, M.S.; Philipson, L.H. SUMO down-regulates GLP-1 stimulated cAMP generation and insulin secretion. Am. J. Physiol. Endocrinol. Metab. 2012. [Google Scholar]
- Rutter, G.A.; Hill, E.V. Insulin vesicle release: Walk, kiss, pause ... then run. Physiology (Bethesda) 2006, 21, 189–196. [Google Scholar] [CrossRef]
- Rorsman, P.; Renstrom, E. Insulin granule dynamics in pancreatic beta cells. Diabetologia 2003, 46, 1029–1045. [Google Scholar] [CrossRef]
- Ehninger, A.; Mziaut, H.; Solimena, M. Emerging role of SUMO in pancreatic beta-cells. Horm. Metab. Res. 2007, 39, 658–664. [Google Scholar] [CrossRef]
- Baggio, L.L.; Drucker, D.J. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157. [Google Scholar]
- MacDonald, P.E.; Ha, X.F.; Wang, J.; Smukler, S.R.; Sun, A.M.; Gaisano, H.Y.; Salapatek, A.M.; Backx, P.H.; Wheeler, M.B. Members of the Kv1 and Kv2 voltage-dependent K+ channel families regulate insulin secretion. Mol. Endocrinol. 2001, 15, 1423–1435. [Google Scholar]
- MacDonald, P.E.; Sewing, S.; Wang, J.; Joseph, J.W.; Smukler, S.R.; Sakellaropoulos, G.; Saleh, M.C.; Chan, C.B.; Tsushima, R.G.; Salapatek, A.M.; et al. Inhibition of Kv2.1 voltage-dependent K+ channels in pancreatic beta-cells enhances glucose-dependent insulin secretion. J. Biol. Chem. 2002, 277, 44938–44945. [Google Scholar]
- Yan, L.; Figueroa, D.J.; Austin, C.P.; Liu, Y.; Bugianesi, R.M.; Slaughter, R.S.; Kaczorowski, G.J.; Kohler, M.G. Expression of voltage-gated potassium channels in human and rhesus pancreatic islets. Diabetes 2004, 53, 597–607. [Google Scholar]
- Jacobson, D.A.; Kuznetsov, A.; Lopez, J.P.; Kash, S.; Ammala, C.E.; Philipson, L.H. Kv2.1 ablation alters glucose-induced islet electrical activity, enhancing insulin secretion. Cell Metab. 2007, 6, 229–235. [Google Scholar]
- Dai, X.Q.; Manning Fox, J.E.; Chikvashvili, D.; Casimir, M.; Plummer, G.; Hajmrle, C.; Spigelman, A.F.; Kin, T.; Singer-Lahat, D.; Kang, Y.; et al. The Kv2.1 channel regulates insulin secretion from rodent and human islets independent of its electrical function. Diabetologia 2012, 55, 1709–1720. [Google Scholar]
- Singer-Lahat, D.; Sheinin, A.; Chikvashvili, D.; Tsuk, S.; Greitzer, D.; Friedrich, R.; Feinshreiber, L.; Ashery, U.; Benveniste, M.; Levitan, E.S.; Lotan, I. K+ channel facilitation of exocytosis by dynamic interaction with syntaxin. J. Neurosci. 2007, 27, 1651–1658. [Google Scholar]
- Gauthier, B.R.; Wollheim, C.B. Synaptotagmins bind calcium to release insulin. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1279–E1286. [Google Scholar] [CrossRef]
- Kanno, T.; Ma, X.; Barg, S.; Eliasson, L.; Galvanovskis, J.; Gopel, S.; Larsson, M.; Renstrom, E.; Rorsman, P. Large dense-core vesicle exocytosis in pancreatic beta-cells monitored by capacitance measurements. Methods 2004, 33, 302–311. [Google Scholar] [CrossRef]
- Jitrapakdee, S.; Wutthisathapornchai, A.; Wallace, J.C.; MacDonald, M.J. Regulation of insulin secretion: Role of mitochondrial signalling. Diabetologia 2010, 53, 1019–1032. [Google Scholar]
- Pigeau, G.M.; Kolic, J.; Ball, B.J.; Hoppa, M.B.; Wang, Y.W.; Ruckle, T.; Woo, M.; Manning Fox, J.E.; Macdonald, P.E. Insulin granule recruitment and exocytosis is dependent on p110γ in insulinoma and human β-cells. Diabetes 2009, 58, 2084–2092. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Vergari, E.; Plummer, G.; Dai, X.; MacDonald, P.E. DeSUMOylation Controls Insulin Exocytosis in Response to Metabolic Signals. Biomolecules 2012, 2, 269-281. https://doi.org/10.3390/biom2020269
Vergari E, Plummer G, Dai X, MacDonald PE. DeSUMOylation Controls Insulin Exocytosis in Response to Metabolic Signals. Biomolecules. 2012; 2(2):269-281. https://doi.org/10.3390/biom2020269
Chicago/Turabian StyleVergari, Elisa, Gregory Plummer, Xiaoqing Dai, and Patrick E. MacDonald. 2012. "DeSUMOylation Controls Insulin Exocytosis in Response to Metabolic Signals" Biomolecules 2, no. 2: 269-281. https://doi.org/10.3390/biom2020269