Homologous Recombination as a Replication Fork Escort: Fork-Protection and Recovery

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
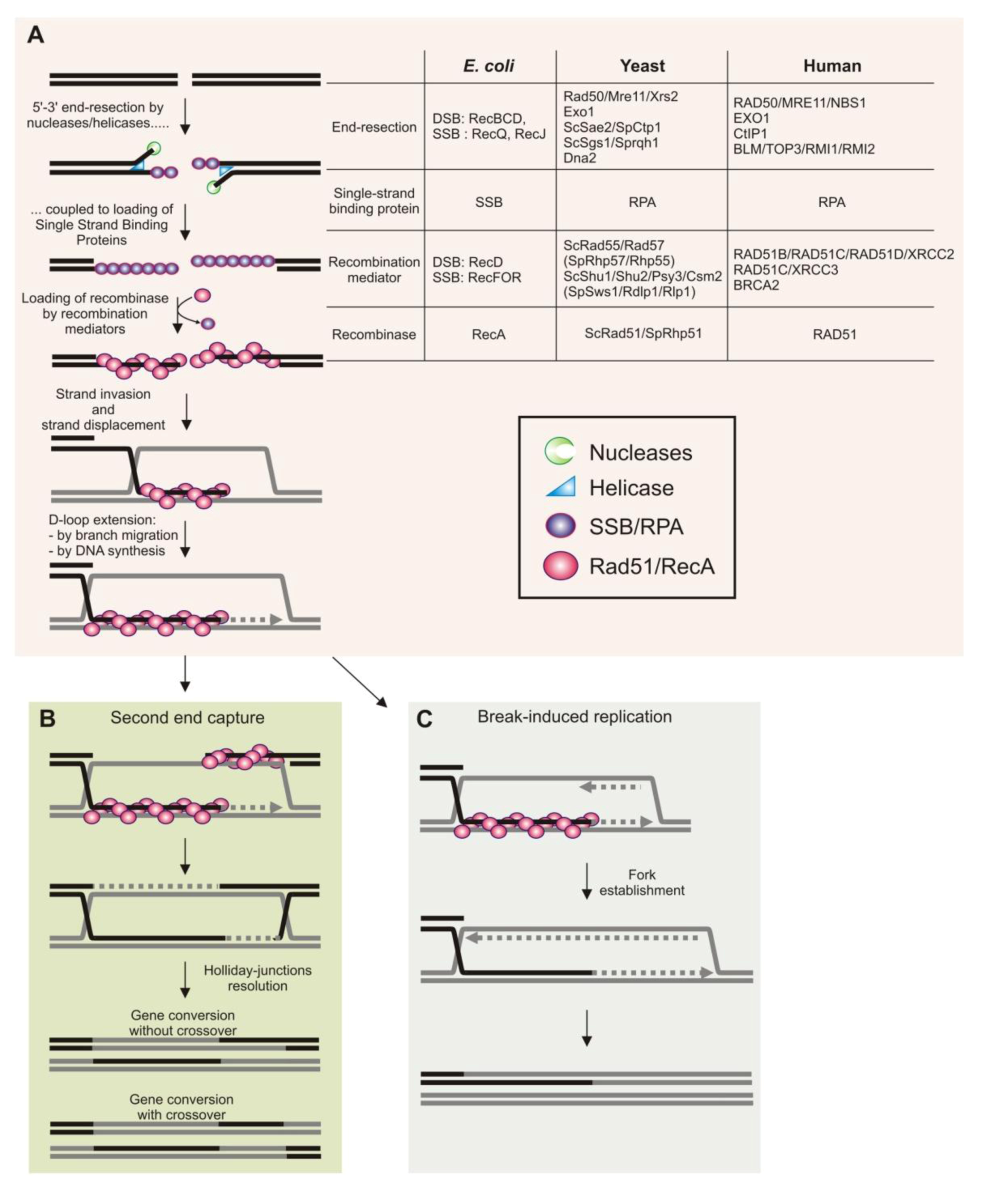
2. Homologous Recombination during DNA Replication in Bacteria
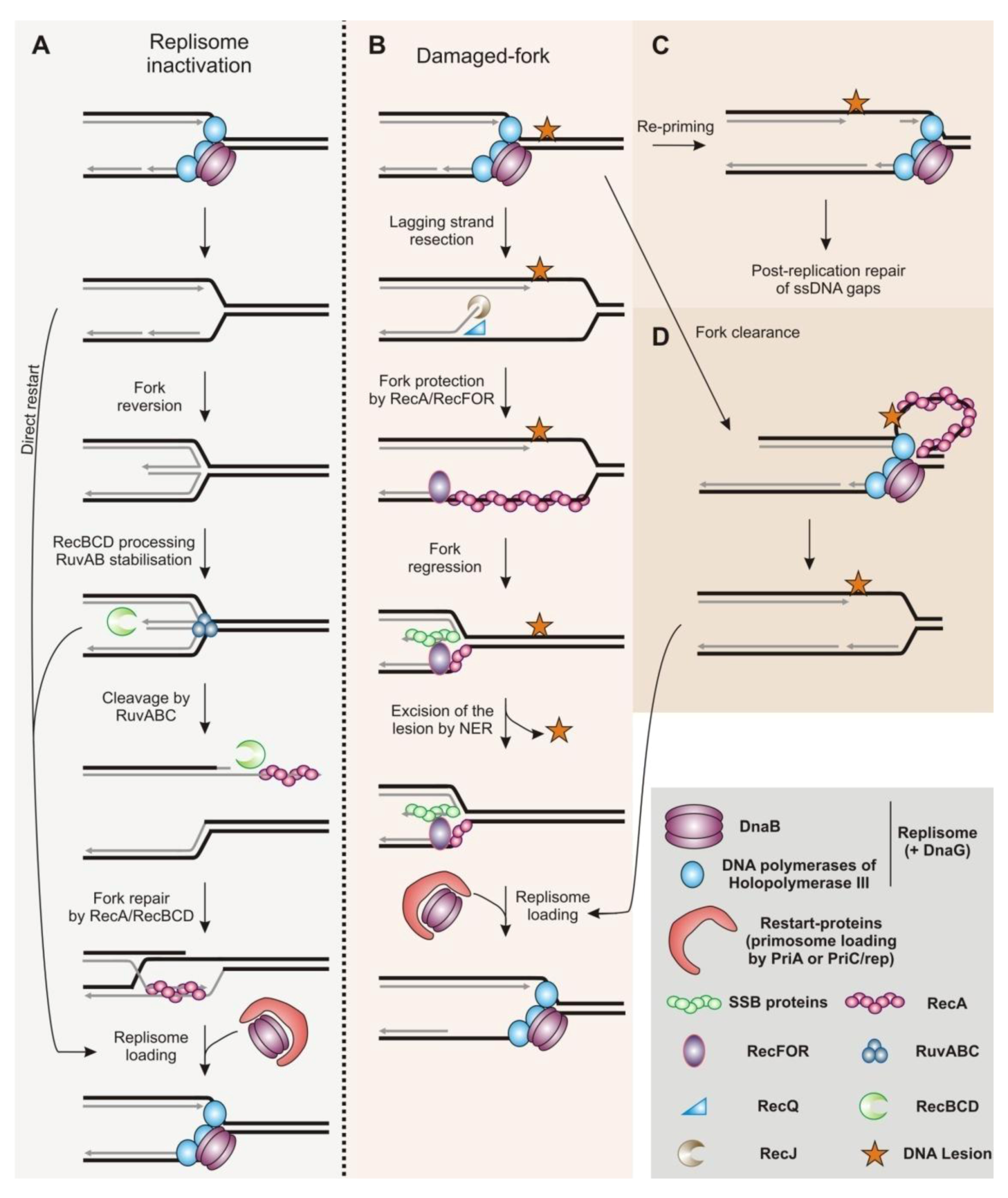
2.1. Restart of Inactivated Replication Forks by Homologous Recombination
2.2. Replication Fork Protection by Homologous Recombination
2.3. Homologous Recombination as an Escort of Fork Progression
3. Homologous Recombination during DNA Replication in Eukaryotes
3.1. Recombination Mediator Proteins in Eukaryotes
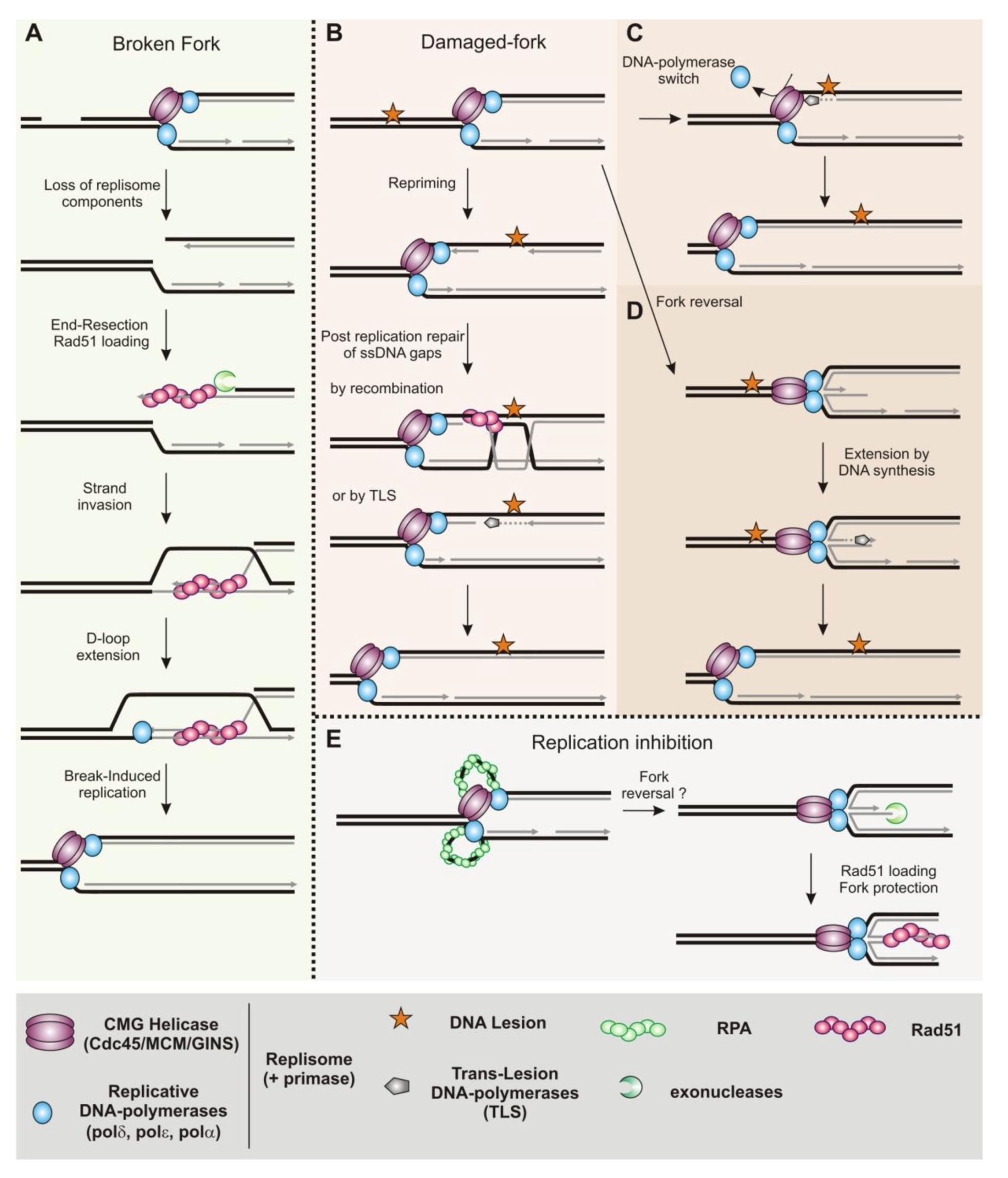
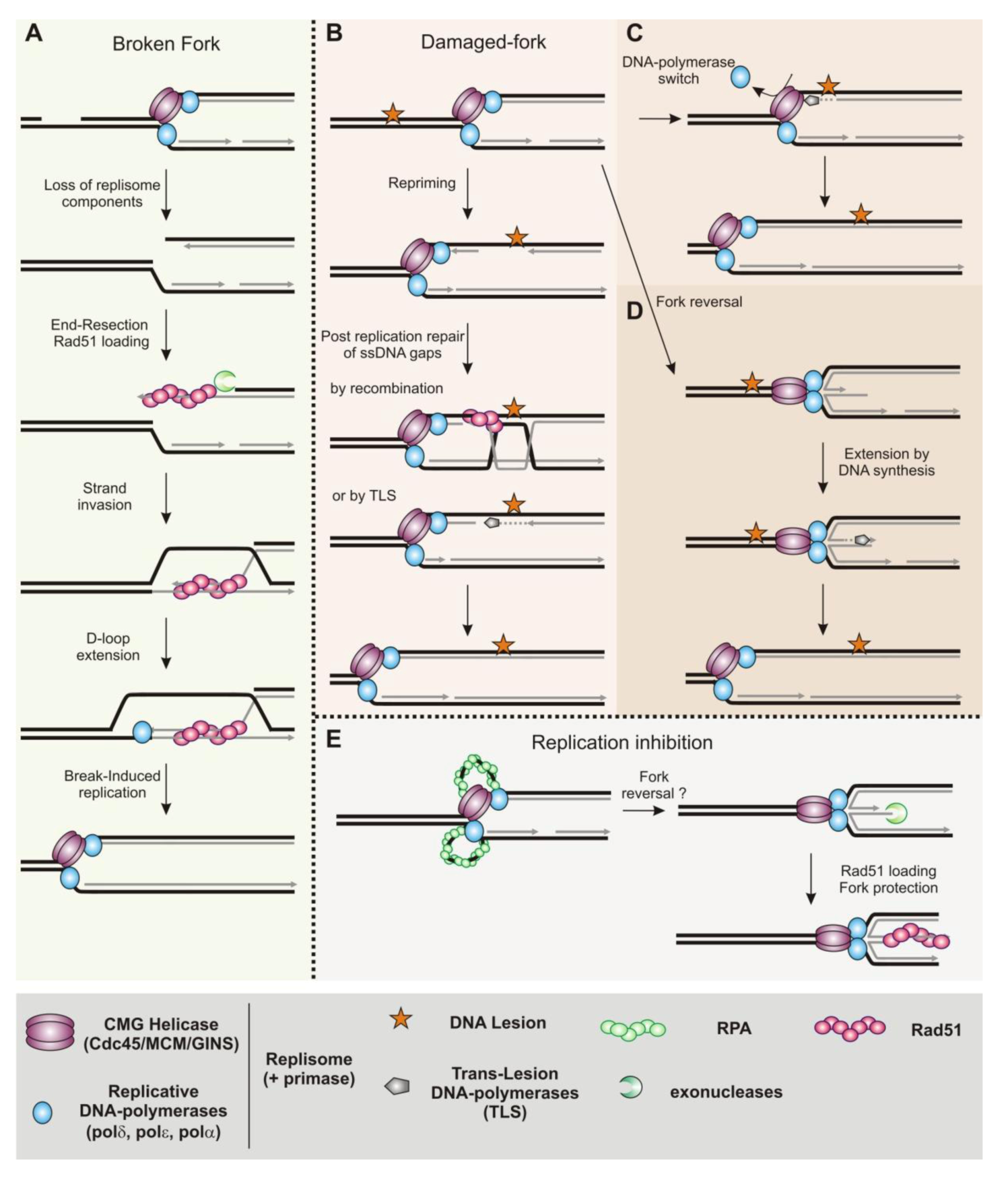
3.2. Homologous Recombination Is Coupled to DNA Replication
3.3. Post-Replication Repair by Homologous Recombination
3.4. Replication Fork Restart and Repair by Homologous Recombination
3.5. A Fork-Protection Function for the Homologous Recombination Machinery
3.6. A Possible Separation of Function
4. Concluding Remarks
Acknowledgments
References
- West, S.C. Molecular views of recombination proteins and their control. Natl. Rev. Mol. Cell Biol. 2003, 4, 435–445. [Google Scholar] [CrossRef]
- Cox, M.M. Regulation of bacterial reca protein function. Crit. Rev. Biochem. Mol. Biol. 2007, 42, 41–63. [Google Scholar] [CrossRef]
- Krejci, L.; Altmannova, V.; Spirek, M.; Zhao, X. Homologous recombination and its regulation. Nucleic Acids Res. 2012, 40, 5795–5818. [Google Scholar]
- Deakyne, J.S.; Mazin, A.V. Fanconi anemia: At the crossroads of DNA repair. Biochem. Biokhimiia 2011, 76, 36–48. [Google Scholar] [CrossRef]
- Michel, B.; Boubakri, H.; Baharoglu, Z.; LeMasson, M.; Lestini, R. Recombination proteins and rescue of arrested replication forks. DNA Repair 2007, 6, 967–980. [Google Scholar] [CrossRef]
- Costanzo, V. Brca2, rad51 and mre11: Performing balancing acts on replication forks. DNA Repair 2011, 10, 1060–1065. [Google Scholar]
- Courcelle, J.; Hanawalt, P.C. Reca-dependent recovery of arrested DNA replication forks. Annu. Rev. Genet. 2003, 37, 611–646. [Google Scholar] [CrossRef]
- Cox, M.M.; Goodman, M.F.; Kreuzer, K.N.; Sherratt, D.J.; Sandler, S.J.; Marians, K.J. The importance of repairing stalled replication forks. Nature 2000, 404, 37–41. [Google Scholar] [CrossRef]
- Heller, R.C.; Marians, K.J. Replication fork reactivation downstream of a blocked nascent leading strand. Nature 2006, 439, 557–562. [Google Scholar]
- Heller, R.C.; Marians, K.J. Replisome assembly and the direct restart of stalled replication forks. Nat. Rev. Mol. Cell Biol. 2006, 7, 932–943. [Google Scholar] [CrossRef]
- Gabbai, C.B.; Marians, K.J. Recruitment to stalled replication forks of the pria DNA helicase and replisome-loading activities is essential for survival. DNA Repair 2010, 9, 202–209. [Google Scholar] [CrossRef]
- Heller, R.C.; Marians, K.J. The disposition of nascent strands at stalled replication forks dictates the pathway of replisome loading during restart. Mol. Cell 2005, 17, 733–743. [Google Scholar]
- Kowalczykowski, S.C. Initiation of genetic recombination and recombination-dependent replication. Trends Biochem. Sci. 2000, 25, 156–165. [Google Scholar] [CrossRef]
- Lusetti, S.L.; Cox, M.M. The bacterial reca protein and the recombinational DNA repair of stalled replication forks. Annu. Rev. Biochem. 2002, 71, 71–100. [Google Scholar] [CrossRef]
- Kuzminov, A. DNA replication meets genetic exchange: Chromosomal damage and its repair by homologous recombination. Proc. Natl. Acad. Sci. USA 2001, 98, 8461–8468. [Google Scholar]
- Liu, J.; Marians, K.J. Pria-directed assembly of a primosome on d loop DNA. J. Biol. Chem. 1999, 274, 25033–25041. [Google Scholar] [CrossRef]
- Ng, J.Y.; Marians, K.J. The ordered assembly of the phix174-type primosome. Ii. Preservation of primosome composition from assembly through replication. J. Biol. Chem. 1996, 271, 15649–15655. [Google Scholar] [CrossRef]
- Liu, J.; Nurse, P.; Marians, K.J. The ordered assembly of the phix174-type primosome. III Prib facilitates complex formation between pria and dnat. J. Biol. Chem. 1996, 271, 15656–15661. [Google Scholar] [CrossRef]
- Xu, L.; Marians, K.J. Pria mediates DNA replication pathway choice at recombination intermediates. Mol. Cell 2003, 11, 817–826. [Google Scholar]
- Nurse, P.; Liu, J.; Marians, K.J. Two modes of pria binding to DNA. J. Biol. Chem. 1999, 274, 25026–25032. [Google Scholar] [CrossRef]
- McGlynn, P.; Al-Deib, A.A.; Liu, J.; Marians, K.J.; Lloyd, R.G. The DNA replication protein pria and the recombination protein recg bind d-loops. J. Mol. Biol. 1997, 270, 212–221. [Google Scholar] [CrossRef]
- Kowalczykowski, S.C.; Krupp, R.A. Effects of escherichia coli ssb protein on the single-stranded DNA-dependent atpase activity of escherichia coli reca protein. Evidence that ssb protein facilitates the binding of reca protein to regions of secondary structure within single-stranded DNA. J. Mol. Biol. 1987, 193, 97–113. [Google Scholar] [CrossRef]
- Roy, R.; Kozlov, A.G.; Lohman, T.M.; Ha, T. Ssb protein diffusion on single-stranded DNA stimulates reca filament formation. Nature 2009, 461, 1092–1097. [Google Scholar]
- Umezu, K.; Kolodner, R.D. Protein interactions in genetic recombination in escherichia coli. Interactions involving reco and recr overcome the inhibition of reca by single-stranded DNA-binding protein. J. Biol. Chem. 1994, 269, 30005–30013. [Google Scholar]
- Umezu, K.; Chi, N.W.; Kolodner, R.D. Biochemical interaction of the escherichia coli recf, reco, and recr proteins with reca protein and single-stranded DNA binding protein. Proc. Natl. Acad. Sci. USA 1993, 90, 3875–3879. [Google Scholar]
- Beernink, H.T.; Morrical, S.W. Rmps: Recombination/replication mediator proteins. Trends Biochem. Sci. 1999, 24, 385–389. [Google Scholar] [CrossRef]
- Kuzminov, A.; Stahl, F.W. Double-strand end repair via the recbc pathway in escherichia coli primes DNA replication. Genes Dev. 1999, 13, 345–356. [Google Scholar] [CrossRef]
- Dillingham, M.S.; Kowalczykowski, S.C. Recbcd enzyme and the repair of double-stranded DNA breaks. Microbiol. Mol. Biol. Rev. 2008, 72, 642–671. [Google Scholar] [CrossRef]
- Yeeles, J.T.; Dillingham, M.S. The processing of double-stranded DNA breaks for recombinational repair by helicase-nuclease complexes. DNA Repair 2010, 9, 276–285. [Google Scholar]
- Sakai, A.; Cox, M.M. Recfor and recor as distinct reca loading pathways. J. Biol. Chem. 2009, 284, 3264–3272. [Google Scholar] [CrossRef]
- Lovett, S.T.; Kolodner, R.D. Identification and purification of a single-stranded-DNA-specific exonuclease encoded by the recj gene of escherichia coli. Proc. Natl. Acad. Sci. USA 1989, 86, 2627–2631. [Google Scholar] [CrossRef]
- Umezu, K.; Nakayama, K.; Nakayama, H. Escherichia coli recq protein is a DNA helicase. Proc. Natl. Acad. Sci. USA 1990, 87, 5363–5367. [Google Scholar] [CrossRef]
- Tseng, Y.C.; Hung, J.L.; Wang, T.C. Involvement of recf pathway recombination genes in postreplication repair in uv-irradiated escherichia coli cells. Mut. Res. 1994, 315, 1–9. [Google Scholar] [CrossRef]
- Courcelle, J.; Hanawalt, P.C. Recq and recj process blocked replication forks prior to the resumption of replication in uv-irradiated escherichia coli. Mol. Gen. Genet. 1999, 262, 543–551. [Google Scholar] [CrossRef]
- Webb, B.L.; Cox, M.M.; Inman, R.B. Recombinational DNA repair: The recf and recr proteins limit the extension of reca filaments beyond single-strand DNA gaps. Cell 1997, 91, 347–356. [Google Scholar]
- Anderson, D.G.; Kowalczykowski, S.C. The translocating recbcd enzyme stimulates recombination by directing reca protein onto ssdna in a chi-regulated manner. Cell 1997, 90, 77–86. [Google Scholar] [CrossRef]
- Baharoglu, Z.; Petranovic, M.; Flores, M.J.; Michel, B. Ruvab is essential for replication forks reversal in certain replication mutants. EMBO J. 2006, 25, 596–604. [Google Scholar] [CrossRef]
- Flores, M.J.; Bierne, H.; Ehrlich, S.D.; Michel, B. Impairment of lagging strand synthesis triggers the formation of a ruvabc substrate at replication forks. EMBO J. 2001, 20, 619–629. [Google Scholar]
- Michel, B.; Ehrlich, S.D.; Uzest, M. DNA double-strand breaks caused by replication arrest. EMBO J. 1997, 16, 430–438. [Google Scholar] [CrossRef]
- Grompone, G.; Seigneur, M.; Ehrlich, S.D.; Michel, B. Replication fork reversal in DNA polymerase III mutants of escherichia coli: A role for the beta clamp. Mol. Microbiol. 2002, 44, 1331–1339. [Google Scholar] [CrossRef]
- Pennington, J.M.; Rosenberg, S.M. Spontaneous DNA breakage in single living escherichia coli cells. Nat. Genet. 2007, 39, 797–802. [Google Scholar] [CrossRef]
- Seigneur, M.; Bidnenko, V.; Ehrlich, S.D.; Michel, B. Ruvab acts at arrested replication forks. Cell 1998, 95, 419–430. [Google Scholar]
- Grompone, G.; Ehrlich, D.; Michel, B. Cells defective for replication restart undergo replication fork reversal. EMBO Rep. 2004, 5, 607–612. [Google Scholar] [CrossRef]
- De Septenville, A.L.; Duigou, S.; Boubakri, H.; Michel, B. Replication fork reversal after replication-transcription collision. PLoS Genet. 2012, 8. [Google Scholar] [CrossRef]
- Seigneur, M.; Ehrlich, S.D.; Michel, B. Ruvabc-dependent double-strand breaks in dnabts mutants require reca. Mol. Microbiol. 2000, 38, 565–574. [Google Scholar] [CrossRef]
- McGlynn, P.; Lloyd, R.G. Genome stability and the processing of damaged replication forks by recg. Trends Genet. 2002, 18, 413–419. [Google Scholar]
- McGlynn, P.; Lloyd, R.G.; Marians, K.J. Formation of holliday junctions by regression of nascent DNA in intermediates containing stalled replication forks: Recg stimulates regression even when the DNA is negatively supercoiled. Proc. Natl. Acad. Sci. USA 2001, 98, 8235–8240. [Google Scholar] [CrossRef]
- Robu, M.E.; Inman, R.B.; Cox, M.M. Reca protein promotes the regression of stalled replication forks in vitro. Proc. Natl. Acad. Sci. USA 2001, 98, 8211–8218. [Google Scholar]
- Flores, M.J.; Bidnenko, V.; Michel, B. The DNA repair helicase uvrd is essential for replication fork reversal in replication mutants. EMBO Rep. 2004, 5, 983–988. [Google Scholar] [CrossRef]
- Flores, M.J.; Sanchez, N.; Michel, B. A fork-clearing role for uvrd. Mol. Microbiol. 2005, 57, 1664–1675. [Google Scholar]
- Setlow, R.B. Cyclobutane-type pyrimidine dimers in polynucleotides. Science 1966, 153, 379–386. [Google Scholar]
- Koehler, D.R.; Courcelle, J.; Hanawalt, P.C. Kinetics of pyrimidine(6-4)pyrimidone photoproduct repair in escherichia coli. J. Bacteriol. 1996, 178, 1347–1350. [Google Scholar]
- Howard-Flanders, P.; Boyce, R.P.; Theriot, L. Three loci in escherichia coli k-12 that control the excision of pyrimidine dimers and certain other mutagen products from DNA. Genetics 1966, 53, 1119–1136. [Google Scholar]
- Howard-Flanders, P.; Theriot, L. Mutants of escherichia coli k-12 defective in DNA repair and in genetic recombination. Genetics 1966, 53, 1137–1150. [Google Scholar]
- Smith, D.W.; Hanawalt, P.C. Repair replication of DNA in ultraviolet irradiated mycoplasma laidlawii b. J. Mol. Biol. 1969, 46, 57–72. [Google Scholar] [CrossRef]
- Villani, G.; Spadari, S.; Boiteux, S.; Defais, M.; Caillet-Fauquet, P.; Radman, M. Replication of chemically modified DNA. Biochimie 1978, 60, 1145–1150. [Google Scholar]
- Truglio, J.J.; Karakas, E.; Rhau, B.; Wang, H.; DellaVecchia, M.J.; Van Houten, B.; Kisker, C. Structural basis for DNA recognition and processing by uvrb. Nat. Struct. Mol. Biol. 2006, 13, 360–364. [Google Scholar]
- Wang, T.C.; Smith, K.C. Mechanisms for recf-dependent and recb-dependent pathways of postreplication repair in uv-irradiated escherichia coli uvrb. J. Bacteriol. 1983, 156, 1093–1098. [Google Scholar]
- Donaldson, J.R.; Courcelle, C.T.; Courcelle, J. Ruvabc is required to resolve holliday junctions that accumulate following replication on damaged templates in escherichia coli. J. Biol. Chem. 2006, 281, 28811–28821. [Google Scholar] [CrossRef]
- Rupp, W.D.; Wilde, C.E., III.; Reno, D.L.; Howard-Flanders, P. Exchanges between DNA strands in ultraviolet-irradiated escherichia coli. J. Mol. Biol. 1971, 61, 25–44. [Google Scholar] [CrossRef]
- Howard-Flanders, P.; Rupp, W.D. Recombinational repair in uv-irradiated escherichia coli. Johns Hopkins Med. J. 1972, 1, 212–225. [Google Scholar]
- Pages, V.; Fuchs, R.P. Uncoupling of leading- and lagging-strand DNA replication during lesion bypass in vivo. Science 2003, 300, 1300–1303. [Google Scholar] [CrossRef]
- Higuchi, K.; Katayama, T.; Iwai, S.; Hidaka, M.; Horiuchi, T.; Maki, H. Fate of DNA replication fork encountering a single DNA lesion during oric plasmid DNA replication in vitro. Genes to Cells: Devoted to Molecular and Cellular Mechanisms 2003, 8, 437–449. [Google Scholar] [CrossRef]
- Yeeles, J.T.; Marians, K.J. The Escherichia coli replisome is inherently DNA damage tolerant. Science 2011, 334, 235–238. [Google Scholar] [CrossRef]
- Khidhir, M.A.; Casaregola, S.; Holland, I.B. Mechanism of transient inhibition of DNA synthesis in ultraviolet-irradiated E. coli: Inhibition is independent of reca whilst recovery requires reca protein itself and an additional, inducible sos function. Mol. Gen. Genet. 1985, 199, 133–140. [Google Scholar] [CrossRef]
- Courcelle, C.T.; Belle, J.J.; Courcelle, J. Nucleotide excision repair or polymerase v-mediated lesion bypass can act to restore uv-arrested replication forks in escherichia coli. J. Bacteriol. 2005, 187, 6953–6961. [Google Scholar] [CrossRef]
- Rudolph, C.J.; Upton, A.L.; Lloyd, R.G. Replication fork stalling and cell cycle arrest in uv-irradiated escherichia coli. Genes Dev. 2007, 21, 668–681. [Google Scholar] [CrossRef]
- Rudolph, C.J.; Upton, A.L.; Lloyd, R.G. Maintaining replication fork integrity in uv-irradiated escherichia coli cells. DNA Repair 2008, 7, 1589–1602. [Google Scholar] [CrossRef]
- Courcelle, J.; Carswell-Crumpton, C.; Hanawalt, P.C. Recf and recr are required for the resumption of replication at DNA replication forks in escherichia coli. Proc. Natl. Acad. Sci. USA 1997, 94, 3714–3719. [Google Scholar] [CrossRef]
- Courcelle, J.; Crowley, D.J.; Hanawalt, P.C. Recovery of DNA replication in uv-irradiated escherichia coli requires both excision repair and recf protein function. J. Bacteriol. 1999, 181, 916–922. [Google Scholar]
- Chow, K.H.; Courcelle, J. Reco acts with recf and recr to protect and maintain replication forks blocked by uv-induced DNA damage in escherichia coli. J. Biol. Chem. 2004, 279, 3492–3496. [Google Scholar]
- Courcelle, C.T.; Landstrom, A.J.; Anderson, B.; Courcelle, J. Cellular characterization of the primosome and rep helicase in processing and restoration of replication following arrest by uv-induced DNA damage in escherichia coli. J. Bacteriol. 2012, 194, 3977–3986. [Google Scholar] [CrossRef]
- Hishida, T.; Han, Y.W.; Shibata, T.; Kubota, Y.; Ishino, Y.; Iwasaki, H.; Shinagawa, H. Role of the escherichia coli recq DNA helicase in sos signaling and genome stabilization at stalled replication forks. Genes Dev. 2004, 18, 1886–1897. [Google Scholar] [CrossRef]
- McInerney, P.; O'Donnell, M. Replisome fate upon encountering a leading strand block and clearance from DNA by recombination proteins. J. Biol. Chem. 2007, 282, 25903–25916. [Google Scholar]
- Courcelle, J.; Hanawalt, P.C. Participation of recombination proteins in rescue of arrested replication forks in uv-irradiated escherichia coli need not involve recombination. Proc. Natl. Acad. Sci. USA 2001, 98, 8196–8202. [Google Scholar] [CrossRef]
- Courcelle, J.; Ganesan, A.K.; Hanawalt, P.C. Therefore, what are recombination proteins there for? BioEssays 2001, 23, 463–470. [Google Scholar] [CrossRef]
- Al-Hadid, Q.; Ona, K.; Courcelle, C.T.; Courcelle, J. Reca433 cells are defective in recf-mediated processing of disrupted replication forks but retain recbcd-mediated functions. Mut. Res. 2008, 645, 19–26. [Google Scholar]
- Renzette, N.; Gumlaw, N.; Nordman, J.T.; Krieger, M.; Yeh, S.P.; Long, E.; Centore, R.; Boonsombat, R.; Sandler, S.J. Localization of reca in escherichia coli k-12 using reca-gfp. Mol. Microbiol. 2005, 57, 1074–1085. [Google Scholar] [CrossRef]
- Simmons, L.A.; Grossman, A.D.; Walker, G.C. Replication is required for the reca localization response to DNA damage in bacillus subtilis. Proc. Natl. Acad. Sci. USA 2007, 104, 1360–1365. [Google Scholar]
- Lecointe, F.; Serena, C.; Velten, M.; Costes, A.; McGovern, S.; Meile, J.C.; Errington, J.; Ehrlich, S.D.; Noirot, P.; Polard, P. Anticipating chromosomal replication fork arrest: Ssb targets repair DNA helicases to active forks. EMBO J. 2007, 26, 4239–4251. [Google Scholar] [CrossRef]
- Costes, A.; Lecointe, F.; McGovern, S.; Quevillon-Cheruel, S.; Polard, P. The c-terminal domain of the bacterial ssb protein acts as a DNA maintenance hub at active chromosome replication forks. PLoS Genet. 2010, 6. [Google Scholar] [CrossRef]
- Mechali, M. Eukaryotic DNA replication origins: Many choices for appropriate answers. Nat. Rev. Mol. Cell Biol. 2010, 11, 728–738. [Google Scholar] [CrossRef]
- Cayrou, C.; Coulombe, P.; Mechali, M. Programming DNA replication origins and chromosome organization. Chrom. Res. 2010, 18, 137–145. [Google Scholar] [CrossRef]
- Kelly, T.J.; Brown, G.W. Regulation of chromosome replication. Annu. Rev. Biochem. 2000, 69, 829–880. [Google Scholar]
- Lambert, S.; Froget, B.; Carr, A.M. Arrested replication fork processing: Interplay between checkpoints and recombination. DNA Repair 2007, 6, 1042–1061. [Google Scholar] [CrossRef]
- Mirkin, E.V.; Mirkin, S.M. Replication fork stalling at natural impediments. Microbiol. Mol. Biol. Rev. 2007, 71, 13–35. [Google Scholar] [CrossRef]
- Szilard, R.; Jacques, P.; Laramée, L.; Cheng, B.; Galicia, S.; Bataille, A.; Yeung, M.; Mendez, M.; Bergeron, M.; Robert, F.; et al. Systematic identification of fragile sites via genome-wide location analysis of gamma-h2ax. Nat. Struct. Mol. Biol. 2010, 17, 299–305. [Google Scholar] [CrossRef]
- Helleday, T. Homologous recombination in cancer development, treatment and development of drug resistance. Carcinogenesis 2010, 31, 955–960. [Google Scholar]
- Kawabata, T.; Luebben, S.W.; Yamaguchi, S.; Ilves, I.; Matise, I.; Buske, T.; Botchan, M.R.; Shima, N. Stalled fork rescue via dormant replication origins in unchallenged s phase promotes proper chromosome segregation and tumor suppression. Mol. Cell 2011, 41, 543–553. [Google Scholar] [CrossRef]
- Woodward, A.M.; Gohler, T.; Luciani, M.G.; Oehlmann, M.; Ge, X.; Gartner, A.; Jackson, D.A.; Blow, J.J. Excess mcm2–7 license dormant origins of replication that can be used under conditions of replicative stress. J. Cell Biol. 2006, 173, 673–683. [Google Scholar] [CrossRef]
- Ge, X.Q.; Jackson, D.A.; Blow, J.J. Dormant origins licensed by excess mcm2–7 are required for human cells to survive replicative stress. Genes Dev. 2007, 21, 3331–3341. [Google Scholar]
- Le Tallec, B.; Dutrillaux, B.; Lachages, A.M.; Millot, G.A.; Brison, O.; Debatisse, M. Molecular profiling of common fragile sites in human fibroblasts. Nat. Struc. Mol. Biol. 2011, 18, 1421–1423. [Google Scholar] [CrossRef]
- Letessier, A.; Millot, G.A.; Koundrioukoff, S.; Lachages, A.M.; Vogt, N.; Hansen, R.S.; Malfoy, B.; Brison, O.; Debatisse, M. Cell-type-specific replication initiation programs set fragility of the fra3b fragile site. Nature 2011, 470, 120–123. [Google Scholar]
- Daboussi, F.; Courbet, S.; Benhamou, S.; Kannouche, P.; Zdzienicka, M.Z.; Debatisse, M.; Lopez, B.S. A homologous recombination defect affects replication-fork progression in mammalian cells. J. Cell Sci. 2008, 121, 162–166. [Google Scholar]
- Branzei, D.; Foiani, M. Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 2010, 11, 208–219. [Google Scholar] [CrossRef]
- Katou, Y.; Kanoh, Y.; Bando, M.; Noguchi, H.; Tanaka, H.; Ashikari, T.; Sugimoto, K.; Shirahige, K. S-phase checkpoint proteins tof1 and mrc1 form a stable replication-pausing complex. Nature 2003, 424, 1078–1083. [Google Scholar] [CrossRef]
- De Piccoli, G.; Katou, Y.; Itoh, T.; Nakato, R.; Shirahige, K.; Labib, K. Replisome stability at defective DNA replication forks is independent of s phase checkpoint kinases. Mol. Cell 2012, 45, 696–704. [Google Scholar]
- Cotta-Ramusino, C.; Fachinetti, D.; Lucca, C.; Doksani, Y.; Lopes, M.; Sogo, J.; Foiani, M. Exo1 processes stalled replication forks and counteracts fork reversal in checkpoint-defective cells. Mol. Cell 2005, 17, 153–159. [Google Scholar] [CrossRef]
- Froget, B.; Blaisonneau, J.; Lambert, S.; Baldacci, G. Cleavage of stalled forks by fission yeast mus81/eme1 in absence of DNA replication checkpoint. Mol. Biol.Cell 2008, 19, 445–456. [Google Scholar]
- Kai, M.; Boddy, M.N.; Russell, P.; Wang, T.S. Replication checkpoint kinase cds1 regulates mus81 to preserve genome integrity during replication stress. Genes Dev. 2005, 19, 919–932. [Google Scholar] [CrossRef]
- Hu, J.; Sun, L.; Shen, F.; Chen, Y.; Hua, Y.; Liu, Y.; Zhang, M.; Hu, Y.; Wang, Q.; Xu, W.; et al. The intra-s phase checkpoint targets dna2 to prevent stalled replication forks from reversing. Cell 2012, 149, 1221–1232. [Google Scholar]
- Nimonkar, A.V.; Sica, R.A.; Kowalczykowski, S.C. Rad52 promotes second-end DNA capture in double-stranded break repair to form complement-stabilized joint molecules. Proc. Natl. Acad. Sci. USA 2009, 106, 3077–3082. [Google Scholar]
- McIlwraith, M.J.; West, S.C. DNA repair synthesis facilitates rad52-mediated second-end capture during dsb repair. Mol. Cell 2008, 29, 510–516. [Google Scholar] [CrossRef]
- Carreira, A.; Hilario, J.; Amitani, I.; Baskin, R.J.; Shivji, M.K.; Venkitaraman, A.R.; Kowalczykowski, S.C. The brc repeats of brca2 modulate the DNA-binding selectivity of rad51. Cell 2009, 136, 1032–1043. [Google Scholar] [CrossRef]
- Jensen, R.B.; Carreira, A.; Kowalczykowski, S.C. Purified human brca2 stimulates rad51-mediated recombination. Nature 2010, 467, 678–683. [Google Scholar]
- Wray, J.; Liu, J.; Nickoloff, J.A.; Shen, Z. Distinct rad51 associations with rad52 and bccip in response to DNA damage and replication stress. Cancer Res. 2008, 68, 2699–2707. [Google Scholar] [CrossRef]
- Lisby, M.; Barlow, J.H.; Burgess, R.C.; Rothstein, R. Choreography of the DNA damage response: Spatiotemporal relationships among checkpoint and repair proteins. Cell 2004, 118, 699–713. [Google Scholar] [CrossRef]
- Sung, P. Yeast rad55 and rad57 proteins form a heterodimer that functions with replication protein a to promote DNA strand exchange by rad51 recombinase. Genes Dev. 1997, 11, 1111–1121. [Google Scholar]
- Ward, J.D.; Barber, L.J.; Petalcorin, M.I.; Yanowitz, J.; Boulton, S.J. Replication blocking lesions present a unique substrate for homologous recombination. EMBO J. 2007, 26, 3384–3396. [Google Scholar] [CrossRef]
- Mankouri, H.W.; Ngo, H.P.; Hickson, I.D. Shu proteins promote the formation of homologous recombination intermediates that are processed by sgs1-rmi1-top3. Mol. Biol Cell 2007, 18, 4062–4073. [Google Scholar]
- Shor, E.; Weinstein, J.; Rothstein, R. A genetic screen for top3 suppressors in saccharomyces cerevisiae identifies shu1, shu2, psy3 and csm2: Four genes involved in error-free DNA repair. Genetics 2005, 169, 1275–1289. [Google Scholar]
- Ball, L.G.; Zhang, K.; Cobb, J.A.; Boone, C.; Xiao, W. The yeast shu complex couples error-free post-replication repair to homologous recombination. Mol. Microbiol. 2009, 73, 89–102. [Google Scholar] [CrossRef]
- Choi, K.; Szakal, B.; Chen, Y.H.; Branzei, D.; Zhao, X. The smc5/6 complex and esc2 influence multiple replication-associated recombination processes in saccharomyces cerevisiae. Mol. Biol Cell 2010, 21, 2306–2314. [Google Scholar] [CrossRef]
- Martin, V.; Chahwan, C.; Gao, H.; Blais, V.; Wohlschlegel, J.; Yates, J.R., III.; McGowan, C.H.; Russell, P. Sws1 is a conserved regulator of homologous recombination in eukaryotic cells. EMBO J. 2006, 25, 2564–2574. [Google Scholar] [CrossRef]
- Liu, J.; Renault, L.; Veaute, X.; Fabre, F.; Stahlberg, H.; Heyer, W.D. Rad51 paralogues rad55-rad57 balance the antirecombinase srs2 in rad51 filament formation. Nature 2011, 479, 245–248. [Google Scholar]
- Papouli, E.; Chen, S.; Davies, A.A.; Huttner, D.; Krejci, L.; Sung, P.; Ulrich, H.D. Crosstalk between sumo and ubiquitin on pcna is mediated by recruitment of the helicase srs2p. Mol. Cell 2005, 19, 123–133. [Google Scholar] [CrossRef]
- Pfander, B.; Moldovan, G.L.; Sacher, M.; Hoege, C.; Jentsch, S. Sumo-modified pcna recruits srs2 to prevent recombination during s phase. Nature 2005, 436, 428–433. [Google Scholar]
- Bashkirov, V.I.; King, J.S.; Bashkirova, E.V.; Schmuckli-Maurer, J.; Heyer, W.D. DNA repair protein rad55 is a terminal substrate of the DNA damage checkpoints. Mol. Cell. Biol. 2000, 20, 4393–4404. [Google Scholar] [CrossRef]
- Herzberg, K.; Bashkirov, V.I.; Rolfsmeier, M.; Haghnazari, E.; McDonald, W.H.; Anderson, S.; Bashkirova, E.V.; Yates, J.R., III.; Heyer, W.D. Phosphorylation of rad55 on serines 2, 8, and 14 is required for efficient homologous recombination in the recovery of stalled replication forks. Mol. Cell. Biol. 2006, 26, 8396–8409. [Google Scholar]
- Haaf, T.; Golub, E.I.; Reddy, G.; Radding, C.M.; Ward, D.C. Nuclear foci of mammalian rad51 recombination protein in somatic cells after DNA damage and its localization in synaptonemal complexes. Proc. Natl. Acad. Sci. USA 1995, 92, 2298–2302. [Google Scholar]
- Tashiro, S.; Kotomura, N.; Shinohara, A.; Tanaka, K.; Ueda, K.; Kamada, N. S phase specific formation of the human rad51 protein nuclear foci in lymphocytes. Oncogene 1996, 12, 2165–2170. [Google Scholar]
- Golub, E.I.; Gupta, R.C.; Haaf, T.; Wold, M.S.; Radding, C.M. Interaction of human rad51 recombination protein with single-stranded DNA binding protein, rpa. Nucleic Acids Res. 1998, 26, 5388–5393. [Google Scholar] [CrossRef]
- Raderschall, E.; Golub, E.I.; Haaf, T. Nuclear foci of mammalian recombination proteins are located at single-stranded DNA regions formed after DNA damage. Proc. Natl. Acad. Sci. USA 1999, 96, 1921–1926. [Google Scholar] [CrossRef]
- Mizuta, R.; LaSalle, J.M.; Cheng, H.L.; Shinohara, A.; Ogawa, H.; Copeland, N.; Jenkins, N.A.; Lalande, M.; Alt, F.W. Rab22 and rab163/mouse brca2: Proteins that specifically interact with the rad51 protein. Proc. Natl. Acad. Sci. USA 1997, 94, 6927–6932. [Google Scholar]
- Tashiro, S.; Walter, J.; Shinohara, A.; Kamada, N.; Cremer, T. Rad51 accumulation at sites of DNA damage and in postreplicative chromatin. J. Cell Biol. 2000, 150, 283–291. [Google Scholar] [CrossRef]
- Chen, J.; Silver, D.P.; Walpita, D.; Cantor, S.B.; Gazdar, A.F.; Tomlinson, G.; Couch, F.J.; Weber, B.L.; Ashley, T.; Livingston, D.M.; et al. Stable interaction between the products of the brca1 and brca2 tumor suppressor genes in mitotic and meiotic cells. Mol. Cell 1998, 2, 317–328. [Google Scholar] [CrossRef]
- Scully, R.; Chen, J.; Plug, A.; Xiao, Y.; Weaver, D.; Feunteun, J.; Ashley, T.; Livingston, D.M. Association of brca1 with rad51 in mitotic and meiotic cells. Cell 1997, 88, 265–275. [Google Scholar]
- Scully, R.; Chen, J.; Ochs, R.L.; Keegan, K.; Hoekstra, M.; Feunteun, J.; Livingston, D.M. Dynamic changes of brca1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell 1997, 90, 425–435. [Google Scholar] [CrossRef]
- Meister, P.; Poidevin, M.; Francesconi, S.; Tratner, I.; Zarzov, P.; Baldacci, G. Nuclear factories for signalling and repairing DNA double strand breaks in living fission yeast. Nucleic Acids Res. 2003, 31, 5064–5073. [Google Scholar] [CrossRef]
- Lisby, M.; Rothstein, R.; Mortensen, U.H. Rad52 forms DNA repair and recombination centers during s phase. Proc. Natl. Acad. Sci. USA 2001, 98, 8276–8282. [Google Scholar]
- Lambert, S.; Mizuno, K.; Blaisonneau, J.; Martineau, S.; Chanet, R.; Fréon, K.; Murray, J.M.; Carr, A.M.; Baldacci, G. Homologous recombination restarts blocked replication forks at the expense of genome rearrangements by template exchange. Mol. Cell 2010, 39, 346–359. [Google Scholar] [CrossRef]
- Lambert, S.; Watson, A.; Sheedy, D.M.; Martin, B.; Carr, A.M. Gross chromosomal rearrangements and elevated recombination at an inducible site-specific replication fork barrier. Cell 2005, 121, 689–702. [Google Scholar] [CrossRef]
- Mizuno, K.; Lambert, S.; Baldacci, G.; Murray, J.M.; Carr, A.M. Nearby inverted repeats fuse to generate acentric and dicentric palindromic chromosomes by a replication template exchange mechanism. Genes Dev. 2009, 23, 2876–2886. [Google Scholar] [CrossRef]
- Zou, H.; Rothstein, R. Holliday junctions accumulate in replication mutants via a reca homolog-independent mechanism. Cell 1997, 90, 87–96. [Google Scholar]
- Tsang, E.; Carr, A. Replication fork arrest, recombination and the maintenance of ribosomal DNA stability. DNA Repair 2008, 7, 1613–1623. [Google Scholar] [CrossRef]
- Karanam, K.; Kafri, R.; Loewer, A.; Lahav, G. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of hr in mid s phase. Mol. Cell 2012, 47, 320–329. [Google Scholar]
- Wong, J.M.; Ionescu, D.; Ingles, C.J. Interaction between brca2 and replication protein a is compromised by a cancer-predisposing mutation in brca2. Oncogene 2003, 22, 28–33. [Google Scholar] [CrossRef]
- Shukla, A.; Navadgi, V.M.; Mallikarjuna, K.; Rao, B.J. Interaction of hrad51 and hrad52 with mcm complex: A cross-talk between recombination and replication proteins. Biochem. Biophys. Res. Commun. 2005, 329, 1240–1245. [Google Scholar] [CrossRef]
- Bailis, J.M.; Luche, D.D.; Hunter, T.; Forsburg, S.L. Minichromosome maintenance proteins interact with checkpoint and recombination proteins to promote s-phase genome stability. Mol. Cell. Biol. 2008, 28, 1724–1738. [Google Scholar] [CrossRef]
- Oyola, S.O.; Bringaud, F.; Melville, S.E. A kinetoplastid brca2 interacts with DNA replication protein cdc45. Int. J. Parasitol. 2009, 39, 59–69. [Google Scholar]
- Alabert, C.; Bianco, J.; Pasero, P. Differential regulation of homologous recombination at DNA breaks and replication forks by the mrc1 branch of the s-phase checkpoint. EMBO J. 2009, 28, 1131–1141. [Google Scholar] [CrossRef]
- Lopes, M.; Foiani, M.; Sogo, J.M. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable uv lesions. Mol. Cell 2006, 21, 15–27. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Ray Chaudhuri, A.; Lopes, M.; Costanzo, V. Rad51 protects nascent DNA from mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struc. Mol. Biol. 2010, 17, 1305–1311. [Google Scholar] [CrossRef] [Green Version]
- Liberi, G.; Maffioletti, G.; Lucca, C.; Chiolo, I.; Baryshnikova, A.; Cotta-Ramusino, C.; Lopes, M.; Pellicioli, A.; Haber, J.E.; Foiani, M. Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of blm recq helicase. Genes Dev. 2005, 19, 339–350. [Google Scholar]
- Branzei, D.; Vanoli, F.; Foiani, M. Sumoylation regulates rad18-mediated template switch. Nature 2008, 456, 915–920. [Google Scholar] [CrossRef]
- Branzei, D.; Sollier, J.; Liberi, G.; Zhao, X.; Maeda, D.; Seki, M.; Enomoto, T.; Ohta, K.; Foiani, M. Ubc9- and mms21-mediated sumoylation counteracts recombinogenic events at damaged replication forks. Cell 2006, 127, 509–522. [Google Scholar] [CrossRef]
- Mankouri, H.W.; Ashton, T.M.; Hickson, I.D. Holliday junction-containing DNA structures persist in cells lacking sgs1 or top3 following exposure to DNA damage. Proc. Nat. Acad. Sci. USA 2011, 108, 4944–4949. [Google Scholar] [CrossRef]
- Branzei, D. Ubiquitin family modifications and template switching. FEBS Lett. 2011, 585, 2810–2817. [Google Scholar]
- Vanoli, F.; Fumasoni, M.; Szakal, B.; Maloisel, L.; Branzei, D. Replication and recombination factors contributing to recombination-dependent bypass of DNA lesions by template switch. PLoS Genet. 2010, 6. [Google Scholar] [CrossRef]
- Moriel-Carretero, M.; Aguilera, A. A postincision-deficient tfiih causes replication fork breakage and uncovers alternative rad51- or pol32-mediated restart mechanisms. Mol. Cell 2010, 37, 690–701. [Google Scholar] [CrossRef]
- Roseaulin, L.; Yamada, Y.; Tsutsui, Y.; Russell, P.; Iwasaki, H.; Arcangioli, B. Mus81 is essential for sister chromatid recombination at broken replication forks. EMBO J. 2008, 27, 1378–1387. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Puddu, F.; Costanzo, V. Rad51- and mre11-dependent reassembly of uncoupled cmg helicase complex at collapsed replication forks. Nat. Struc. Mol. Biol. 2011, 19, 17–24. [Google Scholar] [CrossRef]
- Clemente-Ruiz, M.; Prado, F. Chromatin assembly controls replication fork stability. EMBO Rep. 2009, 10, 790–796. [Google Scholar]
- Long, D.T.; Raschle, M.; Joukov, V.; Walter, J.C. Mechanism of rad51-dependent DNA interstrand cross-link repair. Science 2011, 333, 84–87. [Google Scholar]
- Bjergbaek, L.; Cobb, J.A.; Tsai-Pflugfelder, M.; Gasser, S.M. Mechanistically distinct roles for sgs1p in checkpoint activation and replication fork maintenance. EMBO J. 2005, 24, 405–417. [Google Scholar] [CrossRef]
- Nakahara, M.; Sonoda, E.; Nojima, K.; Sale, J.E.; Takenaka, K.; Kikuchi, K.; Taniguchi, Y.; Nakamura, K.; Sumitomo, Y.; Bree, R.T.; et al. Genetic evidence for single-strand lesions initiating nbs1-dependent homologous recombination in diversification of ig v in chicken b lymphocytes. PLoS Genet. 2009, 5. [Google Scholar] [CrossRef]
- Paek, A.L.; Kaochar, S.; Jones, H.; Elezaby, A.; Shanks, L.; Weinert, T. Fusion of nearby inverted repeats by a replication-based mechanism leads to formation of dicentric and acentric chromosomes that cause genome instability in budding yeast. Genes Dev. 2009, 23, 2861–2875. [Google Scholar]
- Meister, P.; Taddei, A.; Vernis, L.; Poidevin, M.; Gasser, S.M.; Baldacci, G. Temporal separation of replication and recombination requires the intra-s checkpoint. J. Cell Biol. 2005, 168, 537–544. [Google Scholar] [CrossRef]
- Sorensen, C.S.; Hansen, L.T.; Dziegielewski, J.; Syljuasen, R.G.; Lundin, C.; Bartek, J.; Helleday, T. The cell-cycle checkpoint kinase chk1 is required for mammalian homologous recombination repair. Nat. Cell Biol. 2005, 7, 195–201. [Google Scholar] [CrossRef]
- Petermann, E.; Orta, M.L.; Issaeva, N.; Schultz, N.; Helleday, T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different rad51-mediated pathways for restart and repair. Mol. Cell 2010, 37, 492–502. [Google Scholar] [CrossRef]
- Saintigny, Y.; Delacote, F.; Vares, G.; Petitot, F.; Lambert, S.; Averbeck, D.; Lopez, B.S. Characterization of homologous recombination induced by replication inhibition in mammalian cells. EMBO J. 2001, 20, 3861–3870. [Google Scholar]
- Raveendranathan, M.; Chattopadhyay, S.; Bolon, Y.T.; Haworth, J.; Clarke, D.J.; Bielinsky, A.K. Genome-wide replication profiles of s-phase checkpoint mutants reveal fragile sites in yeast. EMBO J. 2006, 25, 3627–3639. [Google Scholar] [CrossRef]
- Hanada, K.; Budzowska, M.; Davies, S.L.; van Drunen, E.; Onizawa, H.; Beverloo, H.B.; Maas, A.; Essers, J.; Hickson, I.D.; Kanaar, R. The structure-specific endonuclease mus81 contributes to replication restart by generating double-strand DNA breaks. Nat. Struc. Mol. Biol. 2007, 14, 1096–1104. [Google Scholar] [CrossRef]
- Bosco, G.; Haber, J.E. Chromosome break-induced DNA replication leads to nonreciprocal translocations and telomere capture. Genetics 1998, 150, 1037–1047. [Google Scholar]
- Llorente, B.; Smith, C.E.; Symington, L.S. Break-induced replication: What is it and what is it for? Cell Cycle 2008, 7, 859–864. [Google Scholar]
- Kraus, E.; Leung, W.Y.; Haber, J.E. Break-induced replication: A review and an example in budding yeast. Proc. Natl. Acad. Sci. USA 2001, 98, 8255–8262. [Google Scholar] [CrossRef]
- Lydeard, J.R.; Jain, S.; Yamaguchi, M.; Haber, J.E. Break-induced replication and telomerase-independent telomere maintenance require pol32. Nature 2007, 448, 820–823. [Google Scholar] [CrossRef]
- Lydeard, J.R.; Lipkin-Moore, Z.; Sheu, Y.J.; Stillman, B.; Burgers, P.M.; Haber, J.E. Break-induced replication requires all essential DNA replication factors except those specific for pre-rc assembly. Genes Dev. 2010, 24, 1133–1144. [Google Scholar]
- Deem, A.; Keszthelyi, A.; Blackgrove, T.; Vayl, A.; Coffey, B.; Mathur, R.; Chabes, A.; Malkova, A. Break-induced replication is highly inaccurate. PLoS Biol. 2011, 9. [Google Scholar] [CrossRef]
- Iraqui, I.; Chekkal, Y.; Jmari, N.; Pietrobon, V.; Freon, K.; Costes, A.; Lambert, S.A. Recovery of arrested replication forks by homologous recombination is error-prone. PLoS Genet. 2012, 8. [Google Scholar] [CrossRef]
- Mizuno, K.; Miyabe, I.; Schalbetter, S.A.; Carr, A.M.; Murray, J.M. Recombination-restarted replication makes inverted chromosome fusions at inverted repeats. Nature 2012. [Google Scholar] [CrossRef]
- Stracker, T.H.; Petrini, J.H. The mre11 complex: Starting from the ends. Nat. Rev. Mol. Cell Biol. 2011, 12, 90–103. [Google Scholar] [CrossRef]
- Lomonosov, M.; Anand, S.; Sangrithi, M.; Davies, R.; Venkitaraman, A.R. Stabilization of stalled DNA replication forks by the brca2 breast cancer susceptibility protein. Genes Dev. 2003, 17, 3017–3022. [Google Scholar] [CrossRef]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for brca2 in blocking stalled replication fork degradation by mre11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef]
- Schlacher, K.; Wu, H.; Jasin, M. A distinct replication fork protection pathway connects fanconi anemia tumor suppressors to rad51-brca1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar]
- Tinline-Purvis, H.; Savory, A.P.; Cullen, J.K.; Dave, A.; Moss, J.; Bridge, W.L.; Marguerat, S.; Bahler, J.; Ragoussis, J.; Mott, R.; et al. Failed gene conversion leads to extensive end processing and chromosomal rearrangements in fission yeast. EMBO J. 2009, 28, 3400–3412. [Google Scholar] [CrossRef]
- Sonoda, E.; Sasaki, M.S.; Buerstedde, J.M.; Bezzubova, O.; Shinohara, A.; Ogawa, H.; Takata, M.; Yamaguchi-Iwai, Y.; Takeda, S. Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J. 1998, 17, 598–608. [Google Scholar] [CrossRef]
- Venkitaraman, A.R. Chromosome stability, DNA recombination and the brca2 tumour suppressor. Curr. Opin. Cell Biol. 2001, 13, 338–343. [Google Scholar] [CrossRef]
- Fachinetti, D.; Bermejo, R.; Cocito, A.; Minardi, S.; Katou, Y.; Kanoh, Y.; Shirahige, K.; Azvolinsky, A.; Zakian, V.A.; Foiani, M. Replication termination at eukaryotic chromosomes is mediated by top2 and occurs at genomic loci containing pausing elements. Mol. Cell 2010, 39, 595–605. [Google Scholar]
- Nitiss, J. DNA topoisomerase ii and its growing repertoire of biological functions. Natl. Rev. Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef]
- Steinacher, R.; Osman, F.; Dalgaard, J.Z.; Lorenz, A.; Whitby, M.C. The DNA helicase pfh1 promotes fork merging at replication termination sites to ensure genome stability. Genes Dev. 2012, 26, 594–602. [Google Scholar] [CrossRef] [Green Version]
- Kojic, M.; Holloman, W.K. Brh2 domain function distinguished by differential cellular responses to DNA damage and replication stress. Mol. Microbiol. 2012, 83, 351–361. [Google Scholar]
- Heyer, W.D.; Li, X.; Rolfsmeier, M.; Zhang, X.P. Rad54: The swiss army knife of homologous recombination? Nucleic Acids Res. 2006, 34, 4115–4125. [Google Scholar] [CrossRef]
- Lambert, S.; Lopez, B.S. Characterization of mammalian rad51 double strand break repair using non-lethal dominant-negative forms. EMBO J. 2000, 19, 3090–3099. [Google Scholar] [CrossRef]
- Liu, P.; Carvalho, C.M.; Hastings, P.; Lupski, J.R. Mechanisms for recurrent and complex human genomic rearrangements. Curr. Opin. Genet. Dev. 2012, 22, 211–220. [Google Scholar] [CrossRef]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef]
- Chabosseau, P.; Buhagiar-Labarchede, G.; Onclercq-Delic, R.; Lambert, S.; Debatisse, M.; Brison, O.; Amor-Gueret, M. Pyrimidine pool imbalance induced by blm helicase deficiency contributes to genetic instability in bloom syndrome. Natl. Commun. 2011, 2, 368. [Google Scholar]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef]
- Evers, B.; Helleday, T.; Jonkers, J. Targeting homologous recombination repair defects in cancer. Trends Pharmacol. Sci. 2010, 31, 372–380. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Costes, A.; Lambert, S.A.E. Homologous Recombination as a Replication Fork Escort: Fork-Protection and Recovery. Biomolecules 2013, 3, 39-71. https://doi.org/10.3390/biom3010039
Costes A, Lambert SAE. Homologous Recombination as a Replication Fork Escort: Fork-Protection and Recovery. Biomolecules. 2013; 3(1):39-71. https://doi.org/10.3390/biom3010039
Chicago/Turabian StyleCostes, Audrey, and Sarah A. E. Lambert. 2013. "Homologous Recombination as a Replication Fork Escort: Fork-Protection and Recovery" Biomolecules 3, no. 1: 39-71. https://doi.org/10.3390/biom3010039