Potential Impact of Oral Inflammations on Cardiac Functions and Atrial Fibrillation

Abstract
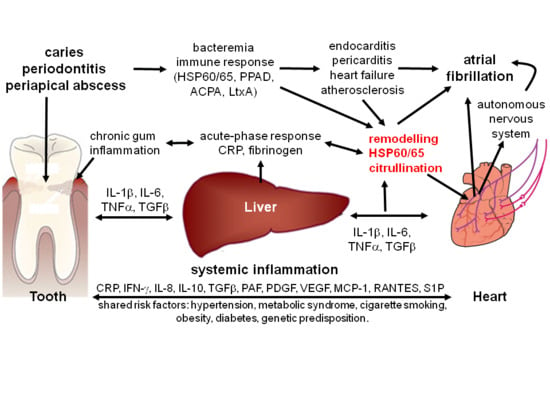
:
1. Introduction
2. Frequent Forms of Chronic Oral Infections Potentially Associated with Atrial Fibrillation
3. Potential Role of Bacteremia
4. Potential Role of Systemic Inflammation
5. Potential Role of Autoimmunity in Atrial Fibrillation
6. Potential Role of the Autonomic Nervous System
7. Potential Role of Bacterial Toxins
8. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chugh, S.S.; Havmoeller, R.; Narayanan, K.; Singh, D.; Rienstra, M.; Benjamin, E.J.; Gillum, R.F.; Kim, Y.H.; McAnulty, J.H.; Zheng, Z.J.; et al. Worldwide epidemiology of atrial fibrillation: A global burden of disease 2010 study. Circulation 2014, 129, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Camm, A.J.; Kirchhof, P.; Lip, G.Y.; Schotten, U.; Savelieva, I.; Ernst, S.; Van Gelder, I.C.; Al-Attar, N.; Hindricks, G.; Prendergast, B.; et al. Guidelines for the management of atrial fibrillation: The task force for the management of atrial fibrillation of the european society of cardiology (ESC). Eur. Heart J. 2010, 31, 2369–2429. [Google Scholar] [PubMed]
- Lloyd-Jones, D.M.; Wang, T.J.; Leip, E.P.; Larson, M.G.; Levy, D.; Vasan, R.S.; D’Agostino, R.B.; Massaro, J.M.; Beiser, A.; Wolf, P.A.; et al. Lifetime risk for development of atrial fibrillation: The framingham heart study. Circulation 2004, 110, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Heeringa, J.; van der Kuip, D.A.; Hofman, A.; Kors, J.A.; van Herpen, G.; Stricker, B.H.; Stijnen, T.; Lip, G.Y.; Witteman, J.C. Prevalence, incidence and lifetime risk of atrial fibrillation: The rotterdam study. Eur. Heart J. 2006, 27, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.J.; Massaro, J.M.; Levy, D.; Vasan, R.S.; Wolf, P.A.; D’Agostino, R.B.; Larson, M.G.; Kannel, W.B.; Benjamin, E.J. A risk score for predicting stroke or death in individuals with new-onset atrial fibrillation in the community: The framingham heart study. JAMA 2003, 290, 1049–1056. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.J.; Larson, M.G.; Levy, D.; Vasan, R.S.; Leip, E.P.; Wolf, P.A.; D’Agostino, R.B.; Murabito, J.M.; Kannel, W.B.; Benjamin, E.J. Temporal relations of atrial fibrillation and congestive heart failure and their joint influence on mortality: The framingham heart study. Circulation 2003, 107, 2920–2925. [Google Scholar] [CrossRef] [PubMed]
- Gage, B.F.; Waterman, A.D.; Shannon, W.; Boechler, M.; Rich, M.W.; Radford, M.J. Validation of clinical classification schemes for predicting stroke: Results from the national registry of atrial fibrillation. JAMA 2001, 285, 2864–2870. [Google Scholar] [CrossRef] [PubMed]
- Christophersen, I.E.; Ravn, L.S.; Budtz-Joergensen, E.; Skytthe, A.; Haunsoe, S.; Svendsen, J.H.; Christensen, K. Familial aggregation of atrial fibrillation: A study in danish twins. Circ. Arrhythm. Electrophysiol. 2009, 2, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.B.; Fritsche, L.G.; Zhou, W.; Teslovich, T.M.; Holmen, O.L.; Gustafsson, S.; Gabrielsen, M.E.; Schmidt, E.M.; Beaumont, R.; Wolford, B.N.; et al. Genome-wide study of atrial fibrillation identifies seven risk loci and highlights biological pathways and regulatory elements involved in cardiac development. Am. J. Hum. Genet. 2018, 102, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Aviles, R.J.; Martin, D.O.; Apperson-Hansen, C.; Houghtaling, P.L.; Rautaharju, P.; Kronmal, R.A.; Tracy, R.P.; Van Wagoner, D.R.; Psaty, B.M.; Lauer, M.S.; et al. Inflammation as a risk factor for atrial fibrillation. Circulation 2003, 108, 3006–3010. [Google Scholar] [CrossRef] [PubMed]
- Spodick, D.H. Arrhythmias during acute pericarditis. A prospective study of 100 consecutive cases. JAMA 1976, 235, 39–41. [Google Scholar] [CrossRef] [PubMed]
- Gaudino, M.; Andreotti, F.; Zamparelli, R.; Di Castelnuovo, A.; Nasso, G.; Burzotta, F.; Iacoviello, L.; Donati, M.B.; Schiavello, R.; Maseri, A.; et al. The -174G/C interleukin-6 polymorphism influences postoperative interleukin-6 levels and postoperative atrial fibrillation. Is atrial fibrillation an inflammatory complication? Circulation 2003, 108 (Suppl. 1), II195–II199. [Google Scholar] [CrossRef] [PubMed]
- Bruins, P.; te Velthuis, H.; Yazdanbakhsh, A.P.; Jansen, P.G.; van Hardevelt, F.W.; de Beaumont, E.M.; Wildevuur, C.R.; Eijsman, L.; Trouwborst, A.; Hack, C.E. Activation of the complement system during and after cardiopulmonary bypass surgery: Postsurgery activation involves c-reactive protein and is associated with postoperative arrhythmia. Circulation 1997, 96, 3542–3548. [Google Scholar] [CrossRef] [PubMed]
- Falk, R.H. Atrial fibrillation. N. Engl. J. Med. 2001, 344, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, P.B.; Brennan, M.T.; Sasser, H.C.; Fox, P.C.; Paster, B.J.; Bahrani-Mougeot, F.K. Bacteremia associated with toothbrushing and dental extraction. Circulation 2008, 117, 3118–3125. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, P.B.; Brennan, M.T.; Thornhill, M.; Michalowicz, B.S.; Noll, J.; Bahrani-Mougeot, F.K.; Sasser, H.C. Poor oral hygiene as a risk factor for infective endocarditis-related bacteremia. J. Am. Dent. Assoc. 2009, 140, 1238–1244. [Google Scholar] [CrossRef] [PubMed]
- Louhelainen, A.M.; Aho, J.; Tuomisto, S.; Aittoniemi, J.; Vuento, R.; Karhunen, P.J.; Pessi, T. Oral bacterial DNA findings in pericardial fluid. J. Oral Microbiol. 2014, 6, 25835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armitage, G.C. Periodontal diagnoses and classification of periodontal diseases. Periodontol. 2000 2004, 34, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Loe, H.; Theilade, E.; Jensen, S.B. Experimental gingivitis in man. J. Periodontol. 1965, 36, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.J. Gingivitis: A prelude to periodontitis? J. Clin. Dent. 1995, 6, 41–45. [Google Scholar] [PubMed]
- König, J.; Holtfreter, B.; Kocher, T. Periodontal health in europe: Future trends based on treatment needs and the provision of periodontal services—Position paper 1. Eur. J. Dent. Educ. 2010, 14 (Suppl. 1), 4–24. [Google Scholar] [CrossRef] [PubMed]
- Burt, B. Position paper: Epidemiology of periodontal diseases. J. Periodontol. 2005, 76, 1406–1419. [Google Scholar] [PubMed]
- Eke, P.I.; Dye, B.A.; Wei, L.; Thornton-Evans, G.O.; Genco, R.J. Prevalence of periodontitis in adults in the united states: 2009 and 2010. J. Dent. Res. 2012, 91, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Rôças, I.N.; Siqueira, J.F. Root canal microbiota of teeth with chronic apical periodontitis. J. Clin. Microbiol. 2008, 46, 3599–3606. [Google Scholar] [CrossRef] [PubMed]
- Odesjö, B.; Helldén, L.; Salonen, L.; Langeland, K. Prevalence of previous endodontic treatment, technical standard and occurrence of periapical lesions in a randomly selected adult, general population. Endod. Dent. Traumatol. 1990, 6, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Eriksen, H.M.; Bjertness, E. Prevalence of apical periodontitis and results of endodontic treatment in middle-aged adults in Norway. Endod. Dent. Traumatol. 1991, 7, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Dugas, N.N.; Lawrence, H.P.; Teplitsky, P.E.; Pharoah, M.J.; Friedman, S. Periapical health and treatment quality assessment of root-filled teeth in two canadian populations. Int. Endod. J. 2003, 36, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Abiko, Y.; Saitoh, M.; Nishimura, M.; Yamazaki, M.; Sawamura, D.; Kaku, T. Role of β-defensins in oral epithelial health and disease. Med. Mol. Morphol. 2007, 40, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.O.; Dommisch, H.; Yin, L.; Dale, B.A. Expression of defensins in gingiva and their role in periodontal health and disease. Curr. Pharm. Des. 2007, 13, 3073–3083. [Google Scholar] [CrossRef] [PubMed]
- Fukui, A.; Ohta, K.; Nishi, H.; Shigeishi, H.; Tobiume, K.; Takechi, M.; Kamata, N. Interleukin-8 and CXCL10 expression in oral keratinocytes and fibroblasts via Toll-like receptors. Microbiol. Immunol. 2013, 57, 198–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinane, D.F.; Lappin, D.F. Immune processes in periodontal disease: A review. Ann. Periodontol. 2002, 7, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Kebschull, M.; Demmer, R.T.; Papapanou, P.N. “Gum bug, leave my heart alone!”—Epidemiologic and mechanistic evidence linking periodontal infections and atherosclerosis. J. Dent. Res. 2010, 89, 879–902. [Google Scholar] [CrossRef] [PubMed]
- Reyes, L.; Herrera, D.; Kozarov, E.; Roldá, S.; Progulske-Fox, A. Periodontal bacterial invasion and infection: Contribution to atherosclerotic pathology. J. Periodontol. 2013, 84, S30–S50. [Google Scholar] [CrossRef] [PubMed]
- Forner, L.; Larsen, T.; Kilian, M.; Holmstrup, P. Incidence of bacteremia after chewing, tooth brushing and scaling in individuals with periodontal inflammation. J. Clin. Periodontol. 2006, 33, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Tomás, I.; Diz, P.; Tobías, A.; Scully, C.; Donos, N. Periodontal health status and bacteraemia from daily oral activities: Systematic review/meta-analysis. J. Clin. Periodontol. 2012, 39, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Kozarov, E.; Sweier, D.; Shelburne, C.; Progulske-Fox, A.; Lopatin, D. Detection of bacterial DNA in atheromatous plaques by quantitative PCR. Microbes Infect. 2006, 8, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Cavrini, F.; Sambri, V.; Moter, A.; Servidio, D.; Marangoni, A.; Montebugnoli, L.; Foschi, F.; Prati, C.; Di Bartolomeo, R.; Cevenini, R. Molecular detection of Treponema denticola and Porphyromonas gingivalis in carotid and aortic atheromatous plaques by fish: Report of two cases. J. Med. Microbiol. 2005, 54, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Okuda, K.; Ishihara, K.; Nakagawa, T.; Hirayama, A.; Inayama, Y. Detection of Treponema denticola in atherosclerotic lesions. J. Clin. Microbiol. 2001, 39, 1114–1117. [Google Scholar] [CrossRef] [PubMed]
- Marcelino, S.L.; Gaetti-Jardim, E.; Nakano, V.; Canônico, L.A.; Nunes, F.D.; Lotufo, R.F.; Pustiglioni, F.E.; Romito, G.A.; Avila-Campos, M.J. Presence of periodontopathic bacteria in coronary arteries from patients with chronic periodontitis. Anaerobe 2010, 16, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Pessi, T.; Karhunen, V.; Karjalainen, P.P.; Ylitalo, A.; Airaksinen, J.K.; Niemi, M.; Pietila, M.; Lounatmaa, K.; Haapaniemi, T.; Lehtimäki, T.; et al. Bacterial signatures in thrombus aspirates of patients with myocardial infarction. Circulation 2013, 127, 1219–1228. [Google Scholar] [CrossRef] [PubMed]
- Chhibber-Goel, J.; Singhal, V.; Bhowmik, D.; Vivek, R.; Parakh, N.; Bhargava, B.; Sharma, A. Linkages between oral commensal bacteria and atherosclerotic plaques in coronary artery disease patients. NPJ Biofilms Microbiomes 2016, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Inaba, H.; Nomura, R.; Nemoto, H.; Takeda, M.; Yoshioka, H.; Matsue, H.; Takahashi, T.; Taniguchi, K.; Amano, A.; et al. Detection of cariogenic Streptococcus mutans in extirpated heart valve and atheromatous plaque specimens. J. Clin. Microbiol. 2006, 44, 3313–3317. [Google Scholar] [CrossRef] [PubMed]
- Gabay, C.; Kushner, I. Acute-phase proteins and other systemic responses to inflammation. N. Engl. J. Med. 1999, 340, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Dye, B.A.; Choudhary, K.; Shea, S.; Papapanou, P.N. Serum antibodies to periodontal pathogens and markers of systemic inflammation. J. Clin. Periodontol. 2005, 32, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Gomes-Filho, I.S.; Freitas Coelho, J.M.; da Cruz, S.S.; Passos, J.S.; Teixeira de Freitas, C.O.; Aragão Farias, N.S.; Amorim da Silva, R.; Silva Pereira, M.N.; Lima, T.L.; Barreto, M.L. Chronic periodontitis and C-reactive protein levels. J. Periodontol. 2011, 82, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Slade, G.D.; Offenbacher, S.; Beck, J.D.; Heiss, G.; Pankow, J.S. Acute-phase inflammatory response to periodontal disease in the US population. J. Dent. Res. 2000, 79, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Noack, B.; Genco, R.J.; Trevisan, M.; Grossi, S.; Zambon, J.J.; De Nardin, E. Periodontal infections contribute to elevated systemic C-reactive protein level. J. Periodontol. 2001, 72, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Slade, G.D.; Ghezzi, E.M.; Heiss, G.; Beck, J.D.; Riche, E.; Offenbacher, S. Relationship between periodontal disease and C-reactive protein among adults in the atherosclerosis risk in communities study. Arch. Intern. Med. 2003, 163, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Boucher, N.E.; Hanrahan, J.J.; Kihara, F.Y. Occurrence of C-reactive protein in oral disease. J. Dent. Res. 1967, 46, 624. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.S.; Blattner, T.C.; Sant’Ana Filho, M.; Grecca, F.S.; Hugo, F.N.; Fouad, A.F.; Reynolds, M.A. Can apical periodontitis modify systemic levels of inflammatory markers? A systematic review and meta-analysis. J. Endod. 2013, 39, 1205–1217. [Google Scholar] [CrossRef] [PubMed]
- Graunaite, I.; Lodiene, G.; Maciulskiene, V. Pathogenesis of apical periodontitis: A literature review. J. Oral Maxillofac. Res. 2012, 2, e1. [Google Scholar] [CrossRef] [PubMed]
- Wollert, K.C.; Drexler, H. The role of interleukin-6 in the failing heart. Heart Fail. Rev. 2001, 6, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Amdur, R.L.; Mukherjee, M.; Go, A.; Barrows, I.R.; Ramezani, A.; Shoji, J.; Reilly, M.P.; Gnanaraj, J.; Deo, R.; Roas, S.; et al. Interleukin-6 is a risk factor for atrial fibrillation in chronic kidney disease: Findings from the cric study. PLoS ONE 2016, 11, e0148189. [Google Scholar] [CrossRef] [PubMed]
- Marcus, G.M.; Whooley, M.A.; Glidden, D.V.; Pawlikowska, L.; Zaroff, J.G.; Olgin, J.E. Interleukin-6 and atrial fibrillation in patients with coronary artery disease: Data from the heart and soul study. Am. Heart J. 2008, 155, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Psychari, S.N.; Apostolou, T.S.; Sinos, L.; Hamodraka, E.; Liakos, G.; Kremastinos, D.T. Relation of elevated C-reactive protein and interleukin-6 levels to left atrial size and duration of episodes in patients with atrial fibrillation. Am. J. Cardiol. 2005, 95, 764–767. [Google Scholar] [CrossRef] [PubMed]
- Bittar, M.N.; Carey, J.A.; Barnard, J.; Fildes, J.E.; Pravica, V.; Yonan, N.; Hutchinson, I.V. Interleukin 6 G-174C polymorphism influences outcome following coronary revascularization surgery. Heart Surg. Forum 2005, 8, E140–E145, discussion E145. [Google Scholar] [CrossRef] [PubMed]
- Galea, R.; Cardillo, M.T.; Caroli, A.; Marini, M.G.; Sonnino, C.; Narducci, M.L.; Biasucci, L.M. Inflammation and C-reactive protein in atrial fibrillation: Cause or effect? Tex. Heart Inst. J. 2014, 41, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.K.; Martin, D.O.; Sprecher, D.; Wazni, O.; Kanderian, A.; Carnes, C.A.; Bauer, J.A.; Tchou, P.J.; Niebauer, M.J.; Natale, A.; et al. C-reactive protein elevation in patients with atrial arrhythmias: Inflammatory mechanisms and persistence of atrial fibrillation. Circulation 2001, 104, 2886–2891. [Google Scholar] [CrossRef] [PubMed]
- Marott, S.C.; Nordestgaard, B.G.; Zacho, J.; Friberg, J.; Jensen, G.B.; Tybjaerg-Hansen, A.; Benn, M. Does elevated C-reactive protein increase atrial fibrillation risk? A mendelian randomization of 47,000 individuals from the general population. J. Am. Coll. Cardiol. 2010, 56, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Chew, D.P.; Bhatt, D.L.; Robbins, M.A.; Penn, M.S.; Schneider, J.P.; Lauer, M.S.; Topol, E.J.; Ellis, S.G. Incremental prognostic value of elevated baseline C-reactive protein among established markers of risk in percutaneous coronary intervention. Circulation 2001, 104, 992–997. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.; Genest, J.; Gotto, A.M.; Kastelein, J.J.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N. Engl. J. Med. 2008, 359, 2195–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Fauchier, L.; Pierre, B.; de Labriolle, A.; Grimard, C.; Zannad, N.; Babuty, D. Antiarrhythmic effect of statin therapy and atrial fibrillation a meta-analysis of randomized controlled trials. J. Am. Coll. Cardiol. 2008, 51, 828–835. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, D.R.; Andrade, C.X.; Chaparro, A.P.; Inostroza, C.M.; Ramirez, V.; Violant, D.; Nart, J. Short-term effects of 2% atorvastatin dentifrice as an adjunct to periodontal therapy: A randomized double blind clinical trial. J. Periodontol. 2015, 80, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Emami, H.; Vucic, E.; Singh, P.; Vijayakumar, J.; Fifer, K.M.; Alon, A.; Shankar, S.S.; Farkouh, M.; Rudd, J.H.; et al. High-dose atorvastatin reduces periodontal inflammation: A novel pleiotropic effect of statins. J. Am. Coll. Cardiol. 2013, 62, 2382–2391. [Google Scholar] [CrossRef] [PubMed]
- Lindhardsen, J.; Ahlehoff, O.; Gislason, G.H.; Madsen, O.R.; Olesen, J.B.; Svendsen, J.H.; Torp-Pedersen, C.; Hansen, P.R. Risk of atrial fibrillation and stroke in rheumatoid arthritis: Danish nationwide cohort study. BMJ 2012, 344, e1257. [Google Scholar] [CrossRef] [PubMed]
- Barnado, A.; Carroll, R.J.; Casey, C.; Wheless, L.; Denny, J.C.; Crofford, L.J. Phenome-wide association studies uncover a novel association of increased atrial fibrillation in males with systemic lupus erythematosus. Arthritis Care Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Irastorza, G.; Crowther, M.; Branch, W.; Khamashta, M.A. Antiphospholipid syndrome. Lancet 2010, 376, 1498–1509. [Google Scholar] [CrossRef]
- Kaya, Z.; Leib, C.; Katus, H.A. Autoantibodies in heart failure and cardiac dysfunction. Circ. Res. 2012, 110, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Tan, H.; Cheng, L.; He, M.; Wei, Q.; Tanguay, R.M.; Wu, T. Expression of heat shock proteins in myocardium of patients with atrial fibrillation. Cell Stress Chaperones 2007, 12, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Schäfler, A.E.; Kirmanoglou, K.; Balbach, J.; Pecher, P.; Hannekum, A.; Schumacher, B. The expression of heat shock protein 60 in myocardium of patients with chronic atrial fibrillation. Basic Res. Cardiol. 2002, 97, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Koutouzis, T.; Haber, D.; Shaddox, L.; Aukhil, I.; Wallet, S.M. Autoreactivity of serum immunoglobulin to periodontal tissue components: A pilot study. J. Periodontol. 2009, 80, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, R.I. Cells in stress: Transcriptional activation of heat shock genes. Science 1993, 259, 1409–1410. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, J.F.; Rôças, I.N. Bacterial pathogenesis and mediators in apical periodontitis. Braz. Dent. J. 2007, 18, 267–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goulhen, F.; Grenier, D.; Mayrand, D. Oral microbial heat-shock proteins and their potential contributions to infections. Crit. Rev. Oral Biol. Med. 2003, 14, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Schett, G.; Seitz, C.S.; Hu, Y.; Gupta, R.S.; Wick, G. Surface staining and cytotoxic activity of heat-shock protein 60 antibody in stressed aortic endothelial cells. Circ. Res. 1994, 75, 1078–1085. [Google Scholar] [CrossRef] [PubMed]
- Leishman, S.J.; Ford, P.J.; Do, H.L.; Palmer, J.E.; Heng, N.C.; West, M.J.; Seymour, G.J.; Cullinan, M.P. Periodontal pathogen load and increased antibody response to heat shock protein 60 in patients with cardiovascular disease. J. Clin. Periodontol. 2012, 39, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Ford, P.J.; Gemmell, E.; Hamlet, S.M.; Hasan, A.; Walker, P.J.; West, M.J.; Cullinan, M.P.; Seymour, G.J. Cross-reactivity of groel antibodies with human heat shock protein 60 and quantification of pathogens in atherosclerosis. Oral Microbiol. Immunol. 2005, 20, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, K.; Ohsawa, Y.; Tabeta, K.; Ito, H.; Ueki, K.; Oda, T.; Yoshie, H.; Seymour, G.J. Accumulation of human heat shock protein 60-reactive T cells in the gingival tissues of periodontitis patients. Infect. Immun. 2002, 70, 2492–2501. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Kiechl, S.; Mayr, M.; Metzler, B.; Egger, G.; Oberhollenzer, F.; Willeit, J.; Wick, G. Association of serum antibodies to heat-shock protein 65 with carotid atherosclerosis: Clinical significance determined in a follow-up study. Circulation 1999, 100, 1169–1174. [Google Scholar] [CrossRef] [PubMed]
- Grundtman, C.; Kreutmayer, S.B.; Almanzar, G.; Wick, M.C.; Wick, G. Heat shock protein 60 and immune inflammatory responses in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 960–968. [Google Scholar] [CrossRef] [PubMed]
- Mandal, K.; Jahangiri, M.; Xu, Q. Autoimmunity to heat shock proteins in atherosclerosis. Autoimmun. Rev. 2004, 3, 31–37. [Google Scholar] [CrossRef]
- Hansson, A.; Madsen-Härdig, B.; Olsson, S.B. Arrhythmia-provoking factors and symptoms at the onset of paroxysmal atrial fibrillation: A study based on interviews with 100 patients seeking hospital assistance. BMC Cardiovasc. Disord. 2004, 4, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czura, C.J.; Tracey, K.J. Autonomic neural regulation of immunity. J. Intern. Med. 2005, 257, 156–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.S.; Chen, L.S.; Fishbein, M.C.; Lin, S.F.; Nattel, S. Role of the autonomic nervous system in atrial fibrillation: Pathophysiology and therapy. Circ. Res. 2014, 114, 1500–1515. [Google Scholar] [CrossRef] [PubMed]
- Pongratz, G.; Straub, R.H. The sympathetic nervous response in inflammation. Arthritis Res. Ther. 2014, 16, 504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- January, C.T.; Wann, L.S.; Alpert, J.S.; Calkins, H.; Cigarroa, J.E.; Cleveland, J.C.; Conti, J.B.; Ellinor, P.T.; Ezekowitz, M.D.; Field, M.E.; et al. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: A report of the american college of cardiology/american heart association task force on practice guidelines and the heart rhythm society. Circulation 2014, 130, e199–e267. [Google Scholar] [CrossRef] [PubMed]
- Gould, P.A.; Yii, M.; McLean, C.; Finch, S.; Marshall, T.; Lambert, G.W.; Kaye, D.M. Evidence for increased atrial sympathetic innervation in persistent human atrial fibrillation. Pacing Clin. Electrophysiol. 2006, 29, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.A.; Hsieh, M.H.; Tai, C.T.; Tsai, C.F.; Prakash, V.S.; Yu, W.C.; Hsu, T.L.; Ding, Y.A.; Chang, M.S. Initiation of atrial fibrillation by ectopic beats originating from the pulmonary veins: Electrophysiological characteristics, pharmacological responses, and effects of radiofrequency ablation. Circulation 1999, 100, 1879–1886. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.; Chen, P.S. Ligament of marshall: Why it is important for atrial fibrillation ablation. Heart Rhythm. 2009, 6, S35–S40. [Google Scholar] [CrossRef] [PubMed]
- Pokushalov, E.; Romanov, A.; Elesin, D.; Bogachev-Prokophiev, A.; Losik, D.; Bairamova, S.; Karaskov, A.; Steinberg, J.S. Catheter versus surgical ablation of atrial fibrillation after a failed initial pulmonary vein isolation procedure: A randomized controlled trial. J. Cardiovasc. Electrophysiol. 2013, 24, 1338–1343. [Google Scholar] [CrossRef] [PubMed]
- Bettoni, M.; Zimmermann, M. Autonomic tone variations before the onset of paroxysmal atrial fibrillation. Circulation 2002, 105, 2753–2759. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Kalusche, D. Fluctuation in autonomic tone is a major determinant of sustained atrial arrhythmias in patients with focal ectopy originating from the pulmonary veins. J. Cardiovasc. Electrophysiol. 2001, 12, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Leinhardt, D.J.; Arnold, J.; Shipley, K.A.; Mughal, M.M.; Little, R.A.; Irving, M.H. Plasma NE concentrations do not accurately reflect sympathetic nervous system activity in human sepsis. Am. J. Physiol. 1993, 265, E284–E288. [Google Scholar] [CrossRef] [PubMed]
- Walkey, A.J.; Wiener, R.S.; Ghobrial, J.M.; Curtis, L.H.; Benjamin, E.J. Incident stroke and mortality associated with new-onset atrial fibrillation in patients hospitalized with severe sepsis. JAMA 2011, 306, 2248–2254. [Google Scholar] [CrossRef] [PubMed]
- Kindem, I.A.; Reindal, E.K.; Wester, A.L.; Blaasaas, K.G.; Atar, D. New-onset atrial fibrillation in bacteremia is not associated with C-reactive protein, but is an indicator of increased mortality during hospitalization. Cardiology 2008, 111, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Im, S.I.; Heo, J.; Kim, B.J.; Cho, K.I.; Kim, H.S.; Heo, J.H.; Hwang, J.Y. Impact of periodontitis as representative of chronic inflammation on long-term clinical outcomes in patients with atrial fibrillation. Open Heart 2018, 5, e000708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okazaki, R.; Iwasaki, Y.K.; Miyauchi, Y.; Hirayama, Y.; Kobayashi, Y.; Katoh, T.; Mizuno, K.; Sekiguchi, A.; Yamashita, T. Lipopolysaccharide induces atrial arrhythmogenesis via down-regulation of L-type Ca2+ channel genes in rats. Int. Heart J. 2009, 50, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Reitz, B.A.; Prager, D.J.; Feigen, G.A. An analysis of the toxic actions of purified streptolysin O on the isolated heart and separate cardiac tissues of the guinea pig. J. Exp. Med. 1968, 128, 1401–1424. [Google Scholar] [CrossRef] [PubMed]
- Zijnge, V.; Kieselbach, T.; Oscarsson, J. Proteomics of protein secretion by aggregatibacter actinomycetemcomitans. PLoS ONE 2012, 7, e41662. [Google Scholar] [CrossRef] [PubMed]
- Stobernack, T.; Glasner, C.; Junker, S.; Gabarrini, G.; de Smit, M.; de Jong, A.; Otto, A.; Becher, D.; van Winkelhoff, A.J.; van Dijl, J.M. Extracellular proteome and citrullinome of the oral pathogen Porphyromonas gingivalis. J. Proteome Res. 2016, 15, 4532–4543. [Google Scholar] [CrossRef] [PubMed]
- Baka, Z.; György, B.; Géher, P.; Buzás, E.I.; Falus, A.; Nagy, G. Citrullination under physiological and pathological conditions. Jt. Bone Spine 2012, 79, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.R.; Fast, W. Histone citrullination by protein arginine deiminase: Is arginine methylation a green light or a roadblock? ACS Chem. Biol. 2006, 1, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Mangat, P.; Wegner, N.; Venables, P.J.; Potempa, J. Bacterial and human peptidylarginine deiminases: Targets for inhibiting the autoimmune response in rheumatoid arthritis? Arthritis Res. Ther. 2010, 12, 209. [Google Scholar] [CrossRef] [PubMed]
- Koziel, J.; Mydel, P.; Potempa, J. The link between periodontal disease and rheumatoid arthritis: An updated review. Curr. Rheumatol. Rep. 2014, 16, 408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quirke, A.M.; Lugli, E.B.; Wegner, N.; Hamilton, B.C.; Charles, P.; Chowdhury, M.; Ytterberg, A.J.; Zubarev, R.A.; Potempa, J.; Culshaw, S.; et al. Heightened immune response to autocitrullinated Porphyromonas gingivalis peptidylarginine deiminase: A potential mechanism for breaching immunologic tolerance in rheumatoid arthritis. Ann. Rheum. Dis. 2014, 73, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Mikuls, T.R.; Payne, J.B.; Yu, F.; Thiele, G.M.; Reynolds, R.J.; Cannon, G.W.; Markt, J.; McGowan, D.; Kerr, G.S.; Redman, R.S.; et al. Periodontitis and Porphyromonas gingivalis in patients with rheumatoid arthritis. Arthritis Rheumatol. 2014, 66, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Bartold, P.M.; Marshall, R.I.; Haynes, D.R. Periodontitis and rheumatoid arthritis: A review. J. Periodontol. 2005, 76, 2066–2074. [Google Scholar] [CrossRef] [PubMed]
- Konig, M.F.; Abusleme, L.; Reinholdt, J.; Palmer, R.J.; Teles, R.P.; Sampson, K.; Rosen, A.; Nigrovic, P.A.; Sokolove, J.; Giles, J.T.; et al. Aggregatibacter actinomycetemcomitans-induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci. Transl. Med. 2016, 8, 369ra176. [Google Scholar] [CrossRef] [PubMed]
- Geraldino-Pardilla, L.; Russo, C.; Sokolove, J.; Robinson, W.H.; Zartoshti, A.; Van Eyk, J.; Fert-Bober, J.; Lima, J.; Giles, J.T.; Bathon, J.M. Association of anti-citrullinated protein or peptide antibodies with left ventricular structure and function in rheumatoid arthritis. Rheumatology 2017, 56, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Fert-Bober, J.; Giles, J.T.; Holewinski, R.J.; Kirk, J.A.; Uhrigshardt, H.; Crowgey, E.L.; Andrade, F.; Bingham, C.O.; Park, J.K.; Halushka, M.K.; et al. Citrullination of myofilament proteins in heart failure. Cardiovasc. Res. 2015, 108, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.B.; Thorolfsdottir, R.B.; Fritsche, L.G.; Zhou, W.; Skov, M.W.; Graham, S.E.; Herron, T.J.; McCarthy, S.; Schmidt, E.M.; Sveinbjornsson, G.; et al. Genome-wide association study of 1 million people identifies 111 loci for atrial fibrillation. bioRxiv 2018, 242149. [Google Scholar] [CrossRef]
- Nielen, M.M.; van Schaardenburg, D.; Reesink, H.W.; van de Stadt, R.J.; van der Horst-Bruinsma, I.E.; de Koning, M.H.; Habibuw, M.R.; Vandenbroucke, J.P.; Dijkmans, B.A. Specific autoantibodies precede the symptoms of rheumatoid arthritis: A study of serial measurements in blood donors. Arthritis Rheum. 2004, 50, 380–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cytokine | Function |
|---|---|
| IL-8, MIP-1, MCP-1, RANTES | Chemotactic |
| IL-1α, IL-1β, TNFα, IL-6, PAF | Pro-inflammatory |
| IL-1RA, IL-4, IL-10 | Anti-inflammatory |
| IFN-γ, IL-2, IL-4, IL-5, IL-7 | Immunoregulatory |
| PDGF, EGF, FGF, IGF, VEGF | Growth factor |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aarabi, G.; Schnabel, R.B.; Heydecke, G.; Seedorf, U. Potential Impact of Oral Inflammations on Cardiac Functions and Atrial Fibrillation. Biomolecules 2018, 8, 66. https://doi.org/10.3390/biom8030066
Aarabi G, Schnabel RB, Heydecke G, Seedorf U. Potential Impact of Oral Inflammations on Cardiac Functions and Atrial Fibrillation. Biomolecules. 2018; 8(3):66. https://doi.org/10.3390/biom8030066
Chicago/Turabian StyleAarabi, Ghazal, Renate B. Schnabel, Guido Heydecke, and Udo Seedorf. 2018. "Potential Impact of Oral Inflammations on Cardiac Functions and Atrial Fibrillation" Biomolecules 8, no. 3: 66. https://doi.org/10.3390/biom8030066